

Abstract
This past year, the National Institutes of Health announced an ambitious translational research initiative with the explicit goal of transforming biomedical research in the United States to emphasize research on human health related issues (1,2). This research announcement stressed the fact that animal models of human disease are often inadequate and hence it was argued that there was a need to focus directly on the study of human populations and biologic samples. The research announcement evoked a mixed response from the scientific community (3) partly because existing experimental approaches and animal models have been extremely successful in defining relevant issues and therapeutic strategies in biomedical research.
Given that it is imprudent to abandon effective experimental paradigms, it is incontrovertible that the ultimate goal of the research initiative, “to translate the remarkable scientific innovations we are witnessing into health gains for the nation” (1) is laudable. However a myopic and primary focus on human disease and on human tissue introduces a plethora of research risks and concerns that could potentially complicate data interpretation and retard scientific progress. While some of these issues are generic when one extrapolates from animal models to the human circumstance, others are more specific to the cardiovascular system in general and to the study of cardiocyte biology in particular. This brief review will highlight some of these.
A mouse is not a man
At the outset an issue that should be acknowledged is that lessons learned from small animal models may not precisely recapitulate the human circumstance. This is particularly striking when evaluating adaptive and maladaptive changes in the cardiocyte in response to external insults such as hypertension or myocardial infarction that in turn define the physiology of congestive heart failure. Certainly, other, non-myocyte related factors influence the cardiac muscle response to pathologic stimuli but in the interests of focus and brevity, this review will focus primarily on the cardiocyte. Three areas are particularly noteworthy, both because there is a robust literature to support their importance in rodent models and also because human and rodent biochemistry in these areas are quite distinct: 1. contractile protein isoform shifts; 2. sarcolemmal and sarcoplasmic reticular calcium handling; and 3. adrenergic receptor biology.
First, with regard to isoform shifting, the discovery that contractile protein isoforms, and in particular the myosin heavy chain, were transcriptionally modifiable in response to external demands placed upon the heart, was critical to our current understanding of cardiac plasticity (4,5). However, this observation was made in rodent models in which the predominant myosin heavy chain isoform in the adult heart, accounting for ∼90% of the total complement, is the αα heterodimer or the “fast” V1 isoform. It is well-described that in response to pressure overload and other pathologic insults, there is a shift so that the ββ or V3 isoform becomes predominant. This shift has energetic implications as it contributes to the depressed myofibrillar ATPase and decreased rates of shortening seen in hypertrophic and failing rodent myocardium. In humans, and large animals, it seems clear that similar myosin isoform shifts occur (6). However, since adult human cardiac muscle is predominately comprised of V3 myosin, the contribution of these shifts in defining the mechanical and energetic properties of the myocardium is likely far less than in rodents. Moreover, recent work in a transgenic mouse model (7) has linked changes in interstitial fibrosis to this myosin isoform shift suggesting that the influences on remodeling may be both more profound and more species specific than previously presumed. Thus, reasoning from mouse to man provides useful insights into the processes that are available to the heart to moderate its functional status, but to assume precise correspondence is potentially misleading.
A similar issue arises when considering the calcium handling machinery of the heart, and in particular the relative contribution of various regulatory moments in calcium handling to cardiac relaxation. In a series of elegant experiments, Bers and others (8,9) have established clear species differences in calcium handling. In the normal adult mouse cardiocyte, the SR-calcium pump contributes ∼95% of the calcium uptake machinery required to ensure relaxation, whereas in human cardiocytes, the mechanisms that account for calcium removal from the contractile apparatus are far more heterogeneous: the SR-calcium pump accounts for ∼65% of the total effect, and the Na-Ca exchanger and the sarcolemmal calcium pump account for ∼33% and 5%, respectively. Given this, it is not surprising that the cellular mechanisms responsible for prolonging the calcium transient in failing human and murine cardiocytes, while qualitatively similar are quantitatively distinct and, again, extrapolating directly from mouse to man is potentially misleading.
