

Abstract
Our objective was to determine if highly active antiretroviral therapy (HAART), previously shown to ameliorate several pathological features of HIV encephalitis (HIVE) in a SCID mouse model, would also reduce additional established pathological features of HIV: cognitive dysfunction, TNF-α production, and reduced MAP-2 expression. SCID mice with HIVE and control mice inoculated with uninfected monocytes were administered HAART or saline. The HIV pathological features evaluated included astrogliosis, viral load, neuronal apoptosis, MAP-2 expression, mouse TNF-α mRNA production and learning acquisition and retention. HAART reduced the HIV induced viral load, and the astro- and microgliosis as previously observed; this effect was extended to HIV-induced increases in TNF-α mRNA production. In contrast, although HIV produced the cognitive deficits previously observed and also decreased MAP-2 expression in the area surrounding injected HIV-infected human monocytes, HAART did not attenuate these effects. Interestingly, there was no neuronal apoptosis evident at the time-point reflecting the above pathology. The results of this study combined with previous reports indicate that HAART reduces TNF-α mRNA, viral load and astrogliosis; however, HAART does not improve HIV-induced cognitive dysfunction or MAP-2 decreases. These results suggest that viral load, astrogliosis, TNF-α and apoptosis are not prominent in the pathogenesis of early functional deficits related to decreased MAP-2 expression or cognitive dysfunction in HIVE in SCID mice.
Introduction
HIV-associated dementia (HAD), which includes both cognitive and motor dysfunction, affects approximately 10% of HIV infected individuals (Gonzalez-Scarano 2005). Since the advent of highly active antiretroviral therapy (HAART) in 1996, the incidence of HAD has decreased by 50% (from 20% to 10%) while the prevalence has increased (6.6:100 in 1994 compared to 10.1 in 2000) (McArthur 2003). Therefore there is a need to gain a better understanding of the specific effects of HAART regimens on HAD pathogenesis.
Due to the uncertainty about which specific features of HAD pathology, termed HIV-encephalitis (HIVE), might correlate with the clinical expression of dementia, HAD pathogenesis is incompletely defined (Kaul 2001, Wesselingh 2001). Common features of HIVE include infected mononuclear phagocytes (i.e. macrophages and microglia) and astrocytes (Takahashi 1996), multinucleated giant cells, diffuse astrogliosis and increased numbers of mononuclear phagocytes, and increased cytokine expression (e.g. TNF-α) (Gonzalez-Scarano 2005). Other features, such as high CNS viral load and neuronal apoptosis, are less consistently correlated with the development of HAD (Everall 1991, Seilhean 1993, Wiley 1994, Asare 1996, Adle-Biassette 1999, Johnson 1996, McArthur 1997).
A SCID mouse model of HIVE was developed to help further elucidate the pathogenesis of HAD (Tyor 1993, Persidsky 1996, Avgeropoulos 1998, Griffin 2004, Cook 2005). The brains of these SCID mice inoculated intracerebrally with HIV-infected human monocytes characteristically have HIV-infected human mononuclear phagocytes, multinucleated giant cells, increased human cytokine expression, and increased levels of astrocytes and microglia, most notably, astrogliosis (Tyor 1993, Persidsky 1996, Griffin 2004, Cook 2005, Griffin 2007). Furthermore, these infected SCID mice with characteristic HIVE neuropathology have cognitive deficits (Avgeropoulos 1998, Griffin 2004, Griffin 2007). The pathological and cognitive deficits in HIVE SCID mice are similar to those seen in humans with HAD. A recent study demonstrated that HAART significantly reduced, but did not eliminate, HIV in the SCID brains and also significantly reduced astrogliosis ratings (Cook 2005).
It is unclear whether the cognitive dysfunction associated with HIV infection is related to neuronal death (e.g., neuronal apoptosis), neuronal physiological dysfunction or some combination of the two (Everall 1991, Seilhean 1993, Wiley 1994, Asare 1996, Adle-Biassette 1999). To explore possible causes of neuronal abnormalities associated with brain HIV infection, and to assess the efficacy of HAART on these possible causes and cognitive behavior, cognitive behavior of SCID mice was evaluated in a 2 × 2 factorial design in groups with or without HIV-infection, and half of each of the two groups receiving either HAART or saline. Cognitive behavior was assessed using a visually cued spatial learning problem in a Morris water maze. After completion of cognitive assessment, brain tissue from the different groups of mice was examined for astrogliosis, viral load, and MAP-2 expression. The effects of the HIV-infection procedure on neuronal death and HIV infection and HAART on TNF- α were also evaluated in separate experiments: 1) comparing neuronal apoptosis in brains of HIV-infected and uninfected mice and 2) measuring TNF- α mRNA in HIV mice treated with HAART or saline. We hypothesized that HAART would attenuate these features of HIVE in SCID mice.
