

Abstract
The synthesis of N-arylamide phosphonates and related arylether and arylamine analogues provided potent, subtype-selective agonists and antagonists of the five known sphingosine 1-phosphate (S1P) receptors (S1P1–5). To this end, the syntheses of phosphoserine mimetics - selectively protected and optically active phosphonoserines - are described. In vitro binding assays showed that the implementation of phosphonates as phosphate mimetics provided compounds with similar receptor binding affinities as compared to their phosphate precursors. Meta-substituted arylamide phosphonates were discovered to be antagonists of the S1P1 and S1P3 receptors. When administered to mice, an antagonist blocked the lymphopenia evoked by a S1P receptor agonist and caused capillary leakage in both lung and kidney.
Keywords: Sphingosine 1-phosphate, VPC23019, VPC44116, FTY720, Immune-modulation
1. Introduction
Sphingosine 1-phosphate (S1P, Figure 1) receptors (S1P1–5) are integral membrane G protein-coupled receptors that were initially referred to as endothelial differentiation gene (EDG) receptors, EDG-1, -5, -3, -6 and -8, respectively.1 These receptors provide control over numerous aspects of cellular physiology when activated by endogenous S1P.2,3 Particular attention has been accorded to the role of the S1P1 receptor in modulating the immune system since the discovery of FTY720, an agonist at the S1P1,3,4,5 receptors (Figure 1). S1P1 receptor agonists have been shown to inhibit the egress of T-lymphocytes from secondary lymphoid tissues, and thus are thought to direct effector T-cells away from sites of inflammation.4
Figure 1.
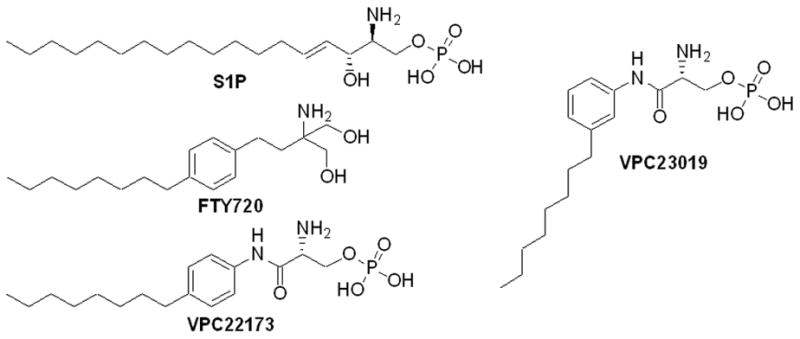
Structures of endogenous sphingosine 1-phosphate (S1P) and the S1P receptor ligands, FTY720, VPC22173 and VPC23019.
FTY720 was developed from synthetic analog studies of myriocin (ISP-1).5 Eventually discovered to be a pro-drug, FTY720 is activated in vivo when phosphorylated by sphingosine kinase type 2 to form FTY720-P.6 The biological activity of FTY720 indicated that S1P receptors are valid targets for the treatment of autoimmune disorders and allograft rejection.7 It is hoped that S1P receptor agonists can modulate immune system function without the toxic liabilities attendant to existing immuno-therapeutics such as calcineurin inhibitors and corticosteroids.7–9
The success of FTY720 in human investigation has prompted synthetic efforts to provide both pharmacological tools to study S1P signaling and therapeutics.10–15 We reported previously the synthesis of aryl-amide containing phosphates (Figure 1, VPC22173 and VPC23019) and profiled their binding affinities at S1P receptors in vitro.14,16 Among these compounds were the first S1P1 receptor antagonists, e.g. VPC23019.16 In an effort to discover compounds with increased resistance to phosphatase-catalyzed hydrolysis (the deactivation pathway of S1P analogs), the synthesis of the corresponding phosphonates is reported herein. Further, the synthesis of related aryl-amine and aryl-ether containing phosphonates is discussed.
To initiate this work, strategies were pursued for the efficient synthesis of chiral phosphonoserines 4a and 4b, which are non-natural amino acids used to study protein phosphorylation.17 Previous syntheses of note include Barton and Vonder-Embse’s synthesis of the fully unprotected phosphonoserine from N-Cbz-glutamic acid in 4 steps and 58% yield, involving the use of white phosphorous (P4).18 Perich and Johns published two syntheses, the most recent in 42% yield and seven steps, from properly protected glutamic acid and using a Barton – McCombie deoxygenation.19 Finally, other methods employed include enzymatic chiral resolutions of racemic materials, 20–22 and the induction of chirality by chiral auxiliaries.23–25 Methods reported in this paper lead to chiral phosphonoserines with protecting groups amenable to our synthetic approach, as well as peptide synthesis, in good yield from commercially available L- or D- serine and R- or S-glycidol.
Derived from protected R- and S- phosphonoserines 4a and 4b, N-aryl-amide phosphonates 12a–f provided similar binding affinities at S1P receptors as their phosphate precursors. Synthesis of the α-fluorophosphonate 13 showed similarly potent binding compared to its corresponding phosphonate 12a. Phosphonate analogues 12c, 12d (VPC44116) and 12f, of our previously described S1P1,3 antagonists,16 proved to retain their activity as antagonists and provided pharmacological tools for in vivo studies. While a new class of arylether phosphonates 18a, 18b and 19 were relatively weak partial agonists or inactive, the aryl-amine 26 retained similar activity to its amide precursor.
2. Results and Discussion
2.1 Chemistry
2.1.1 Synthesis of aryl-amide-phosphonates 12a–f and 13
The production of phosphonate analogues containing an amide linker region was envisaged through the condensation of chiral phosphonoserines (L-or D-2-(N-tert-butoxycarbonyl amine)-4-phosphonyl butyric acids) 4a and 4b with various substituted anilines. The initial efforts (Scheme 1) towards this protected unnatural amino acid began with the synthesis of Garner’s Aldehyde, 1, from commercially available L-serine over five steps.26 A Horner-Wadsworth-Emmons olefination performed from one of two bis-phosphonates installed the α-methylene or α-fluorophosphonates in 2a or 2b, respectively.27 The fluorinated bisphosphonate used to synthesize 2b was derived, as previously described by Prestwich,28 from the commercially available tetraethyl methylenebisphosphonate used to arrive at 2a.
Scheme 1.
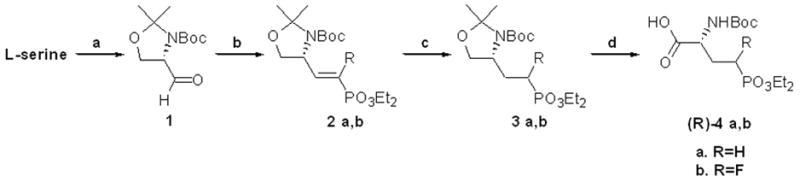
Synthesis of (3R)-4a,b – Method A: a.) i. SOCl2, MeOH, rt, 16h ii. Boc2O, Et3N, CH2Cl2, rt, 12h iii. 2,2-dimethoxypropane, p-TsOH, CH2Cl2, 0 °C to rt, 2h 62% (3 steps) iv. NaBH4, LiCl, 3:2 EtOH/THF, 0 °C to rt, 4h 89% v. DMSO, (COCl)2, CH2Cl2, −78 °C, then Et3N −78 °C to rt, 2–4 h, 97% b.) tetraethyl methylenebisphosphonate, n-BuLi, THF, −78 °C, rt, overnight, 75% (2a) or tetraethyl 2-fluoromethynebisphosphonate, n-BuLi, THF, -78 °C, rt, overnight, 31% (2b) c.) H2, Pd/C, rt., 12h, EtOH, 99% (3a) and 88% (3b) d.) Jones reagent, acetone, 0 °C to rt., 12h then, isopropyl alcohol, celite, rt, 15min., 59% (4a) and 48% (4b).
The resulting olefins 2a and 2b were reduced by hydrogenation over Pd/C to 3a and 3b. Selective acetonide deprotection proved to be low yielding in our initial efforts;29 however, this led to the use of a convenient, simultaneous deprotection and oxidation with the Jones reagent in acetone.30 This un-optimized method led to the protected amino-acids (R)-4a,b in 40% and 18% yields, respectively, in 3 steps from Garner’s Aldehyde.
A second avenue (Scheme 2) led to a shorter and more efficient synthesis of both (S)- and (R)-4a. For example, (R)-(+)-glycidol was benzyl protected,31 and the epoxide was opened by the resultant carbanion of diethyl methylphosphonate and n-BuLi, in the presence of BF3·OEt2, to yield alcohol 6.32 Installation of an azide at the 3-hydroxyl position proved to be the major impediment to this approach, as has been noted by others for similar substrates.33 Mesylate formation followed by azide substitution suffered from a tendency for elimination over a range of temperatures. Therefore, a milder method of azide formation was pursued.
Scheme 2.
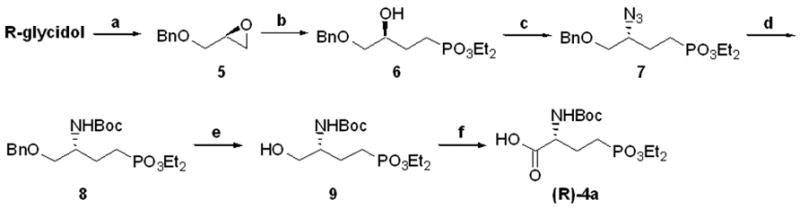
Synthesis of (3R)-[or (3S)-]4 – Method B: a.) BnBr, DMF, 60% NaH, 0 °C to rt, 3h, 78% b.) CH3PO3Et2, n-BuLi, BF3·OEt2, THF, −78 °C to rt, 3h then, NH4Cl, 1h, 96% c.) DPPA, DIAD, 3%-polymer-bound PhPPh2, CH2Cl2, 0 °C to rt, 20h, 96%, d.) Boc2O, H2 (balloon), 20%w/w Lindlar’s catalyst, MeOH, rt, 24h 77% e.) H2 (balloon), Pd/C, EtOH, rt, 24h, 94% f.) TEMPO, bis(acetoxy)iodosobenzene, NaHCO3, 1:1 CH3CN/H2O, rt, 3h., 38% or RuCl3·hydrate, NaIO4, 3:2:2 H2O/CH3CN/CCl4, rt, 3h, 76%.
Diphenylphosphoryl azide (DPPA) under Mitsunobu conditions was successful in assembling the desired azides, 7, despite reported difficulties arising from congested secondary alcohols.34 After extensive isolation efforts, near quantitative yields were found via this procedure. Purification issues led to the investigation of more convenient Mitsunobu reagents to preclude the difficult separation of polar products from the complex reaction mixture (polar phosphonate 7 was nearly inseparable from both triphenyl- and tributylphosphane oxides). The use of commercially available polymer-bound triphenylphosphine yielded equivalent conversions to azide 7. With the use of three equivalents of phosphine, the desired transformation was completed in fewer than 20 hours. The inclusion of a polymer-bound phosphine reagent allowed for standard purification of the crude reaction mixture, after filtration of the resultant phosphane oxide.
A two-step reduction of 7, first in the presence of H2, Boc2O and Lindlar’s catalyst,35 followed by H2, Pd/C, yielded the protected amino alcohol 9 in 72% over two steps. Oxidation of the primary alcohol was performed by means of RuCl3(cat.)/NaIO4 conditions36 (76%) to yield compound (R)-4a in 40% yield over 6 steps from optically active glycidol. (R)- and (S)-4a were further derived to the corresponding diastereomers, (R)- and (S)-4a-Phe-OMe, through a PyBOP mediated condensation with L-phenylalanine methyl ester.38 This was done to ensure high enantiomeric excess of the desired carboxylic acids. These condensation reactions were high yielding and arrived at individual stereoisomers as determined by NMR.
Compounds 12a-f and 13 (Scheme 3) were synthesized by PyBOP initiated condensations37 of (R)- or (S)-4a or (R)-4b with various aniline compounds 10a–d. Aqueous soluble carbodiimide (EDC methiodide) was also investigated as a condensation reagent; however, this method generally produced lower yields. The alkane hydrocarbons of unavailable alkylanilines were installed through Sonogashira couplings with 3-iodo-nitrobenzene,38 followed by concomitant reduction of the nitro group and resulting triple bond. Pd couplings were also successfully performed following the amide formation with m- or p- iodoaniline to complete the desired phosphonates in a linear fashion. The compounds 11a–f were deprotected with bromotrimethylsilane followed by hydrolysis of the ensuing phosphonate silyl-oxy-esters. These conditions conveniently deprotected the N-Boc group, as well, to yield compounds 12a–f. α-Fluorophosphonate 13 was synthesized by the same protocol from 4b and 10a.
Scheme 3.