Finally, the relative contribution of β1 and α adrenergic receptor stimulation to the inotropic potential of the heart is radically different in mouse and in man (10–12). In normal human heart, the general consensus is that the ratio of these two receptor subtypes is ∼90:10 whereas in murine heart the ratio is ∼55:45 and reflecting this, the adenylate cyclase activity of normal human cardiac muscle is 3–4 fold greater than mouse. The change in receptor density and physiology in the context of heart failure has been well described in human heart: the ratio of β1 to α adrenergic receptor shifts to ∼1:1 largely as a result of a functional decline in the former (adenylate cyclase activity is reduced) and the contribution of α adrenergic stimulation to augment dP/dt increases from ∼2% to ∼25–30%, a situation that is quite similar to that seen in normal adult mouse. The changes in receptor density and in adenylate cyclase activity in murine models of heart failure are directionally similar to those seen in the failing human heart (13,14). This certainly validates the observations seen in humans and additionally allows for the molecular dissection of these responses using novel transgenic models with gain and/or loss of function of key components in the adrenergic signaling pathways. However, because of the differences in basal adrenergic tone, these murine models may ultimately fail to recapitulate and precisely model the human situation.
What lessons and caveats can we take home from these species comparisons? First, it is likely that qualitative changes in cardiac biology are evident across species, so that lessons learned in well controlled and easily studied animal models have considerable relevance to the understanding of human cardiac biology. However, quantitative comparisons may be misleading. This is especially true given that many studies in human tissue involve end-stage myopathic tissue whereas studies in mice and other animal models involve the explication of transitional biologic processes. It seems quite likely that pathologic maladaptation in the cardiocyte reflects a spectrum of biochemical changes that effect various moments in excitation:contraction coupling, and that the hierarchic contribution of each of these to cardiac adaptation (and maladaptation) is highly species specific. Ultimately, therefore, in order to understand human cardiac biology it is important to carefully evaluate human cardiac muscle (at various stages of maladaptation), which presents a series of technical challenges and potential pitfalls, some of which are discussed below.
What is a control?
The first and perhaps most central issue when dealing with human cardiac tissue specimens is the definition of a control. In animals, where genetic background can be controlled this is a relatively straightforward issue (although not always completely transparent as it becomes clear that the murine genetic background is strain specific and not homogeneous with regard to modifier genes and QTL biology). However, when dealing with human tissue a variety of obvious and some unintended variables must be confronted.
First, patient selection is an issue. Patient age, gender and co-morbidities (known and unknown), genetic background as well as intercurrent pharmacotherapy, both documented and on occasion serendipitous, are all variables that can profoundly influence cardiac muscle performance. The importance of these considerations becomes obvious when one considers several clinical scenarios that might legitimately be construed as appropriate sources of “control” specimens for the study of cardiac muscle mechanical responses: a young healthy woman taking oral contraceptives; a middle aged man with a history of hypertension taking a beta blocker and an angiotensin converting enzyme inhibitor; a young victim of a motorcycle accident who has sustained massive brain injury and has been maintained in a vegetative state on intravenous pressors for several days. Obviously these three circumstances might have all appeared normal if the sole criteria for making this assessment was limited (as it generally is) to an ante mortem clinical assessment of systolic performance, however each scenario introduces confounding variables, related to sarcolemmal protein function, sarcomeric mass and contractile protein make-up as well as the activity of several cytosolic signaling pathways and the phosphorylation status of several sarcomeric proteins How an investigator confronts this issue is problematic but it is certainly critical that a comprehensive description of relevant clinical variables related to control tissue be provided in any publication. More than this, it is also important for the investigator to consider carefully how these and related clinical variables might affect the dependent variables (protein phosphorylation, sarcoplasmic reticulum calcium load, etc.) that are under investigation.
An appreciation of the subtle impact of these considerations can be seen when analyzing the work of Wolff et al (15) and van den Velden et al (16) both of whom have studied the calcium sensitivity and force generation of skinned cardiac muscle fibers from “control” hearts (used as transplant donors) and patients with end-stage cardiomyopathy. Both groups found that the failing human hearts were more calcium responsive than the donor hearts, a result that is at odds with some, but not all, published literature using small rodents (17). Both groups also found that sarcomeric proteins in the donor hearts were heavily phosphorylated (although the site specificity of this was not described), more so than might have been predicted from animal studies, and both investigative groups postulated that this may well have contributed to the relatively decreased calcium responsiveness seen in these specimens. Whether this hyperphosphorylation is a characteristic feature of control human cardiac muscle in situ is impossible to say on the basis of these studies, but it is certainly noteworthy that several of the donors in the Wolfe study (no details are provided in the van den Velde study) were maintained on inotropic support prior to tissue harvesting and all had sustained brain death with the associated catecholamine surge. In contrast, although no details are provided, it would be common practice for end-stage heart failure patients to have been treated with chronic beta-blocker therapy and also to have a significantly diminished β1:α adrenergic receptor density. Whether a comparison of calcium sensitivity between these groups of heart specimens is legitimate is difficult to say, but it is almost certain that the control group was significantly influenced by in situ circumstances at the time of organ harvesting.
