

Abstract
Using the generalized stepwise mutation model, we propose a method of estimating the relative mutation rates of microsatellite loci, grouped by the repeat motif. Applying ANOVA to the distributions of the allele sizes at microsatellite loci from a set of populations, grouped by repeat motif types, we estimated the effect of population size differences and mutation rate differences among loci. This provides an estimate of motif-type-specific mutation rates up to a multiplicative constant. Applications to four different sets of di-, tri-, and tetranucleotide loci from a number of human populations reveal that, on average, the non-disease-causing microsatellite loci have mutation rates inversely related to their motif sizes. The dinucleotides appear to have mutation rates 1.5–2 times higher than the tetranucleotides, and the non-disease-causing trinucleotides have mutation rates intermediate between the di- and tetranucleotides. In contrast, the disease-causing trinucleotides have mutation rates 3.9–6.9 times larger than the tetranucleotides. Comparison of these estimates with the direct observations of mutation rates at microsatellites indicates that the earlier suggestion of higher mutation rates of tetranucleotides in comparison with the dinucleotides may stem from a nonrandom sampling of tetranucleotide loci in direct mutation assays.
The mutation rate (ν) at genetic loci and the effective population size (N) are two basic parameters for understanding the genetic structure of a population. Numerous population genetic studies addressed the question of estimating these two quantities individually as well as simultaneously (1–3). For populations with large generation time and overlapping generations, direct estimation of effective population size is problematic (4). Likewise, mutation rates at most genetic loci are not large enough to be directly measured, and inference of true mutational events is also complicated by assumptions regarding biological relationships of the observed pedigrees (5, 6).
One alternative is to estimate the mutation rates by indirect procedures that rely on allele frequency distributions in populations (1–3, 5, 7). In these studies it was assumed that (i) population is in a mutation-drift balance, so that the allele frequency distributions in populations could be expressed in terms of Nν, the product of effective population size (N), and the rate of mutation (ν); and (ii) that each mutation yields an allele previously not seen in the population (the infinite allele model). The first assumption is reasonable for most large populations, whereas the second may not apply to all loci. In particular, for microsatellite loci, where polymorphisms are caused by differences in the number of tandem repeats, the infinite allele model does not apply (8–10).
Recent theoretical work suggests that the within-population variance of repeat unit sizes is proportional to the product of two basic parameters, N and ν (11, 12), and this relationship holds even when the pattern of mutational changes at microsatellite loci is an arbitrary, not necessarily single-step symmetric, random walk. Based on this theory, we present an analysis of data on allele size distributions at several microsatellite loci from a number of populations. The aim is to estimate the mutation rates at loci in relative terms when the loci are grouped by their repeat motifs (e.g., di-, tri-, and tetranucleotides) and the alleles are distinguished by their number of repeats only. We show that a two-way ANOVA of the within-population variance of allele sizes from such data provides estimates of mutation rates at microsatellite loci up to a multiplicative constant. The ANOVA model also allows testing of the underlying assumptions of such analysis. We also compare the estimates of the variance of the within-population variance with their theoretical predictions under a general random-walk model of mutations (13).
METHODS
Within-Population Variance at a Microsatellite Locus.
Consider a population of diploid individuals and a locus with alleles indexed by integer numbers representing the numbers of repeats. The expectation of the estimate of the within-population component of genetic variance,
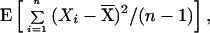 |
where Xi is the size of the allele in the ith chromosome present, 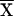 = (1/n)∑iXi, and n is the number of chromosomes sampled, is equal to V/2, where
= (1/n)∑iXi, and n is the number of chromosomes sampled, is equal to V/2, where
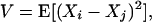 |
1 |
and Xi, Xj are sizes of two alleles randomly selected from the population. We consider the equilibrium value of V in a stepwise mutation model with sampling from the finite allele pool.