Materials and Methods
Animals
CB-17 SCID male mice, 4 weeks of age, (Charles River Laboratory, Wilmington, MA, USA) were single housed in microisolator cages (biosafety level-3 equivalent) for at least one week prior to experimentation. Cages, bedding, food, and water were sterilized before use. All animal protocols were approved by the Medical University of South Carolina’s IACUC and were consistent with the guidelines of the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised 1996).
Method of Infection
Primary human macrophages obtained from Dr. Howard Gendelman, University of Nebraska Medical Center were prepared for injection as HIV-infected, or control uninfected cells as previously described (Cook 2005, Griffin 2007). Briefly, cells cultured for 7 days were divided in half. One of the cultures was infected with 1 ml HIV-1ADA, an R5 strain of HIV, the other with only the media as a control. Immunostaining for p24 of cells collected two weeks after viral infection indicated that approximately 30–50% expressed viral antigen. Both HIV-infected and uninfected cells were resuspended in phosphate buffered saline (PBS) for inoculation.
HAART Cocktail and Intraperitoneal Catheter Surgery
To eliminate the aversive effects of repeated intraperitoneal (i.p.) HAART injections, which could confound the evaluation of cognition, a catheter was surgically implanted into the peritoneal cavity two days prior to inoculation. The procedure was adapted from previous reports (Griffin 2003, Kelley 1997). Briefly, a silastic catheter was inserted through the abdominal wall (1.2 cm length) into the peritoneal cavity and secured by sutures (See Supplemental Materials). The catheter body was routed subcutaneously to the top of the skull and the access port was secured to the skull using a light-cured resin (Groseclose, 1998). AZT (GlaxoWellcome), lamivudine (GlaxoWellcome), and indinavir (derived from Crixivan capsules – Merck) were dissolved under sterile conditions in sterile 0.9% NaCl (Baxter Healthcare Corporation) at a concentration of 2mg/ml and delivered through this catheter three times per day (see Table).
Table 1.
Experimental Design.
| Experiment | HIV Status | Treatment | Measurements | Duration of Experiment |
|---|---|---|---|---|
| 1 | HIV+ | HAART (n=8) | Morris Maze MAP-2 Percent Infected Cells Gliosis | 18 days |
| Vehicle (n=8) | ||||
| HIV− | HAART (n=8) | |||
| Vehicle (n=8) | ||||
| 2 | HIV+ | HAART (n=6) | TNF-α mRNA | 14 days |
| Vehicle (n=7) | ||||
| HIV− | HAART (n=5) | |||
| Vehicle (n=7) | ||||
| 3 | HIV+ | No Treatment (n=3) | Neuronal Apoptosis | 18 days |
| HIV− | No Treatment (n=3) |
Number of days after IC inoculation of cells that mice were sacrificed for pathological analyses.
Apoptosis was analyzed 3 ways (see Methods) including Apoptag staining of brain sections, caspase -3 detection by immunofluorescence in brain sections, and Western blots for caspase-3 expression in brain.
Inoculations and HAART treatment
As previously described (Cook 2005, Griffin 2007) infected (n=16) or uninfected (n=16) macrophages (105 in μl of PBS) were injected into the right frontal lobe under anesthesia (Table). HAART (15mg/kg; n=8 in each group above) or saline (n=8 in each group above) was delivered i.p. immediately after inoculation and continued 3 times daily (0800, 1200, 1600hrs) for the duration of the experiment. Studies involving mouse TNF-α mRNA production were completed on a different set of HIV-infected and control mice that underwent HAART or vehicle treatment for two weeks (Table), as previously described (Cook 2005)
Assessment of Cognition
Cognitive behavior was assessed by determining acquisition (learning) and retention (memory) of the spatial relationship of visual cues in the environment surrounding a Morris water maze. The characteristics of the maze and surrounding environment were previously described (Avgeropoulos 1998). Tests in the Morris water maze began six days after inoculation and continued over a 15-day period. The procedure consisted of four phases: habituation, sensory/motor evaluation, problem acquisition, and problem recall, which are described in detail in previous publications (Avgeropoulos 1998, Griffin 2004). Data for mice which did not meet a learning acquisition criteria of ≥ 20% reduction latency to reach the goal platform [(Day 1 – Day 6)/Day 1] were excluded from the retention test phase of the experiment.