More recent work in human myofilament research has been published by Paulus and associates (18,19) suggesting that titin phosphorylation and other cytoskeletal changes might contribute to the cardiomyocyte abnormalities seen in diastolic heart failure. These studies probably represent an incremental advance over previous work, in that endomyocardial biopsies were used rather than samples harvested from post-mortem tissue, however, these studies remained limited by the lack of a control arm that consisted of tissue obtained from a normal patient population.
Tissue handling
A second parallel and highly relevant issue relates to how the tissue specimen is handled prior to experimental manipulation. Our own experience in this regard is very informative. Several years back we initiated a series of experiments evaluating single cell mechanics using human cardiac muscle biopsy specimens. The experimental approach involved isolating and skinning single cell fragments obtained from a fresh or frozen tissue block and then measuring force generation as a function of calcium concentration, thus providing a measure of maximal force per cross sectional area as well as a measure of calcium sensitivity (EC50) We obtained tissue from a number of sources and were initially struck by the variable quality of the samples, which retrospectively was reflective of the length of time between tissue procurement and immersion in liquid nitrogen. Sometimes this was manifested microscopically by the obvious presence of granular deposits, which deformed and distorted the myofibrillar organization but sometimes the findings were more subtle and the sarcomeric striations though apparently intact were faded (see figure 1 for examples). Regardless, these muscle fragments (both control and experimental) did not generate significant force per cross sectional area nor did they physiologically meaningful EC50 measurements. Of significance, however, these same samples had yielded very useful information about changes in gene expression as a function of disease (20).
Figure 1.
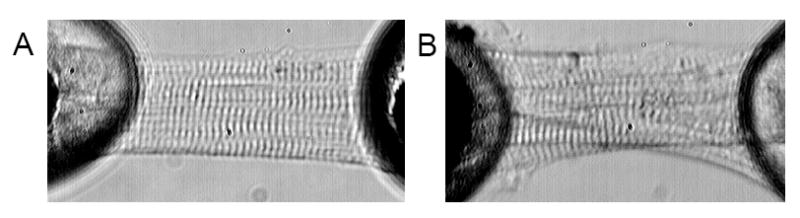
Representative micrographs of cell fragments purified from human cardiac specimens. Panel A shows a cell fragment that was isolated from tissue that had been fast frozen in liquid N2 promptly following surgical excision and generated meaningful F:Ca data. Panel B on the right is a cell fragment that was not preserved as carefully and, despite the intact sarcomere registration, did not generate reliable data. Note the granular deposits and distorted myofibrillar organization.
A second and more subtle issue related to tissue procurement was evident from the analysis of tissue fragments obtained as part of a collaboration with Steve Houser and Ken Margulies at Temple University. This group is exquisitely sensitive to issues related to tissue procurement and has put in place a very careful protocol for tissue collection that includes prompt immersion in liquid nitrogen following muscle excision (21). However, the interests of this group in large part relate to the analysis of ion channel behavior and intracellular calcium handling so obtaining intact and viable cells from cardiac muscle is a primary concern. The cell isolation protocol they have employed involves extensive in situ cardioplegia followed by perfusion with modified Krebs-Henseliet buffer contained BDM and collagenase. Subsequently single cells are isolated from small tissue fragments. The isolated cultured myocytes obtained using this protocol showed a striking reduction in net calcium uptake by the sarcoplasmic reticulum and a marked delay in the calcium transient in cells from myopathic hearts relative to controls (22), findings that have advanced our understanding of the cellular mechanisms underlying human heart failure. When we evaluated single cell mechanics from these same tissue specimens, we were very impressed with the quality of the samples and with the consistency of the mechanical measurements (as well as by the magnitude of the generated Fmax), but we were struck by the fact that the myofibrillar proteins, in contrast to those seen in the studies of van den Velde and Wolff, were largely dephosphorylated. When control and myopathic tissues were contrasted there was little difference in force generation and in EC50, suggesting that mechanical changes in human heart are reflective of post-translational modifications of cardiac myofilament proteins (Figure 2a).
Figure 2.