In particular, we assume that in each generation, the genotypes of all individuals are sampled with replacement from the 2N chromosomes present in the previous generation (Fisher–Wright model), and, furthermore, that each chromosome independently is subject, with probability ν per generation, to a mutation that replaces an allele of size X with an allele of size X + U, where U is an integer-valued random variable with probability generating function ϕ(s) = ∑u=−∞∞suPr[U = u] = E(su), defined for s in the neighborhood of 1. It has been demonstrated in ref. 12, based on the theory of Chakraborty and Nei (14) that
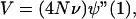 |
2 |
where N is the effective population size, ν is the mutation rate, ψ(s) = [ϕ(s) + ϕ(1/s)]/2 is the probability-generating function of the symmetrized distribution of allele size change following mutation, and ψ"(1) is the second derivative of ψ(s), evaluated at s = 1. The definition of ψ(s) also implies that ψ"(1) is the variance of allele size change caused by each mutation, and hence, it is positive. The equilibrium within-population variance of allelic size Xi is equal to V/2. Note that the above theory is general enough to accomodate arbitrary distributions of allele size change, including multistep and asymmetric cases.
Eq. 1 indicates that V relates to the second moment of the difference of sizes of the two randomly selected alleles, Xi − Xj, which is a random variable with a symmetric distribution, even when each single mutation event produces asymmetric and arbitrary size changes of alleles.
Estimation of Relative Mutation Rates at Different Microsatellite Loci.
Eq. 2 can be used to estimate the relative magnitude of mutation rates in loci of different motif types. Suppose that in populations j = 1, . . . , J, a number of loci of types i = 1, . . . , I, have been investigated. Each type i of loci includes loci k = 1, . . . , Ki (Ki is the number of loci of type i). Let us assume that νi is the mutation rate for each locus of motif type i. The effective size of population j is denoted Nj.
Suppose that for each type-population-locus combination ijk, the variance of the within-population differences of allele sizes (Vijk) has been estimated. Taking natural logarithms of both sides of Eq. 2 we obtain,
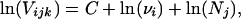 |
3 |
under the assumption that the term ψ"(1) does not vary from one locus to the other. If we denote
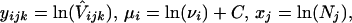 |
then Eq. 3 can be written in the form of a linear model, corresponding to the type I (fixed effects) two-way ANOVA (15),
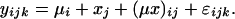 |
4 |
The error term of Eq. 4, ɛijk, represents the uncertainty of the estimate of Vijk and possible variability of mutation rates among loci within each motif type, and the interaction term (μx)ij measures the goodness of fit of the linear model expressed by Eq. 3.
Analysis of the components of variance using the two-way ANOVA can help answer the following questions: (i) is the dependence of mutation rate on locus motif-type significant? and (ii) is the dependence on population size significant?
When the dependence on locus motif type dominates in the analysis, the estimated motif type-specific levels of μi are equal, up to an additive constant C, to logarithms of motif-type-specific average mutation rates. Thus, the motif-type-specific average mutation rates themselves can be determined up to a multiplicative constant.
Estimation of the Variance of the Within-Population Variance.
The usual estimator of the within-population variance,
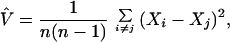 |
5 |
itself has a considerable variance. This variance has been determined for the population model described above, using the coalescence- and probability-generating function approaches (13). For large sample sizes (n ≥ 20),
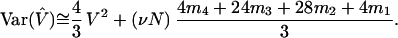 |
6 |
In Eq. 6, V is as given in Eq. 2, while mi, i = 1, 2, 3, 4 are the first four factorial moments of the distribution of allele size change caused by each mutation. In the special case of the symmetric single-step model, we have m1 = 0, m2 = 1, m3 = −3, and m4 = 12, which implies Var(V̂) = (4/3)V2 + (1/3)V. This is well approximated by (4/3)V2, and we will use this latter value as our theoretical variance of V̂.
DATA
Eqs. 3 and 4 suggest that data on allele sizes at multiple motif types of loci in several populations are used preferably to obtain relative average rates of mutations at different motif types of loci and estimate the influence of the differing population sizes as well as the possible interactions. We use two sets of data that satisfy this requirement. The first set is described in refs. 16–18. It includes allele frequencies at eight dinucleotide (FLT1, D13S118, D13S121, D13S71, D13S122, D13S197, D13S193, and D13S124), five trinucleotide (PLA2A, DM, SCA, DRPLA, and HD), and five tetranucleotide (THO1, CSF1R, F13A1, CYP19, and LPL) repeat loci in samples from nine populations (unrelated Caucasians from the Centre d’Étude du Polymorphisme Humain panel; Germans; Brazilian Whites; Brahmins from Uttar Pradesh, India; Sokoto from Nigeria; Benin; Brazilian Blacks; Japanese; and Chinese). The second data set consists of allele size distributions at four trinucleotide (HUMFABP2, PLA2A, D6S366, and AR) and eight tetranucleotide (HPRT, CSF1R, THO1, F13A1, CYP19, LPL, RENA4, and FESFPS) loci, surveyed in four populations (Caucasians, Blacks, Hispanics, and Asians), as reported in refs. 19 and 20.