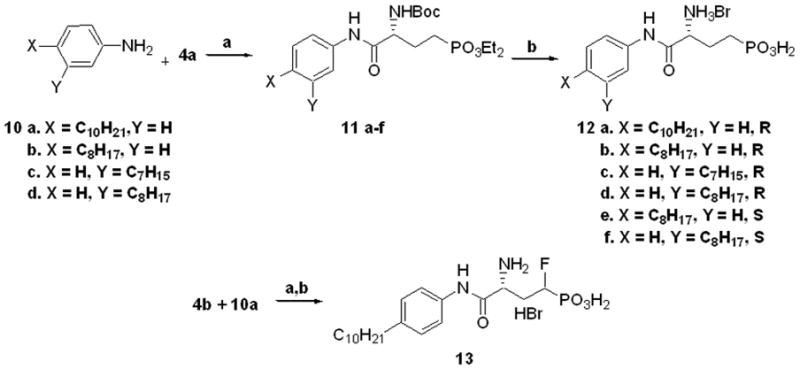
Synthesis of arylamide phosphonates 12 a–f & 13: a) PyBOP, 10 a–d, di-iso- propyl ethylamine (DIEA), CH2Cl2, 24–70% b.) TMSBr, CH2Cl2, rt, 4-6h then, 95:5 MeOH/H2O, rt, 1–4h, 45-100%.
While the synthesis described from glycidol retained the reported efficiency up to half-gram scale, increasing material to greater than one gram proved detrimental to the formation of azide 7. Due to the demand for greater quantities of compounds 13b and 13d, further optimization of our synthesis from serine was undertaken (Scheme 4). Acetonide protected vinyl phosphonate 2 was converted to phosphonoserine 4a in two convenient steps. Compound 2, as displayed in Scheme 1, undergoes reduction within four to six hours under an atmosphere of H2 and in the presence of 10% Pd/C (20w/w %). It was observed that a trace amount of the alcohol 9a was present at this time. The reaction was allowed to stir for one day at room temperature, and it was estimated by TLC that nearly half of the acetonide was hydrolyzed. Following further investigation, the use of an additional half equivalent of 10% Pd/C (10w/w%), added after one day of vigorous stirring, converted the remaining material to the reduced alcohol 9a in less than 72 hours.39 This selective acetonide deprotection proved to be effective with greater than five grams of material and yielded more efficient conversions (>95%) than 1.5 equivalents of p-toluenesulfonic acid in ethanol (65–75 %). The alcohol was then converted to 4a with RuCl3/NaIO4 conditions, as previously described.
Scheme 4.
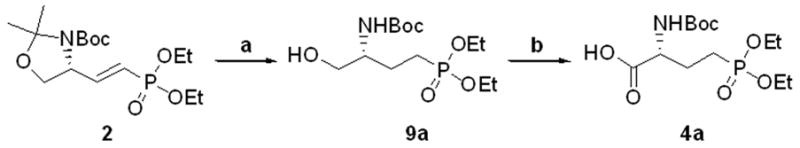
Efficient synthesis of 4a from 2a: a.) H2, 10% Pd/C (20 w/w% followed by 10 w/w%), EtOH, rt, 3d, >95% b.) RuCl3·H2Ox, NaIO4, 2:2:3 CCl4/CH3CN/H2O, rt, 1–3h, 79%.
2.1.2 Synthesis of aryl ether phosphonates 18a, 18b, and 19
Using glycidol as an alternative starting material to serine led conveniently to phenolic ether compounds (Scheme 5). The synthesis began with a Mitsunobu condensation between p-octylphenol and glycidol. Chiral epoxides 14 were then opened, as previously described, to alcohols 15. For these substrates, DPPA/Mitsunobu conditions yielded products with sufficiently disparate polarities from tributylphosphane oxide. The azides, 16, were reduced and deprotected as described above to give enantiomers 18a and 18b. Compound 15a was deprotected to form 19 to confirm the overall effects of an amine at the 3-position of 18b.
Scheme 5.
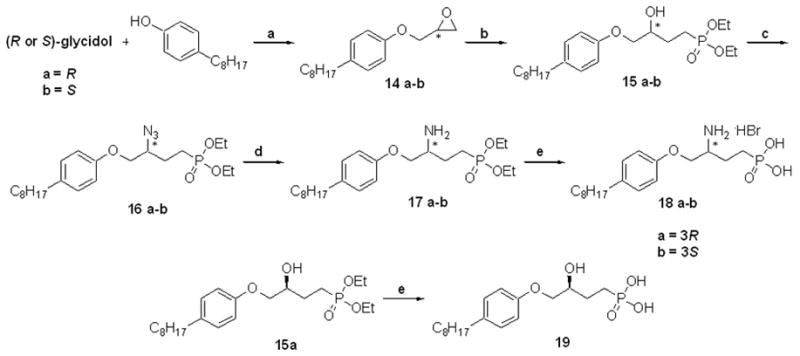
Synthesis of arylether phosphonate analogues 18 a–b and 19: a.) DIAD, PPh3, THF, 0 °C to rt, overnight, 80–84% b.) CH3PO3Et2, n-BuLi, BF3·OEt2, THF, −78 °C to rt, 3h then, NH4Cl, 1h, 72–87% c.) DPPA, DIAD, PPh3, CH2Cl2, 0 °C to rt, 20h, 67–98% d.) H2 (balloon), Pd/C, EtOH, formic acid cat., rt, 3.5h, quantitative e.) TMSBr, CH2Cl2, rt, 4–6h then, 95:5 MeOH/H2O, rt, 2–4h, 79–100%.
2.1.3 Synthesis of aryl amine phosphonate 26
Secondary amine 26 was synthesized to view the effects of reducing the amide bond in 12d (Scheme 6) while retaining a functional group capable of donating a hydrogen bond. After activation of p-octylaniline by mono-tosyl protection, the amine was condensed under Mitsunobu conditions to form epoxide 21. Consecutive nucleophilic ring opening, azide formation with DPPA/Mitsunobu conditions, and reduction yielded amine 24. Deprotection of the N-tosyl group proved difficult. Sluggish reaction times and low yields were discovered with the planned Mg/MeOH deprotection,41 and dissolving metal conditions were used. Solid sodium in NH3(l) unmasked the desired secondary amine in an un-optimized 26% yield. Compound 25 was deprotected with TMSBr to yield the ammonium bromide salt 26 .
Scheme 6.
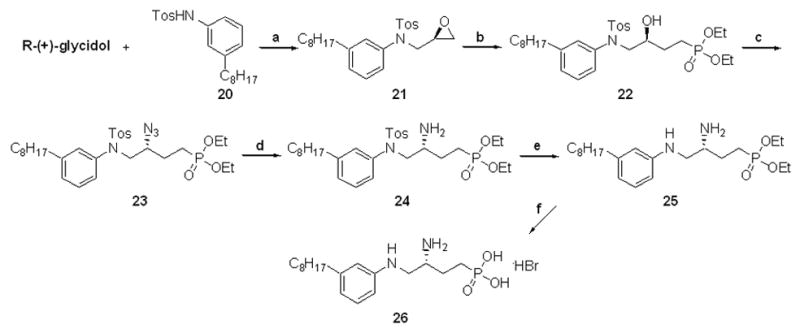
Synthesis of arylamine phosphonate analogue 26: a.) DIAD, PPh3, THF, 0 °C to rt, overnight, 69% b.) CH3PO3Et2, n-BuLi, BF3·OEt2, THF, –78 °C to rt, 2h then, NH4Cl, 2h, 97% c.) DPPA, DIAD, PPh3, CH2Cl2, 0 °C to rt, 20h, 85% (containing 5% OPPh3) d.) H2 (balloon), 20%w/w Pd(OH)2, 20:1 MeOH/conc. HCl, 1h, 100% e.)Na(s), NH3(l), –78 °C, 5 min.then EtOH, 25% (recovered 28% starting material) f.) TMSBr, CH2Cl2, rt, 4–6h then, 95:5 MeOH/H2O, 4h, rt, 95%.
2.2 Biological Evaluation
2.2.1 [γ35S]-GTP Binding Assay
γ35S-GTP dependant receptor binding activity (Table 1) was determined in vitro for S1P, FTY720-P, and all final compounds, as previously communicated.16 Briefly, the expression of individual human S1P receptors and individual G protein subunits was forced in HEK293T cells. The membrane bound G protein α subunits yielded data by binding the labeled, non-hydrolyzable [γ35S]-GTP when activated by an extracellular ligand. Following our discovery of the S1P1,3 antagonist VPC23019 (B), the meta-substituted analogues were analyzed for their ability to antagonize S1P’s endogenous activity in the [γ35S]-GTP assays. The effects on S1P’s endogenous binding constant were determined as previously discussed.16
Table 1.
[γ-35S]-GTP binding assay in HEK293T cells over-expressed with subtype specific S1P receptors.a
| Receptors | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1P1 | S1P2 | S1P3 | S1P4 | S1P5 | ||||||||
| Compound | linker | Head Group | EC50a | Emax | EC50 | Emax | EC50 | Emax | EC50 | Emax | EC50 | Emax |
| FTY720-P | H2CCH2 | phosphate | 1.3 | 1.00 | NA | 0.00 | 0.1 | 0.50 | 4.0 | 0.90 | 4.0 | 0.56 |
| VPC22173 | amide | phosphate | 58.0 | 1.10 | NA | 0.00 | 450.0 | 0.35 | 500.0 | 1.10 | 52.0 | 0.79 |
| VPC2301S | amide | phosphate | NA | 0.00 | NA | 0.00 | NA | 0.00 | 120.0 | 1.13 | 480.0 | 0.57 |
| 12 a | amide | phosphonate | 3.6 | 0.96 | 270.0 | 0.50 | 43.0 | 0.88 | 230.0 | 0.67 | 30.0 | 0.77 |
| b | amide | phosphonate | 27.0 | 0.93 | NA | 0.00 | 270.0 | 0.38 | 2,300.0 | 0.80 | 76.0 | 0.64 |
| c | amide | phosphonate | NA | 0.00 | n/a | n/a | NA | 0.00 | n/a | n/a | n/a | n/a |
| d | amide | phosphonate | NA | 0.00 | NA | 0.00 | NA | 0.00 | 6,100.0 | 1.42 | 33.0 | 0.73 |
| 6 | amide | phosphonate | 1200.00 | NA | NA | 0.00 | 15000 | 0.30 | n/a | n/a | n/a | n/a |
| f | amide | phosphonate | NA | 0.00 | NA | 0.00 | NA | 0.00 | n/a | n/a | n/a | n/a |
| 13 | amide | fluorophosphonate | 2.1 | 0.99 | 490.0 | 0.47 | 23.0 | 0.96 | 170.0 | 0.68 | 19.0 | 0.77 |
| 18 a | ether | phosphonate | 68.0 | 0.70 | NA | 0.00 | NA | 0.00 | 147.0 | 0.71 | 31.0 | 0.41 |
| b | ether | phosphonate | 140.0 | 0.64 | 530.0 | 0.50 | 100.0 | 0.53 | 150.0 | 0.90 | 47.0 | 0.50 |
| 19 | ether | phosphonate | 33.0 | 0.43 | NA | 0.00 | NA | 0.00 | 440,000.0 | 0.45 | NA | 0.00 |
| 26 | amine | phosphonate | 30.0 | 0.56 | NA | 0.00 | NA | 0.00 | 340.0 | 1.18 | 41.0 | 0.61 |
EC50s are nM and determined by the mean of at least three experiments.
Emax values are normalized to the maximal activation of endogenous S1P, at each receptor.
NA = no activation
n/a = not available
Para substituted phosphonates 12a, 12b (VPC44152), 12e, 18a, 18b, and 19 showed various activities as agonists. Phosphonate 12b (VPC44152) was twice as potent as corresponding phosphate A (VPC22173) at S1P1 and S1P3, while less potent at S1P4 and S1P5. Phosphonate 12a gained activity across all receptors with comparison to A and displayed similar potency to FTY720-P and S1P at S1P1. The replacement of the amide linkage with an ether resulted in the loss of activity, at S1P1 and S1P3, for 18a compared to 12b, implicating the importance of available hydrogen-bond donation alpha to the phenyl ring. Interestingly, epimer 18b was considerably less potent than 18a at S1P1 but displayed modest activity at all five S1P receptors. γ-Hydroxyphosphonate 19 was less potent than 18a at S1P1 and functionally inactive at S1P2–5, which is consistent with the two point binding model for S1P receptor interaction.42 Compared with our previously described phosphate agonist A, phosphonates 12a, 12b (VPC44152), and 12e retained similar potency and efficacy. Meta-substituted compounds 12c, 12d (VPC44116), and 12f showed no agonist activity at S1P1 and S1P3 receptors; rather, meta substituted compounds displayed antagonists activity against S1P binding to the S1P1 and S1P3 receptors.