Tissue Collection and Staining Procedures
Immediately following the retention test, mice were administered a final dose of HAART or vehicle injection and sacrificed by cervical dislocation under anesthesia (Table). The tissue collection, sectioning and staining procedures have been previously described (Cook 2005). Briefly, five micron coronal sections were alternately stained using GFAP (1:750; Chemicon), F4/80 (1:20, Caltag), EBM11 (1:50, DAKO), and p24 (1:20, DAKO) and reviewed using light microscopy. Severity of astro- and microgliosis was evaluated by a blinded reviewer (JCE) using a grading scale (0= little to no gliosis, 1= mild gliosis, 2= moderate gliosis, 3= severe gliosis) as previously described (Griffin 2004, Cook 2005).
Additionally, MAP-2 (1:200 dilution; Chemicon) expression was evaluated in the sections using the mouse on mouse (M.O.M.) staining method (Vector). Because MAP-2 staining was reduced in a very limited area surrounding the injected human monocytes (i.e. one slide per brain), the density of the staining was determined from one 40x image taken from the right hemisphere (area of infection and MAP-2 stain reduction). A similar sample was taken from the left hemisphere (no infection) to control for differences in staining between animals. The density of MAP-2 staining was quantified with densitometry (NIH Image) by determining the percent decrease in expression in the right compared to the left hemisphere.
Neuronal Apoptosis Evaluation
Three different types of assays were conducted to evaluate neuronal apoptosis in a separate group of mice inoculated as above with HIV+ (n=3) and HIV− (n=3) monocytes. Neither of the groups was behaviorally tested or treated with HAART (see Table for experimental design). First, Apoptag Peroxidase (Chemicon) staining was performed in tissue sections that were fixed in 1% paraformaldehyde and then post-fixed in ethanol:acetic acid (2:1), according to the manufacturer’s protocol. Then, sections were incubated with TdT enzyme, followed by incubation with anti-digoxigenin peroxidase to stain for fragmented DNA. Peroxidase substrate was applied and the sections were counterstained with methyl green to visualize membrane blebbing and chromatin condensation. Positive controls were generated by pre-treating one slide with Dnase I (Sigma) before quenching endogenous peroxide, again according to the kit’s instructions. Slides were viewed under a light microscope at 20x and 40x (Olympus). Additionally apoptosis was also evaluated by measuring activation of caspase-3 in neurons using double immunofluorescence in sections near the xenograft and from the hippocampus (See Supplemental Methods). Finally, apoptosis was evaluated using western blots for cleaved caspase-3 in brain tissue not used for slides (50–100 mg) (See Supplemental Methods).
TNF alpha Quantification
Intervening tissue sections that were not used for immunocytochemistry were saved and stored at −70° C for RNA extraction. The tissue was homogenized and the RNA was extracted according to the RNAwiz (Ambion) protocol. Approximately 1 μg of RNA was utilized for cDNA synthesis. First strand cDNA synthesis was carried out using the High Capacity cDNA Archive kit (Applied Biosystems) according to manufacturer’s instructions. Samples were run in a thermal cycler (Eppendorf) at 25°C for 10 minutes immediately followed by 37°C for 50 minutes. Semi-quantitative RT-PCR reactions were carried out using forward and reverse primers for mouse TNF-α (5′-CCTGTAGCCCACGTCGTAGC, 3′-TTGACCTCAGCGCTGAGTTG, probe-CTGGAAGACTCCTCCCAGGTATATGGGTTC, product size-374), cDNA template, and Taq Polymerase. Samples were amplified at 95° C for 30 seconds, followed by 37 cycles at 95° C for 15 seconds, 60° C for 30 seconds, and 72° C for 30 seconds. The PCR products were run for 1 hour on a 2% agarose gel at 100 volts. The products were transferred onto a nylon membrane, probed using DIG-labeled probes and then densities of the bands were analyzed using Quantity One (Roche). Additionally, GAPDH levels (3′-CTCAGTGTAGCCCAGGATGC; 5′-ACCACCATGGAGAAGGCTGG; probe- GTGGAAGGACTCATGACCACAGTCCATGC) were determined using the same methodology to control for the amount of RNA recovery. To avoid carry-over contamination, two separate labs are utilized for the procedures described above and dedicated pipettors where used for each step.
Data analysis
Data from the behavioral tests and pathological evaluation were analyzed with 2(Infection) × 2(HAART) factorial analysis of variance (ANOVA)s with repeated measures as appropriate. Post-hoc analysis was conducted using Fisher’s PLSD. Because TNF-α mRNA was not detectable in most non-infected control samples, the scale of measurement did not meet the normal distribution requirements for parametric statistical tests. Thus data from the RT-PCR experiments were analyzed with nonparametric tests and presented as Medians for measures of central tendency. A Median test was used to compare all four groups associated with the above 2 × 2 design and a Kolmogorov-Smirnov test was used to compare the two HIV-infected groups. The null hypothesis was rejected at p values less than 0.05.