As will be shown later in more detail, a two-way ANOVA of these two sets of data indicates that at least for the major population groups of humans, the relative effective sizes of the different populations contribute little to the variation of the within-population variance of allele sizes at different microsatellite loci.
In addition, we used two other sets of data in which the ANOVA could be performed with respect to one-way classification of data.
The first of these was provided by L. B. Jorde (University of Utah). It contains data on 30 tetranucleotide loci in 13 human populations. Ref. 21 lists the names of these loci and the 13 sample populations. We analyzed these data, grouped by three major populations (75 African individuals, 78 Asians, and 90 Europeans) to obtain an estimate of the average within-population variance of allele size for the tetranucleotide loci as a group and to check whether the population effects are significant.
In addition, we also collected allele size data on all the chromosome 19 di-, tri-, and tetranucleotide loci from the Genome Data Base (GDB) for which the allele size distributions are available. These data are specifically related to the study (22) that estimated mutation rates in these groups of loci directly from a sample of selected loci. The data are available for a single population (Caucasians) so that a one-way ANOVA can be used to examine the effect of differences of the relative mutation rates at these motif types of loci. These data include allele frequencies at 116 dinucleotide loci (details available on request), 3 neutral trinucleotide loci (D19S190, D19S261, and SCN1B), 1 disease trinucleotide locus (DM), and 12 tetranucleotide loci (D19S244–D19S47, D19S250–D19S255, and EPOR).
RESULTS
Table 1 shows the summary results of the ANOVA of the natural logarithms of locus- and population-specific variances of allele sizes (lnVijk) for the first two sets of data (16–20). For both sets of data, the component of variation due to population differences is not significant (P > 0.75)—i.e., xj = lnNj is nearly constant over all j in both sets of data. Likewise, the interaction variance component is not significant (P > 0.96) in both analyses. In other words, the linear model of Eq. 4 provides a good fit to both sets of data. In contrast, the ratio MStype/MSwithin is significant (P < 0.001) for both sets of data. In other words, the differences of mutation rates among the di-, tri-, and tetranucleotides in the data from refs. 16–18 and that among the tri- and tetranucleotides in the data from refs. 19 and 20 are sufficient to account for variation in the lnVijk values (Eq. 4).
Table 1.
Two-way ANOVA for the natural logarithm of the within-population variance (V) of microsatellite loci
In view of the absence of significant dependence on populations, the estimate of lnV for the ith motif type of loci, averaged over populations, V̂i = ∑jklnVijk/(KiJ), is also an estimate of μi = lnνi up to an additive constant. In Fig. 1 we plotted the means and standard deviations of the lnV̂i values for the different motif types of loci for the data from refs. 16–18 (Fig. 1a), 19 and 20 (Fig. 1b), and GDB (Fig. 1c).
Figure 1.

○, Estimates of logarithmic mutation rate (lnνi) up to an additive constant C, for microsatellite loci, grouped by their repeat motif types (di-, tri-, and tetranucleotides): (a) data from refs. 16–18, (b) data from ref. 20, and (c) data from GDB. The vertical bars represent the ±1 SD intervals. •, Estimates for disease-causing trinucleotides within the normal range of allele sizes.
Four out of the five trinucleotide repeat loci examined in ref. 17 are disease-associated: DM, SCA, DRPLA, and HD. The four trinucleotide loci in the GDB data include one disease-causing locus (DM). However, allele size frequency data at these loci are obtained only from unaffected healthy individuals, and thus, at each of these disease-causing trinucleotide loci all alleles are without any pathologic phenotypic effect (normal size range). To examine whether, within the normal size ranges of alleles at these loci, the mutation rate is different from the nondisease trinucleotide loci, we estimated lnV̂i values for the disease-associated trinucleotide loci separately. Fig. 1 a and c depict these estimates separately for the neutral and disease-causing trinucleotides.