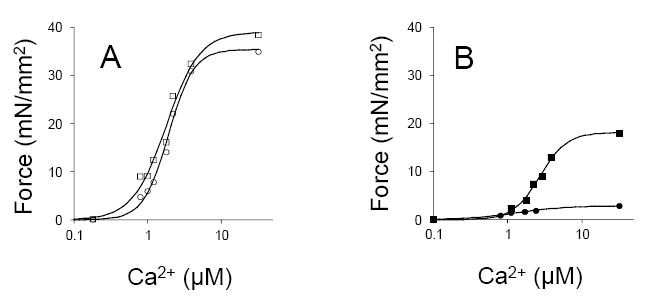
Representative F:Ca relationships in cell fragments obtained from tissue that had undergone extensive cardioplegia and sarcomeric protein dephosphorylation (panel A, on the left) and fragments from tissue that had been fast frozen in the presence of phosphatase inhibitors to preserve the endogenous phosphorylation state (panel B). The square symbols are cells that were isolated from ostensibly normal myocardium whereas the circles indicate cells that were isolated from non-ischemic severely myopathic tissue.
In contrast, we also isolated muscle from patients with and without congestive heart failure undergoing open heart surgery (23, and unpublished). The protocol we employed involved the procurement of a single (∼50 ug) biopsy taken just prior to cardioplegia administration. These samples were then fast frozen in liquid nitrogen and stored in a solution containing phosphatase inhibitors. Single cell mechanics and biochemical data determined in these samples were strikingly different from those seen in the Temple samples: peak force generation was reduced in both control and experimental groups but the heart failure group had depressed function relative to the controls. EC50 was not was not dramatically influenced either by the tissue isolation protocol or by the presence or absence of heart failure. Sarcomeric protein phosphorylation was increased in both control and myopathic specimens relative to that seen in the Temple specimens, but overall was greater in the heart failure samples. Using a similar tissue procurement strategy, Noguchi et al. (24) have shown that in vitro dephosphorylation of cardiac muscle fibers obtained from failing human hearts improves mechanical performance. Of course, our approach and that of the Vermont group could be appropriately criticized because of the failure to acknowledge complex intercurrent cardiac muscle disease and also the heterogeneity of cardiac muscle biology.
However, overall these data suggest that isolated muscle function (Fmax) is highly influenced by the phosphorylation state of the sarcomeric proteins and that protein phosphorylation (or dephosphorylation) is an important mechanism whereby cardiac contractile performance is influenced in human heart failure. Another undeniable conclusion that derives from this series of analyses is that the strategy used for tissue procurement must be tailored to the experimental measurements that are being made. A strategy that is appropriate for genomic or mRNA analysis may not be adequate for evaluation of sarcolemmal or sarcomeric protein biology and strategies that maintain organelle and membrane integrity may well alter important post-translational protein modifications.
Acknowledgement of this important limitation is critical in the evaluation of biologic changes that attend pathologic states in human cardiac muscle and failure to appreciate these issues may well result in misleading conclusions, and leave cardiovascular investigators “lost in translation”.
Acknowledgments
The authors acknowledge support from NIH grants HL62426, HL077195, T32HL67548 and the Eleanor B. Pillsbury Foundation. The authors also wish to thank Steve Houser, Ken Margulies for their advice and Rashad Belin for his contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zerhouni EA. Translational and clinical science—time for a new vision. N Engl J Med. 2005;353(15):1621–3. doi: 10.1056/NEJMsb053723. [DOI] [PubMed] [Google Scholar]
- 2.Zerhouni EA. US biomedical research: basic, translational, and clinical sciences. JAMA. 2005;294(11):1352–8. doi: 10.1001/jama.294.11.1352. [DOI] [PubMed] [Google Scholar]
- 3.Marks AR. Rescuing the NIH before it is too late. J Clin Invest. 2006;116(4):844. doi: 10.1172/JCI28364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahdavi V, Izumo S, Nadal-Ginard B. Developmental and hormonal regulation of sarcomeric myosin heavy chain gene family. Circ Res. 1987;60(6):804–14. doi: 10.1161/01.res.60.6.804. [DOI] [PubMed] [Google Scholar]