To characterize these compounds, Schild regressions were performed as described in earlier work.16 These experiments revealed arylamides 12d and 12f as potent antagonists at the S1P1 and S1P3 receptors. Arylamine 26 displayed antagonist activity at both receptors with a preference for S1P3. The most promising antagonist, 12d (VPC44116), was compared with its phosphate precursor VPC23019. VPC23019 and VPC44116 were nearly indistinguishable in their affinity for the S1P1 and S1P3 receptors (Ki values of about 30nM and 300nM, respectively). This was described in more detail by radioligand displacement experiments, described previously,16 revealing IC50s for the phosphate and phosphonate to be 31 nM and 72 nM, respectively (not shown).
We have demonstrated previously that VPC44116 opposes the protective effect of FTY720 in a mouse model of acute renal injury.43 To characterize this compound further, we injected mice with doses up to 45 mg/kg body weight and measured blood lymphocytes. Lymphopenia (a decrease in circulating lymphocytes below the normal range) is a convenient index of S1P1 receptor agonist action. The quintessential S1P agonist, FTY720, has been proposed to operate as a functional antagonist through receptor desensitization mechanisms44 – a hypothesis that suggests a direct receptor antagonist would behave likewise. Nevertheless, no significant change in circulating lymphocyte numbers was observed at any dose of VPC44116 tested (not shown). However, 12d (VPC44116 (meta)) blocked the lymphopenia evoked by its positional isomer, the agonist 12b (VPC44152 (para)) (Figure 2). A S1P receptor antagonist similar to VPC44116 but containing a hexyl (vs. octyl in VPC44116) group caused vascular leakage in lung when administered to mice.45 To learn whether VPC44116 behaved similarly, we injected VPC44116 into mice followed by Evans blue dye as described previously.43 After sacrifice, we found extravasation of the dye into lung and kidney, but not into heart, liver, testes or intestine (Figure 3).
Figure 2.
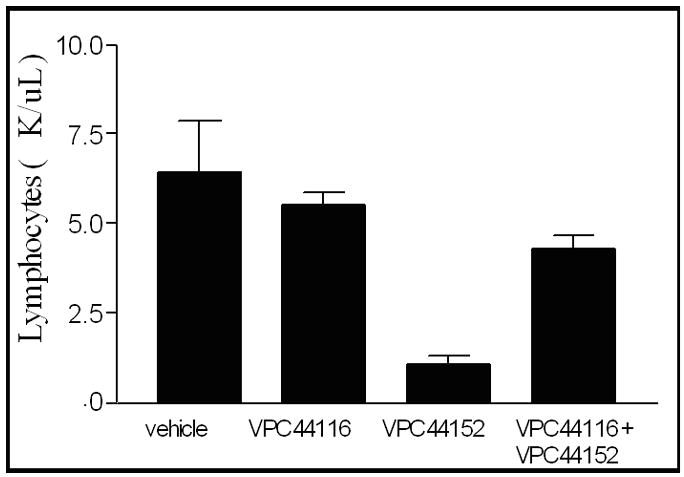
The lymphopenia evoked by the S1P agonist, 12b (VPC44152) is blocked by co-administration of the S1P receptor antagonist 12d (VPC44116). Groups of 3 C57BL/6 x sv129/J mice injected with vehicle (2% hydroxypropyl β-cyclodextrin), VPC44116 (22 mg/kg) and/or VPC44152 (18 mg/kg). After 16 hours, blood was drawn from the orbital sinuses and lymphocytes were measure with a Hemavet blood analyzer. Data are presented as mean ± S.E.
Figure 3.
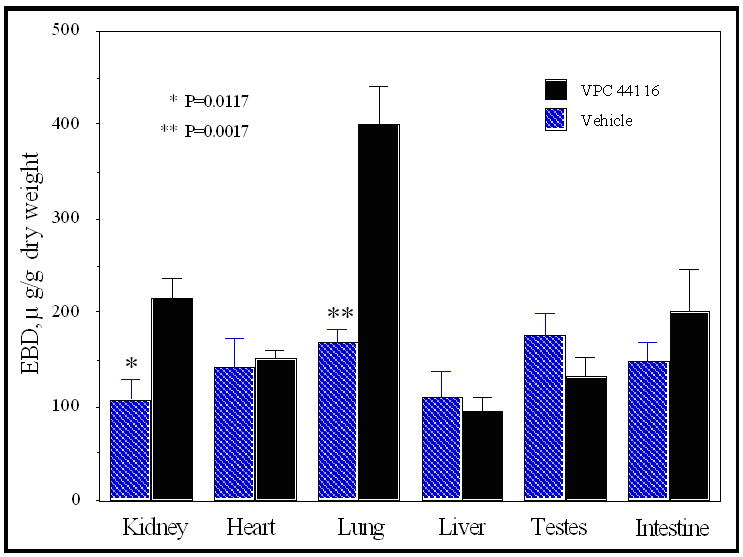
Effect of VPC44116 on vascular permeability. C57BL/6 mice were treated with vehicle (2% hydroxypropyl β-cyclodextrin) or VPC44116 (25 mg/kg) two hours prior to injection 2% Evans Blue dye (EBD) (20 mg/kg) into the jugular vein 30 min. before harvesting tissues. EBD was extracted into formamide, measured in a spectrometer and the amount of extravasated EBD in tissues was calculated from a standard curve. Values are means ± SE; n = 4 for each group. **P < 0.05, **P < 0.01 compared with vehicle treatment.
3. Conclusion
The synthetic methods described provide entry to multiple oxidation states and/or orthogonal protecting groups of phosphonoserines, including an isoelectric α-fluorophosphonate. This is accomplished from two convenient materials of the current chiral pool, L- or D- serine (eight steps and 39% yield) and R- or S- glycidol (six steps and 40% yield). Optically active agonists and antagonists of the S1P receptors are described. Replacing the phosphorous-oxygen bond with a phosphorous-carbon bond provides bioactive agents with putative resistance to degradative phosphatase activity. The phosphonate-containing antagonist, 12d (VPC44116), opposed the lymphopenia evoked by its agonist isomer, 12b (VPC44152), but did not affect numbers of circulating lymphocytes when injected alone. However, injection of VPC44116 alone caused capillary leakage in lung and kidney.
4. Experimental
4.1. General Experimental
All reactions were performed under an inert atmosphere using flame dried glassware. Reaction solvents methylene chloride, diethyl ether, tetrahydrofuran, and toluene were obtained from OptiDry canisters (<50 ppm H2O, Fisher Scientific) and passed through an activated alumina (activity I) column, directly into the reaction flask when possible. Dimethylformamide was obtained from an OptiDry canister without further drying prior to use. All other solvents were used as obtained. All commercially available reagents were purchased from either Aldrich (Milwaukee, WI), Sigma (St. Louis, MO), Acros (Pittsburg, PA), or Advanced Chem Tech (Louisville, KY) and used as obtained unless otherwise stated. The reactions were monitored by analytical thin layered chromatography using Merck silica gel F-254 pre-coated aluminum-backed plates. Rf values refer to column chromatography eluent, unless otherwise noted. When not reported, Rf value ≈ 0.00. Silicycle Ultra Pure Silica Gel (230–400 mesh) or Fisher Scientific Silica Gel 60 Sorbent (230–400 mesh) was used for all normal phase chromatography. All yields refer to chromatographically and spectroscopically pure compounds, unless otherwise indicated.
Optical rotations were measured on a Perkin-Elmer model 343 polarimeter with a sodium lamp at 23 ± 2 °C in the stated solvent; [α]D values are given in 10−1 deg cm2 g−1. 1H and 13C NMR spectra were recorded on UnityInova 300 (75) and 500 (125) MHz spectrometers (Varian). Chemical shifts are reported in δ (ppm) units using 1H (residual) and 13C signals from CDCl3 as an internal standard (7.26 and 77.23 ppm, respectively) unless otherwise specified. Elemental analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) for C, H, and N. Elemental analyses were run in duplicate after thorough drying, before submission and when obtained by the vendor. Low Resolution Electrospray Ionization (ESI) was performed at the University of Virginia Mass Spectometry Laboratory. High-Resolution Mass Spectrometry (HRMS) was performed at the Mass Spectrometry Laboratory at University of Illinois Urbana-Champaign (Micromass Q-T of Ultima).
4.1.1. 4-formyl-2,2-dimethyloxazolidine-3-carboxylic acid tert-butyl ester (1)
L-serine (5.00 g, 0.048 mol) was dissolved in 100 mL of methanol and cooled to <0 °C (brine/ice). Thionyl chloride (20.8 mL, 0.286 mol) was added slowly by syringe. The mixture was stirred overnight and then concentrated and co-evaporated with ether multiple times to eliminate excess thionyl chloride and provide the desired methyl ester that was shown to be >95% pure by 1H NMR. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 4.07 (t, J = 4.0 Hz, 1H), 3.98 (d, J = 4.0 Hz, 2H), 3.83 (s, 3H) ppm.
The amino ester was reconstituted in CH2Cl2 (100 mL) and triethylamine (16.6 mL, 0.119 mol) was added dropwise at 0 °C. To this stirring solution was added di-tert-butyl dicarbonate (11.420 g, 0.052 mol) in one portion. The reaction was stirred until the starting material was consumed, as determined by TLC (1:1 EtOAc/Hex). The reaction mixture was concentrated and dissolved in EtOAc (50 mL) then washed with saturated NaHCO3(aq) (3×25 mL) followed by brine (3×25 mL). The organic layer was dried over Na2SO4(s), filtered and concentrated. 1H NMR (300 MHz, CDCl3, 23°, δ): 5.54 (brd, 1H), 4.35 (m, 1H), 3.89 (ddd, J = 13.8, 11.2, and 3.7 Hz, 2H), 3.76 (s, 3H), 2.76 (bs, 1H), 1.43 (s, 9H) ppm.
The crude oil was dissolved in acetone (120 mL) and 2,2-dimethoxypropane (87 mL, 15 eq.). The solution was stirred at room temperature and BF3·OEt2 (1.2 mL, 9.52 mmol) was added. The reaction mixture turned a yellow-orange hue and was stirred for 2.5 hours. When the reaction was complete, by TLC analysis, the solution was treated with 99% Et3N (1.2 mL) and the solvent was removed. The brown oil was then partitioned between diethyl ether and saturated NaHCO3 (aq). The aqueous layer was extracted with diethyl ether (4×25 mL) and the organic layers were combined, dried (Na2SO4), and concentrated to a yellow oil (> 90% pure by 1H NMR). Rf(1:1 EtOAc/Hexanes) = 0.73. 1H NMR (300 MHz, CD3OD, 23 °C, δ): Major rotamer = 4.37 (dd, J = 7.0, 3.1 Hz, 1H), 4.13 (dt, J = 9.23, 7.0Hz, 2H), 3.74 (S, 3H), 1.52 (s, 3H), 1.48 (s, 3H), 1.40 (s, 9H) ppm; Minor rotamer = 4.48 (dd, J = 6.6, 2.6 Hz, 1H), 4.04 (dt, J = 7.3, 2.9 Hz, 2H), 3.74 (s, 3H), 1.66 (s, 3H), 1.62 (s, 3H) 1.48 (s, 9H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): Major rotamer = 151.38, 80.51, 66.45, 59.46, 52.49, 28.46, 27.60, 25.15, 24.57 ppm; Minor rotamer = 151.38, 80.51, 66.20, 59.38, 52.61, 28.54, 27.60, 26.21, 25.35 ppm.
A mixture of NaBH4 (2.247 g, 59.08 mmol) and LiCl (2.505 g, 59.08 mmol) was prepared in EtOH (42 mL), at 0 °C. A solution of the purified acetonide (7.659 g, 29.54 mmol) dissolved in THF (30 mL) was then added dropwise to the reaction mixture. The mixture was warmed to room temperature and stirred for four hours. After four hours the precipitate was filtered over celite and washed with EtOH. The filtrate was then concentrated and reconstituted in H2O (50 mL) and EtOAc (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3×25 mL). The organic layers were combined and washed with brine (2×25 mL), dried (Na2SO4 (s)), concentrated, and the crude oil was purified by column chromatography (25–50% EtOAc/Hexanes) to give 6.101 g (89%) of the alcohol as a white solid. Rf(1:1 EtOAc/Hexanes) = 0.86. 1H NMR (300 MHz, CDCl3, 35 °C, δ) = 4.07 (m, 2H), 3.75 (s, 1H), 3.68 (m, 2H), 1.52 (s, 6H), 1.49 (s, 9H) ppm.