Two observations are evident from Fig. 1. First, for the loci without any disease implications, the rate of mutation appears to be inversely related with the repeat lengths. The dinucleotide repeats have the largest mutation rate, and the tetranucleotide repeats, the smallest. Under the linear model of Eq. 4 in the absence of significant population and interaction effects, the exponential function of the difference lnV̂i1 − lnV̂i2 measures the relative mutation rate νi1/νi2. Thus, in relation to the tetranucleotide loci, the dinucleotide repeat loci appear to have a 1.48–2.16 times higher mutation rate, and the nondisease trinucleotide loci appear to have a 1.22–1.97 times higher mutation rate, depending on the data set (Table 2). Second, the disease-associated trinucleotide loci appear to have a mutation rate higher than even the dinucleotides. Their mutation rate is 3.86–6.89 times higher than in tetranucleotides. This observation is intriguing, because the data on disease-causing trinucleotide loci contain alleles within the normal range, shorter than the premutation or full-blown mutation alleles. There is no direct data reported thus far regarding the instability of the normal range alleles at these loci.
Table 2.
Summary of the estimated natural logarithm of the within-population variance
| Source of data | Neutral loci
|
Disease loci
|
||||||
|---|---|---|---|---|---|---|---|---|
| Dinucleotide
|
Trinucleotide
|
Tetranucleotide
|
Trinucleotide
|
|||||
| n |
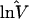 i i
|
n |
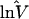 i i
|
n |
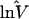 i i
|
n |
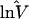 i i
|
|
| Ref. 16 | 8 | 1.51 | 1 | 1.00 | 5 | 0.74 | 4 | 2.09 |
| Ref. 20 | — | — | 4 | 1.25 | 8 | 0.57 | — | — |
| GDB | 115 | 1.38 | 3 | 1.19 | 12 | 0.99 | 1 | 2.92 |
lnV̂i = Σjkln(Vijk)/(KiJ). n, Number of loci.
In Table 2 numerical values of estimates of lnVi are shown along with the number of loci within each locus motif types for the three data sets.
As shown in Table 1, the two-way ANOVA of data from refs. 16–18 and 20 reveal that the relative mutation rates for the different motif types of loci are significantly different from each other. The trend of mean lnVi estimates grouped by motif types of loci in the GDB is the same as the one in the other two data sets, although it is not statistically significant (P = 0.24; see Discussion and Conclusions).
The above observations, based on the ANOVA model of the logarithm of the within-population variance of allele sizes, depend on the normality assumptions. Because for some data sets these are not strictly satisfied (e.g., the GDB dinucleotides have an apparently bimodal distribution), we additionally conducted a nonparametric test. In Fig. 2 we plotted the empirical cumulative distribution functions of the logarithm of the within-population variance, in which data from each population for the same locus are treated as replicate observations, because the effects of population size differences were not significant (Table 1).
Figure 2.

Empirical cumulative distribution functions of microsatellite loci grouped by their repeat motif types: (a) data from refs. 16–18, (b) data from Hammond et al. (20), and (c) data from GDB. In a–c, the lnVi values for the same locus from all populations are treated as replicate observations.
The empirical cumulative distributions in each graph of Fig. 2 indicate that the lnVi values are inversely related with the motif length of the loci. For Fig. 2a, the dinucleotide loci have a significantly larger lnVi than the tetranucleotide loci (P = 6 × 10−6, by the Mann–Whitney U test) and the disease trinucleotide loci have a significantly larger lnVi than the dinucleotides (P = 2 × 10−3, by the Mann–Whitney U test). For data from ref. 20 in Fig. 2b, the trinucleotides have a larger lnVithan the tetranucleotides (P = 7 × 10−4, by the Mann–Whitney U test). The difference between the dinucleotide and tetranucleotide loci in the GDB data in Fig. 2c is in the same direction (di- is larger than tetra-); however, the difference is not significant (P = 0.24, by the Mann–Whitney U test).