- 5.Izumo S, Lompre AM, Matsuoka R, Koren G, Schwartz K, Nadal-Ginard B, Mahdavi V. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy. Interaction between hemodynamic and thyroid hormone-induced signals. J Clin Invest. 1987;79(3):970–7. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–90. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 7.Pandya K, Kim HS, Smithies O. Fibrosis, not cell size, delineates beta-myosin heavy chain re-expression during cardiac hypertrophy and normal aging in vivo. Proc Nat Acad Sci USA. 2006;103:16864–9. doi: 10.1073/pnas.0607700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476(2):279–93. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su Z, LI F, Spitzer KW, Yao A, Ritter M, Barry WH. Comparison of sarcoplasmic reticulum Ca-ATPase in human, dog, rabbit, and mouse ventricular myocytes. JMCC. 2003;35:761–767. doi: 10.1016/s0022-2828(03)00119-6. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee A, Haghani Z, Brady J, Bush L, McBride W, Buja LM, Willerson JT. Differences in myocardial alpha and beta adrenergic receptor numbers in different species. Amer J Physiol. 1983;245:H957–961. doi: 10.1152/ajpheart.1983.245.6.H957. [DOI] [PubMed] [Google Scholar]
- 11.Endoh M, Hiramoto T, Ishihata A, Takanashi M, Inui J. Myocardial alpha 1-adrenoceptors mediate positive inotropic effect and changes in phosphatidylinositol metabolism. Species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circ Res. 1991;68:1179–90. doi: 10.1161/01.res.68.5.1179. [DOI] [PubMed] [Google Scholar]
- 12.Leon-Velarde F, Richalet JP, Chavez JC, Kacimi R, Rivera-Chira M, Palacios JA, Clark D. Inter and intra-species related differences in the regulation of the cardiac autonomic system. Comp Biochem Physiol B Biochem Mol Biol. 1998;119:819–23. doi: 10.1016/s0305-0491(98)00059-5. [DOI] [PubMed] [Google Scholar]
- 13.Perrino C, Naga Prasad SV, Schroder JN, Hata JA, Milano C, Rockman H. Restoration of beta-adrenergic receptor signaling and contractile function in heart failure by disruption of the betaARK1/phosphinositide 3-kinase complex. Circulation. 2005;111:2579–87. doi: 10.1161/CIRCULATIONAHA.104.508796. [DOI] [PubMed] [Google Scholar]
- 14.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman H. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and ventricular rarefaction. J Clin Invest. 2006;116:1547–60. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolff MR, Buck PH, Stoker SW, Greaser ML, Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98(1):167–76. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van den Velden J, Papp Z, Zaremba R, Boontje NM, deJong JW, Owen VY, Burton PB, Goldmann P, Juaquet K, Steinen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57(1):37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 17.deTombe PP. Altered contractile function in heart failure. Cardiovasc Res. 1998;37(2):367–80. doi: 10.1016/s0008-6363(97)00275-7. [DOI] [PubMed] [Google Scholar]
- 18.Borbely A, van den Velden J, Papp Z, Bronzwaer JGF, Edes I, Stienen GJM, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circ. 2005;111(6):774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- 19.van Heerebeek L, Borbely A, Niessen HWM, Bronzwaere JGF, van den Velden J, Stienen GJM, Linke WA, Laarman GJL, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circ. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 20.Okafor C, Liao R, Perreault-Micale C, Li X, Ito T, Stepanek A, Doye A, de Tombe P, Gwathmey JK. Mg-ATPase and Ca+ activated myosin AtPase activity in ventricular myofibrils from non-failing and diseased human hearts—effects of calcium sensitizing agents MCI-154, DPI 201-106, and caffeine. Mol Cell Biochem. 2003;245:77–89. doi: 10.1023/a:1022813726734. [DOI] [PubMed] [Google Scholar]
- 21.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84(4):435–44. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- 22.Piacentino V, 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92(6):651–8. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 23.Jweied EE, McKinney RD, Walker LA, Brodsky I, Geha AS, Massad MG, Buttrick PM, de Tombe PP. Depressed cardiac myofilament function in human diabetes mellitus. Am J Physiol Heart Circ Physiol. 2005;289(6):H2478–83. doi: 10.1152/ajpheart.00638.2005. [DOI] [PubMed] [Google Scholar]
- 24.Noguchi T, Hunlich M, Camp PC, Begin KJ, El-Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P. Thin-filament based modulation of contractile performance in human heart failure. Circulation. 2004;110:982–7. doi: 10.1161/01.CIR.0000139334.43109.F9. [DOI] [PubMed] [Google Scholar]