Results
Brain Pathology
After cognitive testing, brains from infected and uninfected HAART and saline treated mice were examined for astrogliosis, microgliosis, percentage of HIV- infected human cells, and MAP-2 expression (see Methods and Table). As previously reported (Cook 2005), the increases seen in HIV+ mice compared to controls for astrogliosis, microgliosis and percentage of HIV-infected cells were reduced by HAART (data not shown).
MAP-2, a histological correlate of abnormal physiology normally expressed in neuronal dendrites and cell bodies, was also examined in the four groups of mice after cognitive testing was completed (see Methods and Table). MAP-2 expression was reduced in the brain area immediately adjacent to injected human macrophages (Figure 1A–C), an area also exhibiting the most intense staining for astrogliosis and microgliosis (Cook 2005). Human macrophage numbers were similar for all groups and MAP-2 staining in the location of the inoculated human cells was reduced for both uninfected and infected mice. These sections with reduced staining in the area of the graft in the right hemisphere were compared via densitometry with similar anatomical areas of the uninoculated left hemisphere to determine the percent decrease in MAP-2 expression associated with inoculation (Figure 1D). As noted in Figure 1D, MAP-2 expression was reduced to a greater extent in both groups of HIV infected compared to the uninfected groups with HAART having no influence. A 2(Infection) × 2(HAART) ANOVA provided statistical support for the infection-induced reduction in MAP-2 expression [Infection: F(1,12)=7.944, p<0.015] and indicated no effect of HAART [F(1,12) < 1] or its interaction with Infection [F(1,12)< 1]. Thus HIV, directly or indirectly, damaged dendritic structure of neurons, and HAART did not attenuate this effect.
Figure 1. HIV-infection reduces MAP-2 expression regardless of treatment.
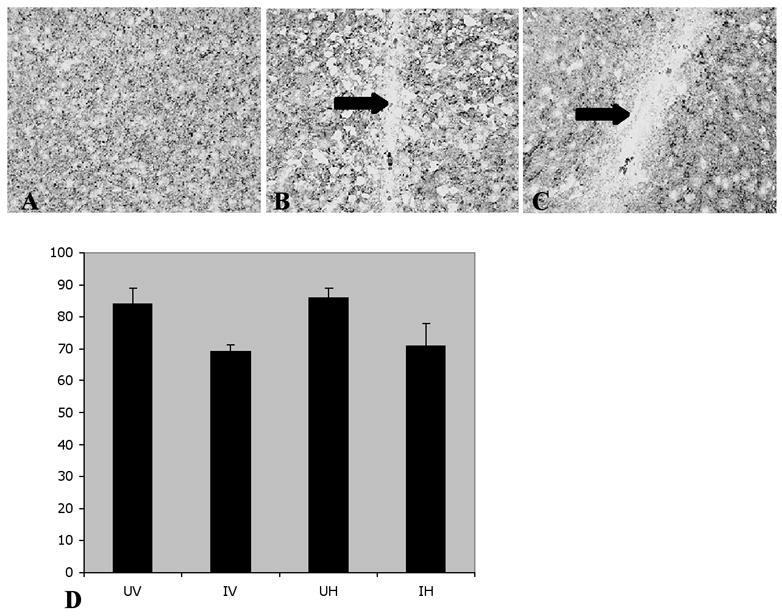
MAP-2 staining in non-injected left hemisphere, an area distant from the graft (A), and near human cells in uninfected (B) and infected brain tissue (C) at 20x magnification. Arrows indicate areas of decreased MAP-2. Areas of reduced density of MAP-2 staining in the right hemisphere were compared to corresponding areas of the left hemisphere to establish percent decrease from normal (100%). The Mean ± SE for UV - uninfected vehicle treated (n=4), IV - infected vehicle treated (n=4), UH - uninfected HAART treated (n=4), and IH - infected HAART treated (n=4) mice are summarized in (D). A 2×2 ANOVA indicated that HIV Infection significantly reduced MAP-2 staining [F(1,12)=7.944, p<0.015], an effect not altered by HAART.