These relative mutation rate estimates have a large variance, because the within-population variance of the allele size distribution has a considerable variance itself. Table 3 shows numerical results concerning this variability. All four sets of data are used in these numerical computations, including data from ref. 21, which provide estimates of variance of V̂i/2 based on 30 tetranucleotide loci. We might note that a one-way ANOVA did not reveal any significant effect of population size (P = 0.88) data from ref. 21. The observed variances of V̂i/2 for each set of loci are the unbiased sample estimates based on V̂i/2. The theoretical values of the variance of within-population variance from the model are computed from the approximation, (4/3)(V̂i/2)2 in Eq. 6.
Table 3.
Estimated variance of the within-population variance (V̂i/2), computed directly from data, and its theoretical value predicted from the model
| Motif type of loci (repeat length) | Number of loci | Data source | Variance of V̂i/2
|
|
|---|---|---|---|---|
| Observed* | Theoretical† | |||
| Di | 8 | Refs. 16–18 | 39.3 | 56.0 |
| 115 | GDB | 36.4 | 52.5 | |
| Tri-neutral | 1 | Refs. 16–18 | — | 10.3 |
| 4 | Ref. 20 | 10.4 | 23.8 | |
| 3 | GDB | 244.7 | 146.3 | |
| Tetra | 5 | Refs. 16–18 | 4.7 | 9.7 |
| 8 | Ref. 20 | 2.0 | 5.8 | |
| 30 | Ref. 21 | 54.5 | 35.8 | |
| 12 | GDB | 33.6 | 38.1 | |
| Tri-disease | 4 | Refs. 16–18 | 81.0 | 158.1 |
| 1 | GDB | — | 457.3 | |
Unbiased sample estimates of variance of V̂i/2.
(4/3)(V̂i/2)2, where V̂i = Σjk Vijk/(KiJ).
Two observations can be made from Table 3. First, the observed variances of the estimates of within-population variance of allele sizes are large but consistent with the theoretical variances. The departures from the theoretical values are not related to the type of loci studied. The large variance is an indicator of highly skewed sampling distribution of estimates of the within-population variance, which may reflect large interlocus variation of mutation rates within each repeat motif type.
DISCUSSION AND CONCLUSIONS
The theory presented here provides an approach to estimation of mutation rates (in relative terms) at microsatellite loci. Applications of this theory to four different sets of data indicate that, as a group, the dinucleotide repeat loci appear to be evolving at a rate 1.5–2 times greater than the tetranucleotide loci. The non-disease-related trinucleotide loci have mutation rates intermediate between the di- and tetranucleotides. In contrast, the disease-related trinucleotides have a mutation rate higher than the dinucleotides, even within the normal allele size range.
Apparently, our conclusions do not agree with those of Weber and Wong (22) who, by direct observations of mutations at 28 chromosome-19 loci, found that the average mutation rate for tetranucleotides is nearly four times higher than that for dinucleotides. Analogous findings were reported in refs. 23 and 24. The paper (22) is widely cited, and the presumed high mutation rates of tetranucleotides are frequently invoked to explain other observations, as in the Discussion of ref. 25.
However, it must be noted that of the 24 in vivo mutations documented in ref. 22, 8 relate to 2 tetranucleotide loci (D19S244 and D19S245). If these two loci are excluded from their analysis the trend of relative mutation rates in their direct assay of mutations becomes the same as ours: the dinucleotides have a higher mutation rate than the trinucleotides and the trinucleotides higher than the tetranucleotides. Similarly, in ref. 23, four out of six observed mutation events are contributed by a single locus, D9S748. Conclusions in Hastbacka et al. (24) concerning the mutation rate of tetranucleotides also are based on fluctuation analysis of a single locus linked with a disease gene.
Results reported in the present paper are consistent over three data sets. In the survey of ref. 20 the loci are selected from different chromosomes. The data of refs. 16–18 include eight dinucleotide loci from chromosome 13, but the tri- and tetranucleotide loci are spread over a number of chromosomes. Data analyzed from GDB represent di-, tri-, and tetranucleotide loci of chromosome 19 alone. This seems to be a sufficiently representative sample.