To a stirring solution of 2.0 M oxalyl chloride, (COCl)2, (11.78 mL, 23.56 mmol) at −78 °C was added drop-wise a solution of DMSO (2.1 mL, 23.56 mmol) in CH2Cl2 (33 mL). This solution was allowed to stir at −78 °C for 15 minutes before a solution of the primary alcohol in CH2Cl2 was added drop-wise. The solution was stirred for 35 minutes before Et3N was added in a drop-wise manner at −78 °C. Following the addition of base, the reaction mixture was allowed to warm to 0 °C and the reaction was quenched with saturated NH4Cl(aq). The mixture was separated and the organic layer was washed with sat. NaHCO3(aq) (2×25 mL) followed by brine (2×25 mL). The organic layer was dried (MgSO4) and concentrated to a clear oil which was used immediately in the following reaction without further purification. Rf(1:3 EtOAc/Hexanes) = 0.35. 1H NMR (300 MHz, CDCl3, 23 °C, δ) = Major rotamer: 9.35 (d, 1H, J = 2.4 Hz), 4.02 (m, 1H), 3.88 (m, 2H), 1.43 (s, 3H), 1.34 (s, 3H), 1.22 (9H) ppm; and Minor rotamer: 9.38 (m, 1H), 4.13 (m, 1H), 3.88 (m, 2H), 1.39 (s, 3H), 1.33–1.27 (m, 12H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ) = Major rotamer: 198.84, 151.07, 94.72, 80.55, 64.53, 63.58, 27.99, 25.51, 23.54 ppm; and Minor rotamer: 199.02, 152.29, 94.05, 80.92, 64.60, 63.16, 28.03, 26.45, 24.47 ppm.
4.1.2.1 4-[2-(diethoxyphosphoryl)vinyl]-2,2-dimethyloxazolidine-3-carboxylic acid tert-butyl ester ((3R)-2a)
To tetraethyl methylenebisphosphonate (4.28 mL, 17.278 mmol) in THF (40 mL) at −78 °C was added 2.5 M n-BuLi in Hexanes (6.28 mL, 15.707 mmol). After 15 minutes of stirring at low temperature, aldehyde 1 (3.601 g, 15.707 mmol) was added in THF (40 mL) and the reaction was allowed to warm to room temperature with continued stirring overnight. The reaction mixture was concentrated to 1 to 2 mLs and purified by column chromatography (500 mL SiO2, 5% MeOH in CHCl3) to yield 5.440 g (95%, two steps) of clear oil. Rf = 0.65. 1H NMR (300 MHz, CDCl3, 23 °C, δ) = 6.63 (dt, 1H, J = 5.9, 17.8 Hz), 5.72 (dt, 1H, J = 15.8, 17.8 Hz), 4.40 (dt, 1H), 4.04 (m, 5H), 3.77 (m, 1H), 1.59 (m, 3H), 1.42 (m, 12H), 1.25 (dt, 6H, J = 1.5, 7.0 Hz) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 157.4, 150.3, 127.3, 119.7, 117.2, 92.6, 67.5, 62.1, 59.6 (d, J = 22.7 Hz), 28.5, 26.7, 25.7, and 16.6 (d, J = 8.1 Hz) ppm. [α]23D = −64.1° (c = 1.00, MeOH). HRMS (ES+) calculated m/z = 364.1889, experimental m/z = 364.1893.
4.1.2.2 4-[2-(diethoxyphosphoryl)vinyl]-2,2-dimethyloxazolidine-3-carboxylic acid tert-butyl ester ((3S)-2a)
The above procedure for the synthesis of (3R)-2a was employed using the epimer of 1 (1.146 g, 5.00 mmol) in THF (15 mL), 2.5M n-BuLi in Hexanes (2.00 mL, 5.00 mmol) and tetraethyl methylenebisphosphonate (1.36 mL, 5.5 mmol) yielded 1.728 g (>95%, two steps) of clear oil. Rf, 1H and 13C NMR were consistent with data reported for (3R)-2a. [α]23D = +65.9° (c = 1.03, MeOH). HRMS (ES+) calculated m/z = 364.1889, experimental m/z = 364.1893.
4.1.3 4-[2-(diethoxyphosphoryl)-2-fluorovinyl]-2,2-dimethyl-oxazolidine-3-carboxylic acid tert-butyl ester (2b)
The above procedure for synthesis of 2a was employed using 1 (229 mg, 1.00 mmol) in THF (2.5 mL), tetraethyl 2-fluoromethylene-bisphosphonate (337 mg, 1.10 mmol) in 2.5 mL of THF, and 2.5M n-BuLi in Hexanes (0.4 mL, 1.00 mmol). The reaction yielded 119 mg (31%) of 2b as a clear liquid. Rf(EtOAc) = 0.36. 1H NMR (300 MHz, CDCl3, 23°C, δ): 5.92 (dt, J = 37.6, 7.9 Hz, 1H), 3.71 (m, 1H), 4.10 (m, 5H), 3.72 (m, 1H), 1.41 (m, 6H), 1.39 (m, 9H), 1.29 (m, 6H) ppm.
4.1.4. General Procedure I: Hydrogenation /or Hydrogenolysis (3a)
To a solution of phosphonate 2a (845 mg, 2.33 mmol) dissolved in 25 mL of anhydrous EtOH was added 10% Pd/C (20% w/w). The reaction flask was repeatedly filled with H2 (balloon) and evacuated. Following three to five repetitions, the reaction was allowed to stir under an H2 atmosphere for 4 h. The mixture was filtered over celite and washed with EtOH. The filtrate, which required no further purification, was concentrated to 844 mg (99%) of clear oil. Rf(EtOAc) = 0.29. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 4.09 (dq, J = 3.3, 7.0 Hz, 4H), 3.94 (m, 1H), 3.68 (m, 2H), 1.80 (m, 4H), 1.59 (s, 3H) 1.54 (s, 3H), and 1.32 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 156.4, 92.5, 89.8, 65.0, 61.9 (d, J = 6.6Hz), 31.2, 28.6, 23.4 (d, J = 20.1Hz), 21.6 (d, J = 20.1 Hz), 16.7 (d, J = 6.0Hz) ppm.
4.1.5. 4-[2-(diethoxyphosphoryl)-2-fluoroethyl]-2,2-dimethyloxazolidine-3-carboxylic acid tert-butyl ester (3b)
General Procedure I was performed on 2b (141 mg, 0.37 mmol) to give 125 mg (88%) of 3b as a clear oil. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 5.24 (d, J = 14.3 Hz, 1H), 4.76 (m, 1H), 4.14 (m, 4H), 4.03 (m, 1H), 3.84 (dd, J = 8.1 Hz, 2H), 3.60 (m, 1H), 2.17 (m, 2H), 1.50 (m, 3H), 1.40 (m, 12H), 1.28 (m, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 1.52.96, 68.24, 66.42, 63.34, 55.68, 28.44, 23.20, 16.55 ppm.
4.1.6. 2-tert-butoxycarbonylamino-4-(diethoxyphosphoryl)butyric acid ((2R)-4a)
In a stirring solution of 3a (844 mg, 2.31 mmol) and 5 mL of acetone, at 0 °C was added Jones’ reagent (1.73 mL, 4.62 mmol). The reaction mixture was allowed to warm to room temperature and the stirring was continued for 12 hours. After this time the mixture was transferred to a larger flask then celite and isopropyl alcohol were added. The mixture was stirred for 15 minutes and the precipitate was filtered, washed with acetone, and made alkaline by the addition of sat. NaHCO3 (aq). The solution was concentrated to remove organic solvents and washed with EtOAc (3 x 25 mL). The aqueous layer was acidified to a pH ~3 by the addition of solid citric acid and extracted with CH2Cl2 (5 x 25 mL). The combined extracts were washed with brine (3 x 15 mL) and dried over solid MgSO4. The solvent was concentrated to 460 mg (59%) of a white solid, which could be recrystallized from Et2O/Hexanes. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 10.41 (brs, 1H), 5.38 (d, J = 7.5 Hz, 1H), 4.32 (m, 1H), 4.10 (t, J = 7.3 Hz, 4H), 1.95 (m, 4H), 1.43 (s, 9H), 1.31 (dt, J = 7.0, 1.5 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23°C, δ): 190.58, 157.35, 62.47, 53.57, 28.49, 25.81, 22.44, 16.50 ppm.
4.1.7. 2-tert-butoxycarbonylamino-4-(diethoxyphosphoryl)-4-fluorobutyric acid ((2R)-4b)
Similar procedures for the synthesis of (2R)-4a were followed using 3b (125 mg, 0.326 mmol), 1 mL of acetone, and Jones’ reagent (0.25 mL, 0.625 mmol) to yield 40 mg (48%) of (2R)-4b as a white solid. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 5.90 (m, 1H), 5.50 (m, 2H), 5.00 (dt, J = 46.4, 8.8 Hz, 1H), 4.31 (m, 1H), 4.17 (m, 4H), 2.38 (m, 2H), 1.41 (s, 9H), 1.32 (dt, J = 7.0, 2.2 Hz, 6H) ppm.
4.1.8.1 2-benzyloxymethyloxirane ((2S)-5)
To a stirring mixture of BnBr (0.72 mL, 6 mmol), 60% NaH (193 mg, 4.8 mmol) suspended in mineral oil, and 5 mL of DMF was added R-glycidol (0.26 mL, 4 mmol) dissolved in 3 mL of DMF, dropwise and at 0 °C. The solution was added via syringe over 30 minutes at which time the reaction was allowed to stir for an additional 3 hours while warming to room temperature. The crude material was diluted with 50 mL of EtOAc and washed with NH4Cl (3×50 mL), NaHCO3 (3×50 mL), LiBr (3×25 mL) and brine (3×50 mL). The organic layer was dried over Na2SO4, filtered and concentrated to clear oil. The crude oil was purified by flash chromatography (~200 mL SiO2, 1:4 EtOAc/ Hexanes) to yield 515 mg (78%) of clear oil. Rf(1:4 EtOAc/Hex.) = 0.31. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.31 (m, 5H), 4.56 (dd, J = 18.7, 12.1 Hz, 2H), 3.74 (dd, J = 11.4, 2.9 Hz, 1H), 3.39 (dd, J = 11.4, 5.9 Hz, 1H), 3.14 (dq, J = 2.9, 0.9 Hz, 1H), 2.73 (t, J = 4.6 Hz, 1H), 2.57 (dd, J = 5.1, 2.6 Hz, 1H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 137.83, 128.21, 128.19, 127.49, 72.96, 70.65, 50.58, 43.85 ppm.
4.1.8.2 2-benzyloxymethyloxirane ((2R)-5)
The above procedures for the formation of (2S)-5 were used to form 1.232 g (75%) of the title compound from S-glycidol (0.66 mL, 10.0 mmol). Rf(1:4 EtOAc/Hex.) = 0.31. 1H and 13C NMR data were consistent with (2S)-5.
4.1.9.1 General Procedure II: Nucleophilic Epoxide Opening ((3S)-6)
To a solution of diethyl methylphosphonate (2.04 mL, 14.1 mmol) dissolved in THF (14 mL), stirring at −78 °C, was added 2.5M n-BuLi (5.65 mL, 14.1 mmol) dropwise. The mixture was stirred for 15 minutes at −78 °C and (3S)-5 (773 mg, 4.71 mmol) dissolved in THF (2.4 mL) followed by BF3·OEt2 (2.32 mL, 18.8 mmol) were added dropwise at −78 °C. Stirring was continued at −78 °C for two additional hours before the reaction was quenched by the dropwise addition of NH4Cl(aq) (~2 mL). The mixture was allowed to warm to room temperature overnight, concentrated to a yellow oil, and purified by column chromatography (~300 mL of SiO2, 1:1 acetone/chloroform) to give 1.424 g (96%) of a faintly yellowed liquid. Rf(1:1 acetone/chloroform) = 0.39. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.34 (m, 5H), 4.55 (s, 2H), 4.10 (m, 4H), 3.85 (m, 1H), 3.49 (dd, J = 9.5, 3.3 Hz, 1H), 3.38 (dd, J = 9.7, 7.3 Hz, 1H), 1.85 (m, 4H), 1.31 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 137.74, 127.81, 127.12, 127.07, 127.00, 73.63, 72.69, 69.20 (d, J = 16.6 Hz), 60.99 (d, J = 6.6 Hz), 26.12 (d, J = 4.5 Hz), 22.03, 20.15, 15.93 (d, J = 6.0 Hz) ppm. [α]23D = −11.9° (c = 1.13, MeOH).