In a separate experiment (see Methods and Table) brains from saline treated and HAART HIV+ and control mice were evaluated for mouse TNF-α (mTNF-α) mRNA. TNF-α a potentially important molecule in the pathogenesis of HAD (Tyor 1995), was detected in all HIV-infected but in very few uninfected mice. Data are expressed as TNF-α mRNA/GAPDH ratios in Figure 2. Because of the zero mTNF-α mRNA values for uninfected mice, data do not meet the measurement scale or the normality requirements for parametric analyses. Hence group data are summarized in Figure 2 as medians and were analyzed with nonparametric statistics. Median analysis of the four groups statistically confirmed the group differences noted in Fig 1 [χ2=10.463 (df,3), p<0.015]. Subsequent analysis of the two infected groups confirmed the reduced mTNF-α mRNA for HAART treated mice [Kolmogorov-Smirnov test: χ2=8.972(df,1), p<0.025]. Thus, HIV infection elevated mTNF-α mRNA, an effect that was reduced but not eliminated by HAART.
Figure 2. HIV induces production of mouse TNF-α mRNA and HAART decreases TNF-α mRNA.
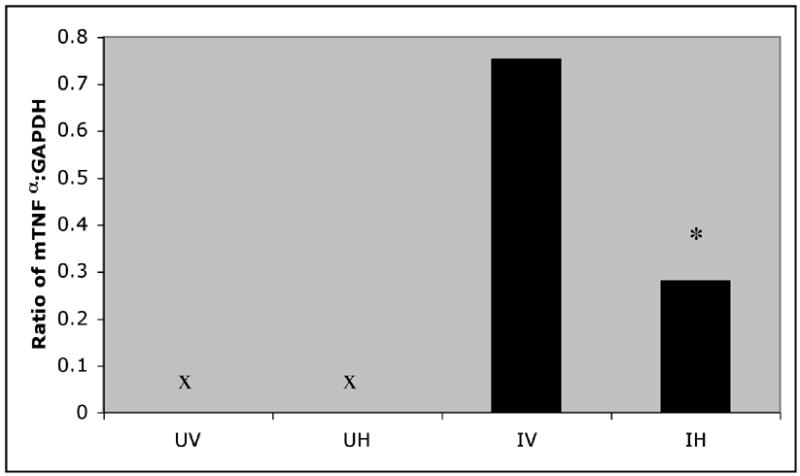
Ratios of mouse TNF-α to GAPDH for brains of UV - uninfected vehicle (n=7), UH - uninfected HAART (n=5), IV - infected vehicle (n=7), and IH - infected HAART (n=6) are summarized as medians. A nonparametric Median test confirmed the group differences noted in the graph: χ2=10.463 (df,3), p<0.015. Follow-up comparison of the IV and IH groups with a Kolmogorov-Smirnov test confirmed the reduced levels for the HAART - treated group: χ2=8.972(df,1), *p<0.025.
Mice in the third experiment (Table) inoculated with HIV-infected (n=3) or uninfected (n=3) monocytes were evaluated for neuronal apoptosis using Apoptag stain, double immunofluorescence for active caspase-3 (n=3), and Western blots for active caspase-3 (n=3). Representative photomicrographs of Apoptag stain are shown in Figure 3 for a positive control mouse (Fig 3A) and HIV-infected mouse (Fig 3B). Comparison of the two photomicrographs provides no evidence of neuronal apoptosis, blebbing or chromatin condensation 18 days after intracerebral inoculation of infected human macrophages (Fig 3B). The additional two methods for assessing apoptosis also provided no evidence for HIV induced apoptosis (data not shown) at this single time point.
Figure 3. Representative photomicrographs of Apoptag staining.

in (A) positive control brain (treated with DNAse I) and (B) HIV infected brain (40X). The absence of brown staining or evidence of blebbing or chromatin condensation associated with the methylgreen staining in the HIV-infected brain (B) indicates the absence of HIV-induced apoptosis.
Cognitive Dysfunction
Potential interactive effects of HIV and HAART on learning and memory were examined in Exp-1 (see Table) using acquisition (learning) and retention (memory) of a spatially cued task in a Morris water maze. Acquisition of the learning problem was defined as a significant reduction in latency to reach a submerged goal platform across six days of testing. Data for the acquisition phase were analyzed with a 2(Infected vs Uninfected) × 2(HAART vs Vehicle) × 2(Day-1 vs Day-6) factorial analysis of variance. All four groups learned the location of the submerged platform, as indicated by a significant reduction in latencies over the six day period (time reduced by 40% – 64% [F(1,33)= 32.04, p<0.001]) (data not shown).