As evident from Eq. 2, the estimated variances generally are proportional to the product of mutation rate and the variance of the symmetrized allele size changes by mutations (i.e., to νiψi"(1). Based on allele size data in populations, there is no direct way of testing whether ψi"(1) significantly varies across loci. However, almost all allele size changes associated with mutations reported by Weber and Wong (22) and others (26–29) are changes by a single repeat, which yields ψ"(1) = 1. This suggests that, perhaps with the exception of the disease-related trinucleotides, the distributions of allele size changes caused by mutations are not drastically different across the di-, tri-, and tetranucleotide loci.
Another assumption of our model is that the allele variation in populations is at equilibrium. In the nonequilibrium case, Eq. 2 is replaced by V = {1 − exp[−t/(2N)]}(4Nν)ψ"(1) (12, 13). Factor {1 − exp[−t/(2N)]} may contribute to the term ɛijk in ANOVA. The fit to the model supports this assumption. Also, the assumption that the effective population size (N) is constant throughout the evolution of the population, which is implicit in the derivation of Eq. 2, is not a limiting feature of our data interpretation. This is so because the linear model of the relationship of the logarithmic variance to the mutation rate also applies to rapidly expanding populations as long as the contrasts of different motif types of loci are made from data obtained from the same set of populations (10, 12).
The relative mutation rates of di-, tri-, and tetranucleotides are significantly different (Table 1) by both ANOVA and nonparametric Mann–Whitney U test for the data in refs. 16–18 and 20. The difference exists but is not statistically significant for the loci collated from the GDB. Fig. 2 provides a possible reason for these findings. The distributions of the tetranucleotides in the GDB appear bimodal and have a disproportionately larger number of tetranucleotide loci that appear to have a higher mutation rate. Because the GDB tetranucleotide loci coincide with the sample assayed by Weber and Wong (22), this is consistent with the fact that these authors recorded a higher mutation rate for these loci compared with the dinucleotide loci. The greater representation of these loci in GDB is also responsible for making their average mutation rate nonsignificantly smaller than that of the dinucleotide loci.
There exists a theoretical explanation that might appear to reconcile the apparently high mutation rates in tetranucleotides observed in direct studies (22–24), with low variances calculated by us. It is sufficient to assume the existence of constraints for the number of DNA repeats in a locus and to assume that these limits are stricter for the tetranucleotides than for other repeat loci. A theoretical model based on similar hypotheses was recently published (30). In that model, constraints on the number of DNA repeats combined with the stepwise mutation model imply frequency distribution of alleles that are uniform or even u-shaped. Inspection of the empirical frequencies reported for 30 tetranucleotide loci (21), however, reveals binomial and Poisson-like tails, more consistent with the absence of constraints.
Acknowledgments
This work was supported by grants GM 41399 (R.C. and D.N.S.), GM 45861 (R.D.), and GM 58545 (R.C., M.K., and L.J.D.) from the National Institutes of Health, and DMS 9409909 (M.K.) from the National Science Foundation and by the Keck’s Center for Computational Biology at Rice University (M.K).
Footnotes
Abbreviation: GDB, Genome Data Base.