4.1.9.2 (4-benzyloxy-3-hydroxybutyl)phosphonic acid diethyl ester ((3R)-6)
Compound (2R)-5 (1.232 g, 7.50 mmol) was converted to (3R)-6 as in the above reaction to yield 1.311 g (55%) of the title compound as a faintly yellowed liquid. Rf(1:1 acetone/chloroform) = 0.39. See (3S)-6 for corresponding 1H and 13C NMR data. [α]23D = +10.4° (c = 1.06, MeOH).
4.1.10.1 (3-azido-4-benzyloxybutyl)phosphonic acid diethyl ester ((3R)-7)
To (3S)-6 (0.992 g, 3.136 mmol) dissolved in CH2Cl2 (4 mL) at 0 °C was added polymer-bound- Ph3P (3.1 g, ~9.3 mmol), followed by DPPA (0.74 mL, 3.450 mmol) then DIAD (0.68 mL, 3.450 mmol) via syringe. The reaction mixture was allowed to warm to room temperature and was stirred overnight. By morning no starting material was present by TLC analysis. The reaction mixture was filtered through celite, which was rinsed with methanol, and the filtrate was concentrated to about 2 g of crude yellow oil. Further purification was acheived by column chromatography (~250 mL of SiO2, 1:9 acetone/chloroform) to yield 952 mg (96%) of clear oil. Rf(1:9 acetone/chloroform) = 0.31. 1H NMR (300 MHz, CDCl3, 23 °C,δ): 7.34 (m, 5H), 4.56 (s, 2H), 4.09 (m, 4H), 3.56 (m, 3H), 1.80 (m, 4H), 1.32 (dt, J = 7.0, 1.1 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 137.78, 128.62, 128.51, 127.98, 127.76, 73.54, 72.58, 61.87, 24.37, 23.33, 21.44, 16.61 (d, J = 6.1 Hz) ppm.
4.1.10.2 (3-azido-4-benzyloxybutyl)phosphonic acid diethyl ester ((3S)-7)
Compound (3R)-6 (741 mg, 2.342 mmol) was converted to (3S)-7 as the above reaction to yield 585 mg (73% isolated yield) of the title compound. Rf(1:9 acetone/chloroform) = 0.31. See (3S)-7 for corresponding 1H and 13C NMR data.
4.1.11.1 (4-benzyloxy-3-tert-butoxycarbonylaminobutyl)phosphonic acid diethyl ester ((3R)-8)
Compound (3R)-7 (1.412 g, 4.137 mmol) was stirred in 32 mL of methanol and Boc2O (0.993 g, 4.551 mmol) followed by Lindlar’s catalyst (285 mg, 20% by weight) were added. The reaction mixture was stirred vigorously and a balloon of H2(g) was affixed. The apparatus was purged numerous times by cycling between vacuum (5×5 min) and H2(g). Finally H2 atmosphere was applied and maintained for 24h at room temperature. At this time no starting material was visible by TLC analysis and the reaction mixture was filtered through Celite 545 with the aid of methanol. The solvent was condensed and the resultant clear oil was purified by column chromatography (~150mL of SiO2, 1:9 acetone/chloroform) to yield 1.324 g (77%) of clear oil. Rf(5% MeOH in CHCl3) = 0.35. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.30 (m, 5H), 4.94 (d, J = 8.5 Hz, 1H), 4.48 (d, J = 4.2 Hz, 2H), 4.06 (m, 4H), 3.74 (m, 1H), 3.45 (d, J = 3.5 Hz, 2H), 1.81 (m, 4H), 1.42 (s, 9H), 1.29 (t, J = 6.9 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 155.63, 137.95, 128.38, 128.22, 127.70, 127.59, 79.25, 73.15, 71.73, 61.54 (d, J = 6.8 Hz), 50.65 (d, J = 19.3 Hz), 28.35, 25.33, 23.34, 21.46, 16.45 (d, J = 6.8 Hz) ppm.
4.1.11.2 (4-benzyloxy-3-tert-butoxycarbonylaminobutyl)phosphonic acid diethyl ester ((3S)-8)
Compound (3S)-7 was converted to (3S)-8 by the above method to yield 605 mg of a clear oil, which was >95% pure by NMR and was carried on to reaction 4.1.12.2 without further purification. Rf(5% MeOH in CHCl3) = 0.35. See (3S)-8 for corresponding 1H and 13C NMR data.
4.1.12.1 (3-tert-butoxycarbonylamino-4-hydroxybutyl)phosphonic acid diethyl ester ((3R)-9)
General Procedure I was utilized for the hydrogenolysis of compound (3R)-8 (1.201 g, 2.891 mmol) to give 2.714 g (94%) of (3R)-9 as a clear oil. No further purification was necessary. Rf(5% MeOH in CHCl3) = 0.09. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 5.33 (d, J = 6.9 Hz), 4.00 (m, 5H), 3.52 (m, 2H), 1.69 (brm, 5H), 1.34 (s, 9H), 1.23 (t, J = 6.9 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 156.20, 79.25, 64.31, 61.76, 57.30, 52.74 (d, JP = 19.3 Hz), 33.31 (d, JP = 15.5 Hz), 28.44, 24.32, 23.17, 16.46 (d, JP = 5.8 Hz) ppm. [α]23D = −10.8° (c ≈ 1.00, MeOH).
4.1.12.2 (3-tert-butoxycarbonylamino-4-hydroxybutyl)phosphonic acid diethyl ester ((3S)-9)
The crude material from (3S)-8 (605 mg, 1.4 mmol) was converted to (3S)-9 by General Procedure I. The resultant oil was purified by flash chromatography (100 mL SiO2, 5% methanol in chloroform) to yield 347 mg (72% over two steps) as clear oil. Rf(5% MeOH in CHCl3) = 0.09. See (3R)-9 for corresponding 1H and 13C NMR data. [α]23D = +10.4° (c = 1.00, MeOH).
4.1.12.3 (3-tert-butoxycarbonylamino-4-hydroxybutyl)-phosphonic acid diethyl ester ((3R)-9) from (3R)-2a
To a solution of compound 2a (5.211 g, 14.340 mmol) in EtOH (145 mL) was added 10% Pd/C (2.8 g, 20% w/w) [caution: Pd/C may spark on contact with ground glass joint; funnel and joint were rinsed with EtOH prior to attaching ground glass apparatus.] The round-bottomed flask was affixed with a three-way adapter w/stopcock. The adapter was fitted with a balloon of H2 and a vacuum hose. The system was purged of air and filled with H2 repeatedly in five minute increments. After 3 cycles the vacuum was removed and mixture was opened to H2 atmosphere. After 1.5 days the reaction had preceded ~60% by TLC and a second portion of Pd/C (1.4 g, 10% w/w) was added as well as more EtOH (50 mL). Following another 1.5 days the reaction had progressed to one spot by TLC analysis. The mixture was filtered over celite and washed with MeOH (4×50 mL). The solvent was evaporated and NMR analysis showed the resultant clear oil (4.72 g, 14.34 mmol) to be >95% pure. 1H and 13C NMR are consistent with data reported for 9a from 8a. [α]23D = −10.2° (c = 1.00, MeOH). HRMS (ES+) calculated m/z = 326.1733, experimental m/z = 326.1737.
4.1.12.3 (3-tert-butoxycarbonylamino-4-hydroxybutyl-phosphonic acid diethyl ester ((3S)-9) from (3S)-2a
The above procedure was repeated with (3S)-2a (1 g, 2.75 mmol), 10% Pd/C (1.5 g then 0.75 g) in EtOH (75 mL then 25 mL) to yield 0.905 g ( > 95%) of clear oil. The physical data was consistent with that of compound (3S)-9 derived from (3S)-8. HRMS (ES+) calculated m/z = 326.1733, experimental m/z = 326.1729.
4.1.13.1 TEMPO Oxidation: ((2R)-4a)
To a mixture of (3R)-9 (181 mg, 0.72 mmol) and iodosobenzenediacetate (430 mg, 1.335 mmol) and NaHCO3 (1.44 mmol) stirring in 22 mL of 1:1 MeCN/H2O was added 2,2,6,6-tetramethyl-1-piperidinyloxy (20 mg, 0.128 mmol). The mixture was stirred vigorously at room temperature for three hours then diluted with 10 mL of chloroform and extracted with Na2CO3. Ethyl acetate was added to the aqueous layer and the solution was acidified with 1N HCl and extracted (5×15 mL) with EtOAc. The organic layers were combined, dried over Na2SO4(s), filtered and evaporated to an amorphous solid. The desired material was used without further purification in the next series of reactions. Rf(AcOH/MeOH/CH2Cl2) = 0.40. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 9.91 (brs, 1H), 5.41 (m, 1H), 4.31 (m, 1H), 4.08 (m, 4H), 2.02 (m, 4H), 1.41 (s, 9H), 1.29 (m, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 173.79, 135.57, 80.15, 62.53 (d, JP = 6.0Hz), 57.58, 53.52, 28.41, 25.66, 22.40, 20.51, 16.46 (d, JP = 6.0 Hz) ppm. HRMS (ES+) calculated m/z = 340.1525, experimental m/z = 340.1514.
4.1.13.2 RuCl3/NaIO4 Oxidation: ((2S)-4a)
To (3S)-9 (231 mg, 0.710 mmol) in 1.4 mL of CCl4, 1.4 mL of MeCN, and 2.2 mL of H2O was added NaIO4 (456 mg, 2.130 mmol) followed by RuCl3 hydrate (3 mg, 0.016 mmol). By TLC analysis all the alcohol was consumed in 5 minutes (presumably to the aldehyde; Rf(1:9 MeOH/CHCl3) ≈ 1). After 1.5 hours, only a baseline spot was visible by TLC and the reaction was diluted with H2O and extracted with CHCl3 (5×15 mL). The organic layers were combined and dried over MgSO4, then concentrated and purified by flash chromatography (~75 mL of SiO2, 10 to 25% MeOH in CH2Cl2) to yield 184 mg (76%) of an amorphous white solid. Rf(AcOH/MeOH/CH2Cl2) = 0.40. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 11.32 (s, 1H), 5.39 (d, J = 7.2 Hz), 4.23 (m, 1H), 4.03 (dq, J = 7.2, 2.4 Hz, 4H), 1.93 (m, 4H), 1.35 (s, 9H), 1.23 (t, J = 7.0 Hz, 6H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 173.81, 155.59, 79.94, 62.30, 53.32 (d, J = 19.8 Hz), 28.29, 25.55, 22.30, 20.40, 16.33 (d, J = 6.1 Hz) ppm. HRMS (ES+) calculated m/z = 340.1525, experimental m/z = 340.1525.
4.1.14.1 General Procedure III: PyBOP Condensation (11a)
To a solution of the protected amino acid (2R)-4a (50 mg,0.147 mmol) in dry CH2Cl2 (4 mL) was added PyBOP (77 mg, 0.147 mmol) and DIEA (0.03 mL, 0.147 mmol), followed by p-decylaniline (34 mg, 0.147 mmol). The reaction was stirred at room temperature for 12 hours then concentrated and purified by column chromatography (0 to 20% acetone in chloroform) to 35 mg of clear oil. Rf(1:1 EtOAc/Hex.)=0.15. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 9.16 (s, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 5.69 (d, J = 6.5 Hz, 1H), 4.44 (m, J = 7.3, 1.2 Hz, 1H), 4.12 (m, 4H), 2.54 (t, J = 7.7 Hz, 2H), 2.07 (m, J = 7.7 Hz, 2H), 1.88 (m, 2H), 1.56 (p, J = 6.9 Hz, 2H), 1.44 (s, 9H), 1.32 (dt, J = 11.9, 7.3 Hz, 6H), 0.87 (t, J = 6.9 Hz, 3H) ppm.
4.1.14.2 [3-tert-butoxycarbonylamino-3-(4-octylphenylcarbamoyl)propyl]-phosphonic acid diethyl ester (11b)
General Procedure III was used to convert (2R)-4a (50 mg, 0.147 mmol) and p-octylaniline (0.05 mL, 0.147 mmol) to the title compound. The crude material was purified through flash chromatography (~ 50 mL of SiO2, 1:9 Acetone/ CHCl3) to give 54 mg (70%) of clear oil. Rf(1:9 Acetone/ CHCl3) = 0.2. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 9.22 (s, 1H), 7.45 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 5.77 (d, J = 8.1 Hz, 1H), 4.44 (m, 1H), 4.10 (m, 4H), 2.53 (t, J = 7.9 Hz, 2H), 2.09 (m, 2H), 1.87 (m, 2H), 1.55 (p, J = 6.8 Hz, 2H), 1.43 (s, 9H), 1.31 (dt, J = 10.6, 7.0 Hz, 6H), 0.86 (t, J = 7.0, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 169.75, 155.96, 139.21, 135.76, 128.99, 120.12, 80.30, 62.29 (d, J = 22.2 Hz), 35.63, 32.13, 31.78, 29.72, 29.51, 28.59, 26.52 (d, J = 5.0 Hz), 23.08, 22.91, 21.20, 16.67 (d, J = 7.6 Hz), 14.35 ppm.