Retention of the acquired spatial learning problem was assessed by comparing the latency to locate the goal platform on the final day of acquisition (Day 6) with the latency obtained on the retention test conducted after a seven day hiatus from training. Latencies on the retention test were improved in comparison to the last day of acquisition for UV mice (Figure 4), suggesting a memory consolidation process during the training hiatus. In contrast, for HIV-infected mice, latencies during the retention test did not differ significantly from those on the last day of acquisition. A 2(Infection Status) × 2(Treatment) ANOVA on the percent change scores (i.e., Acquisition Day 6 - Retention day latency)/Day 6 × 100; Figure 4) indicated a significant Infection factor [F(1,25)=6.35, p<0.019], suggesting that HIV interferes with memory consolidation. However, the Infection × HAART interaction was not significant [F(1,25)=0.025, p<0.875] indicating that the HAART regimen used in this experiment did not improve the cognitive deficit associated with HIVE.
Figure 4. HIV infected mice exhibit cognitive deficits regardless of treatment.
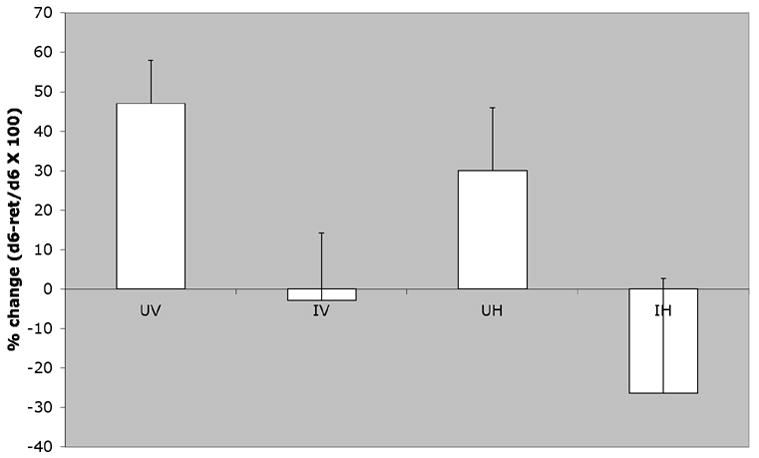
Percent change in time required to reach the goal in a spatial learning task after a 6-day hiatus for UV - uninfected vehicle treated (n=7), IV - infected vehicle treated (n=8), UH - uninfected HAART treated (n=7), and IH - infected HAART treated (n=8) mice (Mean ± SE). Performance was enhanced after the break in training for uninfected mice (UV and UH), but not infected mice [Infection: F(1,25)=6.35, p<0.019]. The HAART factor and its interaction with Infection were not significant.
Discussion
This is the first study of HAART effects on HIVE pathology, cognitive dysfunction, and neuronal pathology under the same experimental paradigm in this HIV infection SCID mouse model. The study confirms previous reports of viral load, astrogliosis, elevated TNF alpha and the cognitive deficits in this model (Tyor 1993, Persidsky 1996, Avgeropoulos 1998, Griffin 2004), and that HAART reduces viral load and astrogliosis (Cook 2005). However in contrast to these positive aspects of HAART, under the conditions of this experiment the treatment did not prevent cognitive deficits or the HIV-induced reductions in MAP-2 expression.
The present study confirms our previous report (Cook 2005) that HAART reduced astrogliosis and the percentage of HIV infected cells in brains of SCID mice. This study also extends the therapeutic effect of HAART to include a reduction in TNF-α, a proinflammatory cytokine that may play a role in HAD pathogenesis. Although there is differential control of TNF-α transcription and translation (Keffer 1991), studies comparing mRNA with protein levels have consistently shown a positive correlation (Iida 2001, Miller 1998). In addition, SCID mice with HIVE were reported to have elevated TNF- α mRNA (Persidsky 1997) which can be significantly reduced by treatment with a TNF- α inhibitor (Persidsky 2001). Furthermore, the observed elevation of TNF-α in HIVE SCID mice is consistent with post-mortem examination of HIV infected humans diagnosed with HIVE (Tyor 1992, Wesselingh 1993). In the present study HAART reduced, but did not completely eliminate, mTNF-α mRNA production, an effect likely related to the HAART-induced reduction, but not elimination, of viral load. While the conclusion that the TNF- α mRNA levels determined in this study reflect TNF-α protein is not certain, the preponderance of clinical and experimental reports noted above suggest that similar changes would be noted for the TNF-α protein. Future studies will examine brain cytokine protein levels using ELISA’s.
Consistent with previous reports (Zink 2002, Persidsky 2001), HIV reduced MAP-2 expression in the SCID mouse model of HIVE. Since MAP-2 is an indicator of dendritic outgrowth necessary for normal neuronal function, it serves as a marker for abnormal neurophysiology. In this light, the abnormal neurophysiology noted for HIV infected mice is consistent with their cognitive dysfunction observed in this and previous studies (Avgeropoulos 1998, Griffin 2004, Griffin 2007). This relationship of HIV induced cognitive dysfunction and reduction in MAP-2 expression is strengthened by the fact that HAART did not attenuate either. Thus, despite the HAART-induced decreases in viral load, mTNF-α mRNA and astrogliosis, the physiological and behavioral dysfunctions remained.