References
- 1.Nei M. Am J Hum Genet. 1977;29:225–232. [PMC free article] [PubMed] [Google Scholar]
- 2.Zouros E. Genetics. 1979;92:623–646. doi: 10.1093/genetics/92.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty R, Neel J V. Proc Natl Acad Sci USA. 1989;86:9407–9411. doi: 10.1073/pnas.86.23.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nei M. Molecular Population Genetics and Evolution. Amsterdam: North–Holland; 1975. [PubMed] [Google Scholar]
- 5.Rothman E D, Neel J V, Hoppe F M. Am J Hum Genet. 1981;33:617–628. [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty R, Stivers D N. J Forens Sci. 1996;41:667–673. [PubMed] [Google Scholar]
- 7.Chakraborty R. Ann Hum Biol. 1981;8:221–230. doi: 10.1080/03014468100004991. [DOI] [PubMed] [Google Scholar]
- 8.Valdes A M, Slatkin M, Freimer N B. Genetics. 1993;133:737–749. doi: 10.1093/genetics/133.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shriver M D, Jin L, Chakraborty R, Boerwinkle E. Genetics. 1993;134:983–993. doi: 10.1093/genetics/134.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Rienzo A, Peterson A C, Garza J C, Valdes A M, Slatkin M, Freimer N B. Proc Natl Acad Sci USA. 1994;91:3155–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slatkin M. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimmel M, Chakraborty R, Stivers D, Deka R. Genetics. 1996;143:549–555. doi: 10.1093/genetics/143.1.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimmel, M. & Chakraborty, R. (1996) Theor. Popul. Biol., in press. [DOI] [PubMed]
- 14.Chakraborty R, Nei M. Genet Res. 1982;39:303–314. [Google Scholar]
- 15.Sokal R R, Rohlf F J. Biometry. New York: Freeman; 1981. [Google Scholar]
- 16.Deka R, Jin L, Shriver M D, Yu L M, DeCroo S, Hundrieser J, Bunker C H, Ferrell R E, Chakraborty R. Am J Hum Genet. 1995;56:461–474. [PMC free article] [PubMed] [Google Scholar]
- 17.Deka R, Shriver M D, Yu L M, Ferrell R E, Chakraborty R. Electrophoresis. 1995;16:1659–1664. doi: 10.1002/elps.11501601275. [DOI] [PubMed] [Google Scholar]
- 18.Deka R, Majumder P P, Shriver M D, Stivers D N, Zhong Y, Yu L M, Barrantes R, Yin S-J, Miki T, Hundrieser J, Bunker C H, McGarvey S T, Sakallah S, Ferrell R E, Chakraborty R. Genome Res. 1996;6:142–154. doi: 10.1101/gr.6.2.142. [DOI] [PubMed] [Google Scholar]
- 19.Edwards A, Hammond H A, Jin L, Caskey C T, Chakraborty R. Genomics. 1992;12:241–253. doi: 10.1016/0888-7543(92)90371-x. [DOI] [PubMed] [Google Scholar]
- 20.Hammond H A, Jin L, Zhong Y, Caskey C T, Chakraborty R. Am J Hum Genet. 1994;55:175–189. [PMC free article] [PubMed] [Google Scholar]
- 21.Jorde L B, Bamshad M J, Watkins W S, Zenger R, Fraley A E, Krakowiak P A, Carpenter K D, Soodyall H, Jenkins T, Rogers A R. Am J Hum Genet. 1995;57:523–538. doi: 10.1002/ajmg.1320570340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber J L, Wong C. Hum Mol Genet. 1993;2:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]
- 23.Zahn L M, Kwiatkowski D J. Genomics. 1995;28:140–146. doi: 10.1006/geno.1995.1124. [DOI] [PubMed] [Google Scholar]
- 24.Hastbacka J, de la Chapelle A, Kaitila I, Sistonen P, Weaver A, Lander E. Nat Genet. 1992;2:204–211. doi: 10.1038/ng1192-204. [DOI] [PubMed] [Google Scholar]
- 25.Morell R, Liang Y, Asher J H, Weber J L, Hinnant J T, Winata S, Arhya I N, Friedman T B. Hum Mol Genet. 1995;4:85–91. doi: 10.1093/hmg/4.1.85. [DOI] [PubMed] [Google Scholar]
- 26.Petrukhin K E, Speer M C, Cayanis E, Bonaldo N F, Tantravali U, Soares M B, Fischer S G, Warburton D, Gilliam T C, Ott J. Genomics. 1993;15:76–85. doi: 10.1006/geno.1993.1012. [DOI] [PubMed] [Google Scholar]
- 27.Bowcock A, Osborne-Lawrence S, Barnes R, Chakravarti A, Washington S, Dunn C. Genomics. 1993;15:375–386. doi: 10.1006/geno.1993.1071. [DOI] [PubMed] [Google Scholar]
- 28.Kwiatkowski D J, Hanski E P, Weimar K, Ozelius L, Gusella J F, Haines J. Genomics. 1992;12:229–240. doi: 10.1016/0888-7543(92)90370-8. [DOI] [PubMed] [Google Scholar]
- 29.Mahtani M M, Willard H F. Hum Mol Genet. 1993;2:431–437. doi: 10.1093/hmg/2.4.431. [DOI] [PubMed] [Google Scholar]
- 30.Nauta M J, Weissing F J. Genetics. 1996;143:1021–1032. doi: 10.1093/genetics/143.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]