4.1.14.3 [3-tert-butoxycarbonylamino-3-(3-heptylphenylcarbamoyl)propyl]-phosphonic acid diethyl ester (11c)
General Procedure III was used to convert (2R)-4a (50 mg, 0.147 mmol) and m-heptylaniline39 (0.28 mg, 0.147 mmol) to the title compound. The crude material was purified through flash chromatography (~50 mL of SiO2, 1:3 Acetone/ CHCl3) to give 35 mg (46%) of clear oil. Rf(1:3 Acetone/ CHCl3)=0.55. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 9.22 (s, 1H), 7.40 (s, 1H), 7.39 (d, J = 9.2 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 6.91 (d, J = 7.3 Hz, 1H), 5.68 (d, J = 7.3 Hz, 1H), 4.46 (m, 1H), 4.11 (m, 4H), 2.55 (t, J = 7.7 Hz, 2H), 2.07 (m, 2H), 1.89 (m, 2H), 1.58 (p, J = 7.3 Hz, 2H), 1.44 (s, 9H), 1.32 (dt, J = 11.9, 6.9 Hz, 6H), 0.86 (t, J = 6.9 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 169.72, 144.14, 138.00, 128.90, 124.61, 123.12, 119.93, 117.28, 78.50, 36.21, 62.15, 36.21, 31.99, 31.67, 29.53, 29.38, 28.52, 26.44, 22.58, 21.18, 16.64, 14.29 ppm.
4.1.14.4 [3-tert-butoxycarbonylamino-3-(4-octylphenylcarbamoyl)propyl]-phosphonic acid diethyl ester (11d)
General Procedure III was used to convert (2R)-4a (1.122 g, 3.307 mmol) and m-octylaniline (0.815 g, 3.968 mmol) to the title compound. The crude material was purified through flash chromatography (~ 300 mL of SiO2, 1:9 acetone/CHCl3) to give 1.134 g (65%) of translucent oil. Rf (3:7 acetone/CHCl3) = 0.58. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 9.28 (s, 1H), 7.41 (s, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.18 (t, J = 8.1 Hz, 1H), 6.90 (d, J = 7.7 Hz, 1H), 5.74 (d, J = 8.1 Hz, 1H), 4.46 (m, 1H), 4.12 (ddt, J = 24.6, 7.3, 3.5 Hz, 4H), 2.54 (t, J = 7.3 Hz, 2H), 2.07 (sep., J = 6.5 Hz, 2H), 1.90 (dq, J = 18.4, 13.5, 6.5 Hz, 2H), 1.57 (p, J = 7.7 Hz, 2H), 1.43 (m, 9H), 1.31 (dt, J = 10.0, 7.3 Hz, 6H), 1.28 (m, 12H), 0.86 (t, J = 6.5 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 169.77, 144.07, 138.04, 128.85, 124.53, 120.33, 119.94, 118.11, 117.29, 80.20, 62.36, 62.04, 54.79, 36.20, 32.06, 31.65, 30.07, 29.66, 29.56, 29.43, 29.13, 28.51, 26.46, 23.03, 22.83, 22.78, 21.16, 16.64, 16.55, 14.28 ppm.
4.1.14.5 [3-tert-butoxycarbonylamino-3-(4-octylphenylcarbamoyl)propyl]-phosphonic acid diethyl ester (11e)
General Procedure III was used to convert (2S)-4a (50 mg, 0.147 mmol) and p-octylaniline (0.05 mL, 0.147 mmol) to the title compound. The crude material was purified through flash chromatography (~ 50 mL of SiO2, 1:9 Acetone/ CHCl3) to give 54 mg (70%) of clear oil. Rf(1:9 Acetone/ CHCl3) = 0.2. See 11b for corresponding 1H and 13C NMR data.
4.1.14.6 [3-tert-butoxycarbonylamino-3-(3-octylphenylcarbamoyl)propyl]-phosphonic acid diethyl ester (11f)
General Procedure III was used to convert (2S)-4a (34 mg, 0.100 mmol) and m-octylaniline39 (0.02 mL, 0.100 mmol) to the title compound. The crude material was purified through flash chromatography (~50 mL of SiO2, 1:1 EtOAc/ Hex.) to give 23 mg (45%) of an off-white solid. Rf(1:1 EtOAc/Hex.)=0.10. See 11d for corresponding 1H and 13C NMR data.
4.1.15.1 General Procedure IV: Phosphonate Diester Deprotection (12a)
The phosphonate diester 11a (35 mg, 0.063 mmol) was dissolved in 0.65 mL of CH2Cl2 to which bromotrimethylsilane (0.08 mL, 0.630 mmol) was added via syringe. The solution was allowed to stir for four to six hours. The off-white solution was then concentrated and reconstituted in > 0.65 mL of 95% methanol in water. The new solution was allowed to stir for four hours to ensure hydrolysis and was concentrated and co-evaporated with methanol and diethyl ether until a pasty solid was attained. The compound was triturated to a fine white solid on addition of water. The solid was filtered through a fine fritted funnel and washed with water (3×) followed by cold pentane (3×) to yield 31 mg of the desired compound as a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.51 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 4.12 (t, J = 6.4 Hz, 1H), 2.58 (t, J = 7.7 Hz, 2H), 2.24 (m, 2H), 1.86 (m, 2H), 1.60 (p, J = 6.7 Hz, 2H), 1.28 (m, 12H), 0.90 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 141.04, 130.04, 121.40, 114.14, 56.36, 36.47, 33.21, 32.88, 30.89, 30.86, 30.74, 30.60, 30.41, 23.88, 14.59 ppm. MS (ESI+) m/z 399 [M+H]+.
4.1.15.2 [3-amino-3-(4-octylphenylcarbamoyl)propyl]-phosphonic acid (12b – VPC44152)
General Procedure IV was used to globally deprotect compound 11b in formation of the title compound as 47 mg (100%) of an off-white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.51 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.3 Hz, 2H), 4.13 (t, J = 6.5 Hz, 1H), 2.59 (t, J = 7.9 Hz, 1H), 2.24 (m, 2H), 1.88 (m, 2H), 1.60 (p, J = 6.2 Hz, 2H), 1.31 (m, 10H), 0.89 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 140.90, 136.48, 129.69, 121.25, 66.69, 57.13, 36.32, 33.02, 32.72, 30.56, 30.41, 30.27, 23.71, 22.89, 22.46, 14.43 ppm. MS (APCI) m/z = 371.9 [M+1]+, 370.80 [M, 100%]+. Elemental CHN: calculated C = 47.90%, H = 7.15%, N = 6.21%; found C = 48.21%, H = 7.25%, N = 6.02%.
4.1.15.3 [3-amino-3-(3-heptylphenylcarbamoyl)propyl]-phosphonic acid (12c)
General Procedure IV was used to globally deprotect compound 11c in formation of the title compound as 25 mg (84%) of a white solid. 1H NMR (300 MHz, (CD3)2SO, 23 °C, δ): 8.26 (m, 1H), 7.42 (m, 1H), 7.26 (t, J = 6.8 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 3.98 (m, 1H), 2.55 (m, 2H), 2..03 (m, 2H), 1.60 (m, 4H), 1.26 (m, 8H), 0.85 (m, 3H) ppm. 13C NMR (300 MHz, (CD3)SO, 23 °C, δ): 166.79, 143.28, 137.83, 128.94, 124.37, 119.27, 116.95, 53.25, 35.17, 31.27, 30.87, 28.60, 28.54, 25.36, 24.32, 22.51, 22.11, 13.99 ppm. MS (ESI+) m/z 357 [M+H]+.
4.1.15.4 [3-amino-3-(3-octylphenylcarbamoyl)propyl]-phosphonic acid (12d – VPC44116)
General Procedure IV was used to globally deprotect compound 12d in formation of the title compound as 28 mg (90%) of a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.45 (m, 2H), 7.23 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.6 Hz, 1H), 4.15 (m, 1H), 2.59 (t, J = 7.3 Hz, 2H), 2.25 (m, 2H), 1.89 (m, 2H), 1.61 (m, 2H), 1.31 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 167.99, 139.32, 130.01, 126.19, 121.28, 119.44, 118.72, 55.39, 55.20, 37.04, 33.17, 32.77, 30.74, 30.47, 29.72, 27.11, 25.42, 23.87, 23.61, 14.58 ppm. MS (APCI) m/z = 371.9 [M+1]+, 370.80 [M, 100%]+. HRMS (ES+) calculated m/z = 371.2100, experimental m/z = 371.2108. Elemental CHN: calculated C = 47.90%, H = 7.15%, N = 6.21%; found C = 48.11%, H = 7.14%, N = 6.14%.
4.1.15.5 [3-amino-3-(4-octylphenylcarbamoyl)propyl]-phosphonic acid (12e)
General Procedure IV was used to globally deprotect compound 11e in formation of the title compound as 25 mg (96%) of a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.47 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 4.25 (m, 1H), 3.95 (t, J = 6.8 Hz, 1H), 2.57 (t, J = 7.5 Hz, 2H), 2.22 (m, 2H), 1.97 (m, 1H), 1.84 (m, 2H), 1.58 (m , 2H), 1.26 (m, 10H), 0.87 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 140.90, 136.48, 129.69, 121.25, 66.69, 57.13, 36.32, 33.02, 32.72, 30.56, 30.41, 30.27, 23.71, 22.89, 22.46, 14.43 ppm. MS (ESI+) m/z 371 [M+H]+.
4.1.15.6 [3-amino-3-(3-octylphenylcarbamoyl)propyl]-phosphonic acid (12f)
General Procedure IV was used to globally deprotect compound 11f in formation of the title compound as 20 mg (100%) of a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.44 (s, 1H), 7.42 (d, J = 10.0 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 4.08 (t, J = 6.6 Hz, 1H), 2.60 (t, J = 7.3 Hz, 2H), 2.23 (m, 2H), 1.89 (m, 2H), 1.61 (quin., J = 6.8Hz, 2H), 1.32 (m, 10H), 0.89 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 167.99, 139.32, 130.01, 126.19, 121.28, 119.44, 118.72, 55.39, 55.20, 37.04, 33.17, 32.77, 30.74, 30.47, 29.72, 27.11, 25.42, 23.87, 23.61, 14.58 ppm. MS (ESI+) m/z 371 [M+H]+.
4.1.15.7 [3-amino-3-(4-decylphenylcarbamoyl)-2-fluoropropyl]-phosphonic acid (13)
Compounds (3R)-4b and p-decylaniline were converted to the title compound by application of first; General Procedure III to yield 22 mg (24%) of a clear liquid, Rf(1:1 EtOAc/Hexanes)=0.21. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 8.78 (d, J = 38.9, 1H), 7.43 (d, J = 7.7 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H), 5.56 (d, J = 22.6 Hz, 1H), 4.99 (m, 1H), 4.56 (m, 1H), 4.22 (m, 4H), 2.55 (t, J = 7.9 Hz, 2H), 2.40 (m, 2H), 1.56 (p, J = 7.0 Hz, 2H), 1.45 (s, 9H), 1.36 (dt, J = 7.2, 7.0 Hz, 6H), 1.25 (m, 12H), 0.87 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 129.01, 120.11, 100.16, 63.98, 63.47, 51.81, 51.50, 25.59, 32.10, 31.74, 29.83, 29.81, 29.71, 29.53, 29.45, 28.50, 28.48, 22.89, 16.63, 14.33 ppm. General Procedure IV was then implemented to give 19 mg (100%) of the title compound as a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.50 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 5.02 (m, 1H), 4.26 (m, 1H), 4.11 (m, 1H), 2.59 (t, J = 7.6 Hz, 2H), 2.39 (m, 1H), 1.60 (p, J = 7.0 Hz, 2H), 1.29 (m, 12H), 0.90 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 166.71, 140.61, 135.83, 129.57, 120.97, 36.04, 32.64, 32.29, 30.32, 30.21, 30.05, 29.89, 23.36, 16.81, 14.37 ppm. MS (ESI+) m/z 417 [M+H]+.