Neuronal apoptosis, contrary to expectations, was not evident 18 days after inoculation of HIV-infected monocytes. This result suggests that concomitant apoptosis is not a necessary condition for HIV-associated neuronal pathology; however, it does not exclude the possibility that neuronal death occurs at a different time after infection, or perhaps through non-apoptotic mechanisms. The present study also suggests that viral load, at least early in HIVE, is not correlated with neuronal dysfunction. This finding is consistent with human studies that failed to show a correlation between CNS viral load and HAD (Glass 1995, Johnson 1996) but contrasts with other studies that have suggested there may be a CNS viral load effect on HAD (Ellis 1997, McArthur 1997). Although the present study establishes that neuronal dysfunction was not related to existing viral load, it does not exclude the possibility that viral load at other time points might be related to HIVE in the SCID mouse model.
Overall, the results of this study suggest that neuronal dysfunction in the SCID mouse model of HIVE, as indicated by MAP-2 expression and impaired cognition, is important in the early pathogenesis and may translate to the human condition. Moreover, the noted cognitive deficits on the spatial learning task in the present study are consistent with the previously reported short- and long-term potentiation impairment in the hippocampus of HIV infected mice (Zink 2002), a brain region important in spatial memory and learning. Since reduced MAP-2 and weakened electrophysiological properties reflect impaired membrane qualities and disruption of secondary messenger systems (Zink 2002), the combined studies provide a strong basis for a synaptic irregularity associated with the HIV related cognitive deficit in this model. Our interpretation is that early cognitive dysfunction in HAD is related more to a neurotoxin(s)-induced physiological dysfunction of neurons rather than frank neuronal death. It is also plausible that as HIVE is prolonged or becomes more severe, additional factors contribute to neuronal death, which would not be apparent in this model. Nevertheless, the early physiological dysfunction of neurons likely provides a window of opportunity to reverse cognitive dysfunction in HIV infected mice (and perhaps patients) using treatments that target this neurotoxin(s).
Taken together, the experiments of this study suggest that indicators of cognitive and neuronal dysfunction associated with HIVE can be expressed even when HAART attenuates astrogliosis, TNF-α expression, and viral load. Although the results suggest that viral load, TNF alpha and astrogliosis are not primary contributors to cognitive deficits at the time they were observed, they do not rule out the contribution of these HIV-related phenomena at other stages of either the neuronal and/or cognitive dysfunction. It is also possible that certain viral proteins, such as tat, even at low levels are neurotoxic and might play an important role in the pathogenesis of HAD (Nath 2002). Although much attention has been focused on the significance of high viral load, TNF-α, apoptosis, and astrogliosis in the pathogenesis of HAD, the present study suggests that more attention might be directed at other pathologies and neurotoxins that have been implicated in dementia.
Supplementary Material
Acknowledgments
The authors would like to thank Elaine Terry for her outstanding technical support. This project was supported by a grant from the National Institutes of Health (R01 MH62697-05).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Adle-Biassette H, Chretien F, Wingertsmann L, et al. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- Asare E, Dunn G, Glass J, et al. Neuronal pattern correlates with the severity of human immunodeficiency virus-associated dementia complex. Usefulness of spatial pattern analysis in clinicopathological studies. Am J Pathol. 1996;148:31–38. [PMC free article] [PubMed] [Google Scholar]
- Avgeropoulos N, Kelley B, Middaugh L, et al. SCID mice with HIV encephalitis develop behavioral abnormalities. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:13–20. doi: 10.1097/00042560-199805010-00003. [DOI] [PubMed] [Google Scholar]
- Cook J, Dasgupta S, Terry E, et al. Highly active antiretroviral therapy of HIV-encephalitis in immunodeficient mice. Ann Neurol. 2005;57:795–803. doi: 10.1002/ana.20479. [DOI] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, et al. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Hsia K, Spector SA, et al. Cerebrospinal fluid human immunodeficiency virus type 1 RNA levels are elevated in neurocognitively impaired individuals with acquired immunodeficiency syndrome. Ann Neurol. 1997;42:679–688. doi: 10.1002/ana.410420503. [DOI] [PubMed] [Google Scholar]