4.1.16.1 General Procedure V: Standard Mitsunobu Conditions (14a)
To a solution of p-octylphenol (1.000 g, 4.847 mmol) in 5 mL of anhydrous THF was added (R)-(+)- glycidol (0.35 mL, 5.331 mmol) and Ph3P (1.398 g, 5.331 mmol). The reaction mixture was cooled to 0 °C and DIAD (1.05 mL, 5.331 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for 24 hours. A white precipitate formed upon addition of Et2O and was filtered off through a fine fritted funnel. The filtrate was concentrated and purified by column chromatography (~200 mL SiO2, CHCl3) to give 1.023 g (80%) of 14a as colorless oil. Rf(CHCl3)=0.66. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.09 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 4.19 (dd, J = 11.2, 3.3 Hz, 1H), 3.95 (dd, J = 11.0, 5.5 Hz, 1H), 3.35 (p, J = 4.8 Hz, 1H), 2.90 (t, J = 4.2 Hz, 1H), 2.75 (dd, J = 4.8, 2.6 Hz, 1H), 2.54 (t, J = 7.5 Hz, 2H), 1.56 (m, 2H), 1.28 (m, 10H), 0.88 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 156.70, 135.92, 129.51, 114.63, 69.02, 50.44, 45.01, 35.26, 32.10, 31.93, 29.70, 29.49, 22.89, 14.33 ppm.
4.1.16.2 2-(4-octylphenoxymethyl)-oxirane (14b)
The title compound was formed, through General Procedure V, starting with 1.000 g of (S)-(-)-glycidol, as 1.073 g (84%) of a clear oil following column chromatography (~200 mL SiO2, CHCl3) Rf(CHCl3)=0.66. 1H and 13C NMR were consistent with those of epimer 14a.
4.1.17.1 [3-hydroxy-4-(4-octylphenoxy)-butyl]-phosphonic acid diethyl ester (15a)
General Procedure II was used to transform 14a (1.023 g, 3.899 mmol) to 1.400 g (87%) of 15a as a clear oil after column chromatography (~300 mL SiO2, 1:4 acetone/chloroform). Rf(1:4 acetone/chloroform) =0.33. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.09 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 4.11 (m, 4H), 4.02 (m, 1H), 3.90 (m, 2H), 2.53 (t, J = 7.7 Hz, 2H), 1.92 (m, 4H), 1.56 (p, J = 8.1 Hz, 2H), 1.30 (m, 16H), 0.87 (t, J = 6.9 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 156.70, 135.89, 129.54, 114.54, 71.77, 70.08, 62.02, 35.26, 32.11, 31.95, 29.70, 29.49, 26.59, 23.12, 22.89, 21.23, 16.69, 14.33 ppm.
4.1.17.2 [3-hydroxy-4-(4-octylphenoxy)-butyl]-phosphonic acid diethyl ester (15b)
General Procedure II was used to transform 14b (1.073 g, 4.089 mmol) to 1.215 g (72%) of 15b as a clear oil after column chromatography (~300 mL SiO2, 1:4 acetone/chloroform). Rf(1:4 acetone/chloroform) =0.33.
4.1.18.1 General Procedure VI: Azide Formation with Free Phosphine (16a)
To a stirring solution of 15a (500 mg, 1.206 mmol) in 1.6 mL of CH2Cl2 at 0 °C was added Ph3P (348 mg, 1.327 mmol). The solution was stirred for 15 minutes and then DIAD (0.26 mL, 1.327 mmol) and DPPA (0.29 mL, 1.327 mmol) were added consecutively, dropwise. The reaction was allowed to warm to room temperature and stirred overnight. Mixture was concentrated and purified by chromatography (~150 mL SiO2, 1:9 Acetone/CHCl3) followed by (~100 mL SiO2, 50% to 80% Et2O in petroleum ether) to yield 518 mg (98%) of non-viscous clear liquid. Rf(1:9 Acetone/CHCl3)=0.44. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.04 (d, J = 8.6 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 4.05 (m, 4H), 3.95 (m, 2H), 3.75 (m, 1H), 2.48 (t, J = 7.9 Hz, 2H), 1.80 (m, 4H), 1.51 (p, J = 7.3 Hz, 2H), 1.27 (t, J = 7.0 Hz, 6H), 1.23 (m, 10H), 0.82 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 156.11, 135.80, 129.29, 114.30, 70.45, 61.67, 60.95, 35.01, 31.84, 31.68, 29.44, 29.22, 24.16, 23.10, 22.63, 21.21, 16.41, 14.06 ppm.
4.1.18.2 [3-azido-4-(4-octylphenoxy)-butyl]-phosphonic acid diethyl ester (16b)
General Procedure VI was used to form the title compound from alcohol 15b. Purification consisted of two chromatography steps. First a column (~250 mL of SiO2, 1:9 Acetone/CHCl3) was run to yield the desired product with OPPh3. The crude material was then run through a short plug (~50 mL of SiO2) with CHCl3 as the eluent. The phosphine oxide did not more over this system and the plug yielded 354 mg (67%) of a clear liquid. Data were consistent with that of 16a.
4.1.19.1 [3-amino-4-(4-octylphenoxy)-butyl]-phosphonic acid diethyl ester (17a)
General Procedure I was used to reduce the azide 16a to 488 mg of amine 17a as a yellow liquid. Further purification was not necessary and the product was carried on to the final deprotection. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 6.97 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 8.1 Hz, 2H), 5.43 (m, 2H), 3.99 (m, 4H), 3.90 (m, 2H), 2.43 (t, J = 7.9 Hz, 2H), 1.89 (m, 4H), 1.47 (p, J = 7.0 Hz, 2 H), 1.21 (m, 16H), 0.79 (t, J = 6.7 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 168.83, 156.23, 135.57, 129.18, 114.35, 69.57, 61.82, 51.05, 49.84, 34.96, 31.81, 31.66, 29.41, 29.20, 24.83, 22.73, 22.58, 20.85, 16.34, 14.03 ppm.
4.1.19.2 [3-amino-4-(4-octylphenoxy)-butyl]-phosphonic acid diethyl ester (17b)
General Procedure I was used to reduce the azide 16b to 333 mg of amine 17b as a light yellow liquid. Further purification was not necessary and the product was carried on to the final deprotection. Data were similar to that obtained for 17a.
4.1.20.1 [3-amino-4-(4-octylphenoxy)-butyl]-phosphonic acid (18a)
General Procedure IV was used to deprotect compound 17a in formation of the title compound as 500 mg (97%) of an off-white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.12 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.22 (dd, J = 10.4, 3.5 Hz, 1H), 4.07 (dd, J = 10.7, 6.1 Hz, 1H), 3.68 (m, 1H), 2.55 (t, J = 7.3 Hz, 2H), 2.09 (m, 2H), 1.88 (m, 2H), 1.57 (p, J = 7.7 Hz, 2H), 1.30 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (300 MHz, CD3OD, 23 °C, δ): 13764, 130.64, 115.70, 101.52, 68.14, 36.14, 33.18, 33.07, 30.73, 30.58, 30.39, 25.28, 24.70, 23.87, 14.58 ppm. MS (ESI+) m/z 358 [M+H]+.
4.1.20.2 [3-amino-4-(4-octylphenoxy)-butyl]-phosphonic acid (18b)
General Procedure IV was used to deprotect compound 17b in formation of the title compound as 278 mg (79%) of an off-white solid. 1H NMR, 13C NMR, and MS (ESI+) were consistent with those obtained for compound 18a. Elemental CHN: calculated C = 60.49%, H = 9.02%, N = 3.92%; found C = 60.21%, H = 9.00%, N = 3.68%.
4.1.21. [3-hydroxy-4-(4-octylphenoxy)-butyl]-phosphonic acid (19)
General Procedure IV was used to deprotect compound 15a (25 mg, 0.060 mmol) in formation of the title compound. Further purification was performed by recrystallization from EtOAc and Hexanes to yield 22 mg (79%) of a white solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.07 (m, 2H), 6.85 (m, 2H), 3.90 (m, 3H), 2.53 (m, 2H), 1.95 (m, 2H), 1.78 (m, 2H), 1.29 (m, 10H), 0.89 (m, 3H) ppm. MS (ESI-) m/z 357 [M-H]−.
4.1.22. 4-methyl-N-(3-octylphenyl)-benzenesulfonamide (20)
To a stirring solution of m-octylaniline40 (1.000 g, 4.87 mmol) in 5 mL of pyridine at 0 °C was added tosyl chloride (928 mg, 4.87 mmol). The reaction mixture was warmed to room temperature. After one hour, the mixture was diluted with EtOAc and washed with 1N HCl (3×), NaHCO3 (2×), and brine (2×). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (~150 mL SiO2, 1:19 Acetone/Chloroform) to yield 1.700 g (97%) of a white solid. Rf (1:19 Acetone/Chloroform) = 0.70. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.72 (d, 2H), 7.59 (bs, 1H), 7.19 (d, J = 7.8 Hz, 2H), 7.03 (q, 4H), 2.51 (t, J = 7.2Hz, 2H), 2.34 (s, 3H), 1.54 (p, J = 7.6 Hz, 2H), 1.27 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 144.2, 140.5, 136.7, 134.7, 130.1, 129.7, 127.9, 122.4, 35.8, 32.4, 31.9, 30.0, 29.8, 23.2, 22.0, 14.6. MS (ESI) m/z 360 [M+H]+.
4.1.23. 4-methyl-N-(3-octylphenyl)-N-oxiranylmethylbenzenesulfonamide (21)
General Procedure V was used to couple 20 (0.809 g, 2.25 mmol) with (R)-(+)-glycidol (0.23 mL, 3.375 mmol) to form 644 mg (69%) of the title compound 21 after flash chromatography (~150 mL SiO2, 1:4 EtOAc/Hexanes), as a clear oil. Rf(1:4 EtOAc/Hex.) = 0.36. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.45 (d, J = 8.1 Hz, 2H), 7.20 (m, 2H), 7.07 (m, 1H), 7.02 (m, 1H), 6.89 (m, 1H), 6.82 (m, 1H), 3.65 (ddd, J = 35.2, 14.3, 5.3 Hz, 2H), 3.08 (m, 1H), 2.63 (m, 1H), 2.49 (t, J = 7.5 Hz, 2H), 2.37 (s, 3H), 2.16 (m, 1H), 1.48 (p, J = 6.8 Hz, 2H), 1.25 (m, 10H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (300 MHz, CDCl3, 23 °C, δ): 144.01, 143.51, 139.60, 138.83, 129.39, 127.68, 126.10, 53.65, 50.24, 45.78, 35.56, 31.84, 31.24, 29.41, 29.15, 22.64, 21.47, 14.10 ppm.
4.1.24. {3-hydroxy-4-[(3-octylphenyl)-(toluene-4-sulfonyl)-amino]-butyl}-phosphonic acid diethyl ester (22)
General Procedure II completed the synthesis of 853 mg (97%) of compound 22 from 21 as a clear oil following column chromatography (SiO2, 5 to 20% Acetone in CHCl3). Rf(1:4 EtOAc/Hex.) = 0.05. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.41 (dd, J = 8.1, 6.5 Hz, 2H), 7.18 (d, J = 7.7 Hz, 2H), 7.13 (d, J = 8.1, 1H), 7.04 (d, J = 7.7 Hz, 1H), 6.85 (m, 1H), 6.73 (m, 1H), 4.00 (m, 4H), 3.61 (m, 2H), 3.47 (m, 2H), 2.45 (t, J = 8.1Hz, 2H), 1.76 (m, 4H), 1.45 (m, 2H), 1.24 (m, 16H), 0.83 (t, J = 6.9 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 143.63, 139.60, 138.92, 134.77, 129.50, 129.44, 128.93, 128.31, 127.88, 126.04, 68.80 (d, J = 13.5 Hz), 61.74 (d, J = 5.8 Hz), 56.23 (d, J = 7.7 Hz), 35.66, 31.93, 31.35, 29.49, 29.30, 28.98, 27.10 (d, J = 4.8 Hz), 22.70, 21.59, 20.48, 16.47 (d, J = 5.8 Hz), 14.18 ppm.
4.1.25. {3-azido-4-[(3-octylphenyl)-(toluene-4-sulfonyl)-amino]-butyl}-phosphonic acid diethyl ester (23)
General Procedure VI transformed alcohol 22 (302 mg, 0.532 mmol) into azide 23. The crude material was purified twice by chromatography (~150 mL of SiO2, 10%Acetone/CHCl3) followed by (~100 mL of SiO2, 7:3 EtOAc/Hexanes) to yield 267 mg (85%) as a clear oil. [Another 5% of the desired product was isolated with residual phosphine oxide.] Rf(1:9 Acetone/CHCl3) = 0.43. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.38 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 7. Hz, 2H), 7.07 (d, J = 7.7 Hz, 1H), 6.86 (d, J = 13.2 Hz, 1H), 6.78 (m, 1H), 4.02 (m, 4H), 3.53 (m, 3H), 2.48 (t, J = 7.5 Hz, 2H), 2.37 (s, 3H), 1.75 (m, 4H), 1.46 (p, J = 7.0 Hz, 2H), 1.25 (m, 16H), 0.84 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 144.36, 143.80, 139.38, 134.55, 132.04, 129.48, 129.05, 128.45, 127.87, 125.73, 61.75 (d, J = 6.6 Hz), 61.32 (d, J = 16.1 Hz), 54.74, 35.68, 31.92, 31.33, 29.49, 29.28, 28.97, 28.56, 25.30 (d, J = 4.5 Hz), 22.70, 21.59, 16.49 (d, J = 6.0 Hz), 14.16 ppm.