- Everall IP, Luthert PJ, Lantos PL. Neuronal loss in the frontal cortex in HIV infection. Lancet. 1991;337:1119–1121. doi: 10.1016/0140-6736(91)92786-2. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Griffin WC, Middaugh LD. Acquisition of lever pressing for cocaine in C57BL/C6 mice: effects of prior Pavlovian conditioning. Pharm Biochem Behav. 2003;76(3–4):543–549. doi: 10.1016/j.pbb.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Griffin WC, III, Middaugh LD, Cook JE, Tyor WR. The severe combined immunodeficient (SCID) mouse model of human immunodeficiency virus encephalitis: deficits in cognitive function. J Neurovirol. 2004;10:109–115. doi: 10.1080/13550280490428333. [DOI] [PubMed] [Google Scholar]
- Griffin WC, Middaugh LD, Tyor WR. Chronic cocaine exposure in a SCID mouse model of HIV encephalitis. Brain Research. 2007;1134:214–219. doi: 10.1016/j.brainres.2006.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groseclose CH, Draughn R, Tyor WR, Middaugh LD. Technique for long-term intracranial cannulation in mice. J Neurosci Meth. 1998;79:31–36. doi: 10.1016/s0165-0270(97)00158-1. [DOI] [PubMed] [Google Scholar]
- Iida KT, Shimano H, Kawakami Y, et al. Insulin up-regulates tumor necrosis factor-alpha production in macrophages through an extracellular-regulated kinase-dependent pathway. J Biol Chem. 2001;276:32531–7. doi: 10.1074/jbc.M009894200. [DOI] [PubMed] [Google Scholar]
- Johnson RT, Glass JD, McArthur JC, Chesebro BW. Quantitation of human immunodeficiency virus in brains of demented and nondemented patients with acquired immunodeficiency syndrome. Ann Neurol. 1996;39:392–395. doi: 10.1002/ana.410390319. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor : a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BM, Bandy AE, Middaugh LD. A novel, chronic and detachable indwelling jugular catheterization procedure for mice. Physiol Behav. 1997;62:163–167. doi: 10.1016/s0031-9384(97)00029-2. [DOI] [PubMed] [Google Scholar]
- McArthur JC, McClernon DR, Cronin MF, et al. Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Ann Neurol. 1997;42:689–698. doi: 10.1002/ana.410420504. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, et al. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- Miller L, Hunt JS. Regulation of TNF-α production in activated mouse macrophages by progesterone. J Immunol. 1998;160:5098–5104. [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, McComb R, et al. Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol. 1996;149:1027–1053. [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Buttini M, Limoges J, Bock P, Gendelman HE. An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J NeuroVirol. 1997;3:401–416. doi: 10.3109/13550289709031186. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, Rasmussen J, Zheng J, Gearing A, Gendelman HE. Reduction in glial immunity and neuropathology by a PAF antagonist and an MMP and TNF-alpha inhibitor in SCID mice with HIV-1 encephalitis. J Neuroimmunol. 2001;114:57–68. doi: 10.1016/s0165-5728(00)00454-9. [DOI] [PubMed] [Google Scholar]
- Seilhean D, Duyckaerts C, Vazeux R, et al. HIV-1-associated cognitive/motor complex: absence of neuronal loss in the cerebral neocortex. Neurology. 1993;43:1492–1499. doi: 10.1212/wnl.43.8.1492. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Wesselingh SL, Griffin DE, et al. Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol. 1996;39:705–711. doi: 10.1002/ana.410390606. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Glass JD, Griffin JW, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Power C, Gendelman HE, Markham RB. A model of human immunodeficiency virus encephalitis in scid mice. Proc Natl Acad Sci U S A. 1993;90:8658–8662. doi: 10.1073/pnas.90.18.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyor WR, Wesselingh SL, Griffin JW, McArthur JC, Griffin DE. Unifying hypothesis for the pathogenesis of HIV-associated dementia complex, vacuolar myelopathy, and sensory neuropathy. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:379–388. [PubMed] [Google Scholar]
- Wesselingh SL, Power C, Glass JD, et al. Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Thompson KA. Immunopathogenesis of HIV-associated dementia. Curr Opin Neurol. 2001;14:375–379. doi: 10.1097/00019052-200106000-00018. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Achim C. Human immunodeficiency virus encephalitis is the pathological correlate of dementia in acquired immunodeficiency syndrome. Ann Neurol. 1994;36:673–676. doi: 10.1002/ana.410360422. [DOI] [PubMed] [Google Scholar]
- Zink WE, Anderson E, Boyle J, et al. Impaired spatial cognition and synaptic potentiation in a murine model of human immunodeficiency virus type 1 encephalitis. J Neurosci. 2002;22:2096–2105. doi: 10.1523/JNEUROSCI.22-06-02096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.