4.1.26. {3-amino-4-[(3-octylphenyl)-(toluene-4-sulfonyl)-amino]-butyl}-phosphonic acid diethyl ester (24)
Compound 23 (267 mg, 0.450 mmol) was dissolved in 50 mL of a 20:1 MeOH/HCl solution. To this solution was added ~0.5 g of Pd(OH)2 and the apparatus was assembled and experiment run analogous to that of General Procedure I. When no starting material remained (~four hours by TLC) the mixture was filtered over celite and washed with 2% Et3N in MeOH. The filtrate was concentrated to yield 268 mg (100%) of the title compound as yellow oil. No further purification was required. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.26 (d, J = 7.9 Hz, 2H), 7.11 (d, J = 7.9 Hz, 2H), 7.06 (d, J = 7.7 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.72 (d, J = 7.7 Hz, 1H), 6.60 (m, 1H), 4.02 (bs, 2H), 3.87 (m, 4H), 3.30 (m, 2H), 2.63 (m, 1H), 2.35 (t, J = 7.7 Hz, 2H), 2.27 (s, 3H), 1.59 (m, 4H), 1.33 (m, 2H), 1.12 (m, 16H), 0.72 (t, J = 6.6 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 144.36, 143.95, 138.95, 137.75, 129.45, 128.99, 128.49, 128.07, 127.68, 125.79, 61.96 (d, J = 7.1 Hz), 56.05, 48.67 (t, J = 21.7), 35.48, 31.76, 31.22, 29.33, 29.13, 26.26 (d, J = 7.6 Hz), 22.52, 22.22, 21.29, 20.35, 16.08 (d, J = 6.6 Hz), 13.86 ppm.
4.1.27. [3-amino-4-(3-octylphenylamino)-butyl]-phosphonic acid diethyl ester (25)
A solution of Na(s) (70 mg, 3.06 mmol) in 25 mL of NH3(l) was contained in a 2-neck flask fitted with a cold finger cooled to −78 °C and stir bar. Compound 24 (182 mg, 0.306 mmol) diluted in a minimal amount of THF was quickly added to the solution and the reaction was allowed to proceed for no longer than 5 minutes, at which time ethanol was added under vigorous stirring and the reaction was allowed to warm to room temperature. The reaction mixture was concentrated to dryness, dissolved in EtOAc, washed with NaHCO3 (3×) and brine (1×). The organic layer was dried then columned (~75 mL of SiO2, 5% MeOH in CHCl3). The desired product was found to be a clear oil, in 25 mg (25%) quantities. Starting material (28%) was also recovered. Rf(5% MeOH in CHCl3) = 0.17. 1H NMR (300 MHz, CDCl3, 23 °C, δ): 7.07 (t, J = 7.5 Hz, 1H), 6.53 (t, J = 7.0 Hz, 1H), 6.44 (m, 2H), 4.08 (m, 4H), 3.70 (m, 1H), 3.11 (m, 3H), 2.50 (t, J = 7.9 Hz, 2H), 1.83 (m, 4H), 1.57 (p, J = 7.3 Hz, 2H), 1.31 (m, 16H), 0.87 (t, J = 7.0 Hz, 3H) ppm. 13C NMR (300 MHz, CDCl3, 23 °C, δ): 157.35, 148.46, 129.30, 118.14, 113.44, 110.41, 62.05, 61.72, 58.20, 47.13, 36.41, 34.55, 32.10, 31.72, 29.92, 29.73, 29.64, 29.49, 28.18 (d, J = 8.6 Hz), 22.88, 16.69 (d, J = 7.1 Hz), 14.32 ppm.
4.1.28. [3-amino-4-(3-octylphenylamino)-butyl]-phosphonic acid (26)
The deprotection of 25 (23 mg, 0.056 mmol) was carried out by General Procedure IV to give 20 mg (83%) of the title compound as a colorless solid. 1H NMR (300 MHz, CD3OD, 23 °C, δ): 7.36 (dd, J = 7.9, 3.7 Hz, 1H), 7.26 (t, J = 1.7 Hz, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 3.70 (m, 3H), 2.65 (t, J = 7.5 Hz, 2H), 2.14 (m, 2H), 1.95 (m, 2H), 1.64 (p, J = 7.5 Hz, 2H), 1.31 (m, 10H), 0.89 (t, J = 7.0 Hz, 3H) ppm. MS (ESI+) m/z 357 [M+H]+.
Acknowledgments
This research was funded by grants from the NIH (R01 GM067958, F31 GM064101).
Footnotes
Initial synthesis and binding affinities of compounds 12a,b, and d were presented: Foss, F. W. Jr.; Clemens, J. J.; Davis, M. D.; Lynch, K. R.; Macdonald, T. L. Abstracts of Papers, 228th National Meeting of the American Chemical Society, Philadelphia, PA 2004.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Lynch KR. Biochim et Biophys Acta. 2002;1582:70–71. doi: 10.1016/s1388-1981(02)00138-5. [DOI] [PubMed] [Google Scholar]
- 2.Hla T. Pharmacol Rev. 2003;47:401–407. doi: 10.1016/s1043-6618(03)00046-x. [DOI] [PubMed] [Google Scholar]
- 3.Watterson K, Sankala H, Milstien S, Spiegel S. Prog in Lipid Res. 2003;42:344–357. doi: 10.1016/s0163-7827(03)00015-8. [DOI] [PubMed] [Google Scholar]
- 4.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 5.For a convenient review: Im D-S. TRENDS in Pharmacol Sci. 2003;24:12–4. doi: 10.1016/s0165-6147(02)00012-3.
- 6.Kharel Y, Lee S, Snyder AH, Sheasley-O’Neill SL, Morris MA, Setiady Y, Zhu R, Zigler MA, Burcin TL, Ley K, Tung KSK, Engelhard VH, Macdonald TL, Lynch KR. J Biol Chem. 2005;280:36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 7.Rosen J, Sanna G, Alfonso C. Immunol Rev. 2003;195:160–177. doi: 10.1034/j.1600-065x.2003.00068.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson RM, Henry ML, Elkhammas EA, Davies EA, Bumgardner GL, Pelletier RP, Rajab A. Am J Surg. 2003;186:306–311. doi: 10.1016/s0002-9610(03)00219-8. [DOI] [PubMed] [Google Scholar]
- 9.Lei G, Suzuki AS, Goto T, Kokubo T, Miyamoto M, Kimura H. Transplant Proc. 2000;32:1628. doi: 10.1016/s0041-1345(00)01443-3. [DOI] [PubMed] [Google Scholar]
- 10.For a review of initial studies: Kiuchi M, Adachi K, Kohara T, Minoguchi M, Hanano T, Aoki Y, Mishina T, Arita M, Nakao N, Ohtsuki M, Hoshino Y, Teshima K, Chiba K, Sasaki S, Fujita T. J Med Chem. 2000;43:2946–2961. doi: 10.1021/jm000173z.
- 11.Kiuchi M, Adachi K, Tomatsu A, Chino M, Tadeda S, Tanaka Y, Maeda Y, Sato N, Mitsutomi N, Sugahara K, Chiba K. Bioorg Med Chem. 2005:425–432. doi: 10.1016/j.bmc.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Hale JJ, Neway W, Mills SG, Hajdu R, Keohane CA, Rosenbach M, Milligan J, Shei GJ, Chrebet G, Bergstrom H, Card D, Koo GC, Koprak SL, Jackson JJ, Rosen H, Mandala S. Bioorg & Med Chem Let. 2004;14(12):3351–3355. doi: 10.1016/j.bmcl.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 13.Lim HS, Park JJ, Ko K, Lee MH, Chung SK. Bioorg Med Chem. 2004;14:2499–2503. doi: 10.1016/j.bmcl.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Bioorg Med Chem Lett. 2003;13:3401–3404. doi: 10.1016/s0960-894x(03)00812-6. [DOI] [PubMed] [Google Scholar]
- 15.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Bioorg Med Chem Lett. 2004;14:4903–4906. doi: 10.1016/j.bmcl.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. J Biol Chem. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro S, Buechler D, Enz A, Pombo-Villar E. Tetrahedron Lett. 1994;35:1173–1176. [Google Scholar]
- 18.Barton DHR, Vonder Embse RA. Tetrahedron. 1998;54:12475–12496. [Google Scholar]
- 19.Glenn T, Perich JW, Johns RB. Aust J Chem. 1992;45:1225–40. [Google Scholar]
- 20.Antczak K, Szewczyk J. Phos Sulf. 1985;22:247–251. [Google Scholar]
- 21.Rozhko LF, Klochkov SG, Ragulin VV, Tsvetkov EN. Russ J Gen Chem. 1999;69:1093–1096. [Google Scholar]
- 22.Yokomatsu T, Sato M, Shibuya S. Tetrahedron Asymm. 1996;7:2743–2745. [Google Scholar]
- 23.Jiao XY, Chen WY, Hu BF. Synth Comm. 1992;22:1179–1186. [Google Scholar]
- 24.Shapiro G, Buechler D, Ojea V, Pombo-Villar E, Ruiz M, Weber HP. Tetrahedron Lett. 1993;34:6255–6258. [Google Scholar]
- 25.Villanueva JM, Collignon N. Tetrahedron. 1983;39:1299–1305. [Google Scholar]
- 26.Williams L, Zhang Z, Shao F, Carroll PJ, Joullié MM. Tetrahedron. 1996;52:11673–11694. [Google Scholar]
- 27.For a review of vinyl phosphonate formations see: Minami T, Motoyoehiya J. Synthesis. 1992:333–349.
- 28.Xu Y, Qian L, Prestwich GD. Org Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- 29.Selective cleavage of the acetonide in 3a or 3b while retaining N-tert-butoxy-carbamate was unsuccessful by: Dowex 50×8 in EtOH; Camphorsulfonic acid in MeOH; and 0.5M HCl/THF; and low yielding by cat. p-TsOH in H2O/MeOH; and trifluoroacetic acid/CH2Cl2 followed by Boc2O, 10% Na2CO3 in Dioxane/H2O.
- 30.Yonezawa Y, Shimizu K, Yoon K, Shin C. Synthesis. 2000;5:634–636. [Google Scholar]
- 31.Tse B. J Am Chem Soc. 1996;118:7094–7100. [Google Scholar]
- 32.Li Z, Racha S, Dan L, El-Subbagh H, Abushanab E. J Org Chem. 1993;58:5779–5783. [Google Scholar]
- 33.Sun C, Bittman R. J Org Chem. 2004;69:7694–7699. doi: 10.1021/jo0487404. [DOI] [PubMed] [Google Scholar]
- 34.Lal B, Pramanik BN, Manhas MS, Bose AK. Tetrahedron Lett. 1977;23:1977–1980. [Google Scholar]
- 35.Reddy PG, Pratap RV, Kumar GDK, Mohanty SK, Baskaran S. Eur J Org Chem. 2002:3740–3743. [Google Scholar]
- 36.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- 37.Coste J, Le-Nguyen D, Castro B. Tetrahedron Lett. 1990;31:205–208. [Google Scholar]
- 38.Jones L, II, Schumm JS, Tour JM. J Org Chem. 1997;62:1388–1410.The authors further suggest Urgaonkar S, Verkade JG. J Org Chem. 2004;69:5752–5755. doi: 10.1021/jo049325e.
- 39.Reaction times were longer for runs where Pd/C accumulated on the walls of the reaction flask.
- 40.Preparations of meta-substituted anilines were performed as described in Ref. 38 (Jones II, L.; Schumm, J. S.; Tour, J. M.).
- 41.Nyasse B, Grehn L, Ragnarsson U. Chem Commun. 1997:1017–1018. [Google Scholar]
- 42.Parrill AL, Wang D, Bautista DL, Van Brocklyn JR, Lorincz Z, Fischer DJ, Baker DL, Liliom K, Spiegel S, Tigyi G. J Biol Chem. 2000;275:39379–39384. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- 43.Awad AS, Ye H, Huang L, Li L, Foss FW, Macdonald TL, Lynch KR, Okusa MD. Am J Phys. 2006;290:1516–1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 44.Matloubian M, Lo CG, Cinamon G, Lesneski MG, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 45.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H. Nature Chem Biol. 2006;2:434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]