

Abstract
Formation of herpes simplex virus-1 capsids requires the presence of intact scaffolding proteins. The C terminus of the abundant scaffolding protein associates with the major capsid shell protein VP5 through hydrophobic interactions. After cleavage by the viral encoded protease, which removes their C-terminal 25 aa, the scaffolding proteins are released from the capsid. We have used electron cryomicroscopy and computer image processing to determine, to 13 Å, the three-dimensional structures of capsids containing either cleaved or uncleaved scaffolding proteins. Detailed comparisons show that the structures of the outer icosahedral shells are almost identical in the two capsid types. Differences are apparent in the radial distribution of the density inside the capsid shell (within a radius of 460 Å) which represents the scaffolding core. However, in both capsid types, the bulk of this internal density exhibits no icosahedral symmetry. Close examination revealed localized regions of icosahedrally arranged extra density at the interface between the outer shell and the scaffold of protease-minus capsids. Rod-like densities extending inwards for ≈40 Å from the capsid shell are present under four of the six quasi-equivalent triplex positions. Under triplexes Tb, Tc, and Te, the major additional densities appear as pairs with the rods in each pair situated 37 Å apart. We propose that these rods are formed by the C-termini of the scaffolding proteins and represent the sites of interaction between the capsid shell and scaffold.
The herpes simplex virus-1 (HSV-1) capsid is the largest and most complex icosahedral virus particle yet studied structurally. The T = 16 icosahedral capsid shell is 160 Å thick and has a diameter of 1,250 Å. Historically, capsids have been considered to exist in three forms, with B capsids (containing scaffolding proteins) being the progenitors of C (containing viral DNA) and A (empty) capsids. This situation is now known to be more complex with B capsids being classifiable into at least two classes, large cored and small cored (the typical form seen in virus infected cells), depending on the state of their scaffold. Moreover, Newcomb and coworkers (1) recently described a procedure for in vitro assembly of HSV capsids, which generates capsid-sized, spherical particles of distinctive morphology (2). These have been designated procapsids, and in thin section they appear as double shells similar to large cored B capsids. Upon storage at room temperature, procapsids undergo major conformational rearrangements to adopt the more angular appearance typical of mature capsids. The authors propose that procapsids are the progenitors of all other capsid types, but the exact relationships between the various forms have not yet been elucidated.
Compositionally, the most complex capsid type is the small cored B capsid that contains seven structural proteins (3), four of which (VP5, VP19C, VP23, and VP26) constitute the capsid shell. The three remaining proteins (VP21, VP24, and VP22a) are involved in formation and processing of the scaffold. Newly assembled capsids are believed to contain ≈100 copies of the protease encoded by the gene UL26 and >1,000 copies of the scaffolding protein (preVP22a) (4), which is encoded by an overlapping gene, UL26.5 (5, 6). PreVP22a interacts with the major capsid protein, VP5, through its C-terminal 25 aa. (7), and this interaction is essential for capsid assembly (8, 9). The viral protease cleaves itself at two sites to yield VP21 and VP24 and also cleaves the C-terminal 25 aa of preVP22a to yield VP22a (5, 6). Cleavage of preVP22a is an essential maturational step that breaks the link between the scaffold and the capsid shell and converts large cored B capsids to the small cored form (10). If proteolytic cleavage of the scaffolding protein is prevented, the large cored phenotype is maintained. This observation is the case both in HSV mutants in which the protease is inactive (10, 11), or absent (12), or in a baculovirus-based capsid assembly model system in which the protease is inactive (13) or omitted (14, 15).
Although scaffolding proteins play important roles in the assembly of many eukaryotic and prokaryotic viruses, their mode of action is poorly understood. In particular, the nature of the interactions between the scaffold and shell is largely unknown. With the improved resolution of three-dimensional (3D) reconstruction by using electron cryomicroscopy and image processing, it becomes possible to detect structural changes associated with the proteolytic cleavage. Here we report the identification of the sites of contact between the HSV-1 scaffold and capsid shell based on the 3D structural comparison of small cored and large cored B capsids.
MATERIALS AND METHODS
Capsid Preparation.
Wild-type B capsids were purified using established procedures from baby hamster kidney cells infected at 37°C for 16 h with 5 pfu/cell of HSV-1 strain 17. Protease-minus capsids were isolated from Spodoptera frugiperda (Sf21) cells coinfected, as described (14), with 5 pfu/cell of each of five different recombinant baculoviruses expressing VP5, VP19C, VP23, preVP22a, and VP26. The protein compositions of the purified B capsids and protease-minus capsids were confirmed by SDS/PAGE.
Electron Cryomicroscopy.
Capsids were embedded in vitreous ice suspended across the holes in holey carbon grids for electron cryomicroscopic observation (16). Microscope alignment, specimen assessment, and focusing were performed using a Gatan (Pleasanton, CA) slow-scan charge-coupled device camera installed on a JEOL 4000 microscope (17). Focal pairs were obtained by imaging each suitable specimen area twice at different defocus values on Kodak SO163 films with a dosage of 6 electrons/Å2/image (16).
Data Processing.
Selected micrographs were digitized on a Perkin–Elmer 1010M microdensitometer with a step-size corresponding to a specimen resolution of 4.67 Å/pixel. The image quality was assessed quantitatively by evaluating the contrast transfer function rings visualized in the incoherently averaged Fourier transforms of particle images by using the contrast transfer function determination option in ice (18, 19). Only focal pairs with the contrast transfer function first zero of their close-to-focus micrographs within the range between  –
–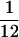 Å−1 were used for further analysis. Based on this analysis, 8 and 10 focal pairs were selected respectively for wild-type B capsid and protease-minus capsid reconstruction.
Å−1 were used for further analysis. Based on this analysis, 8 and 10 focal pairs were selected respectively for wild-type B capsid and protease-minus capsid reconstruction.
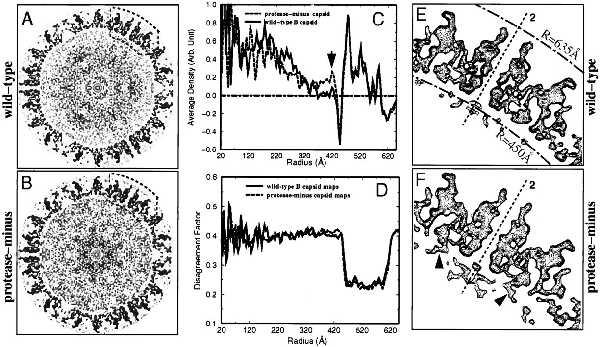
Data processing was carried out on a Silicon Graphics (Mountain View, CA) Power Challenge with 24 processors by using a scheme described (20). The final reconstructions were calculated by Fourier–Bessel synthesis by using Fourier data truncated at 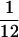 Å−1. To evaluate the reliability of the reconstructions, we randomly split the original images into two half sets and calculated the phase differences or Fourier ring correlation coefficients as a function of spatial frequency between the two reconstructions obtained from the half sets. The effective resolution was based on the phase difference being <45° and the Fourier ring correlation coefficients being twice those of random noise (16). To evaluate the extent of icosahedral symmetry, we calculated a disagreement factor (see Fig. 2D) as a function of radius for two independent reconstructions of each capsid type. Graphics rendering was carried out by using explorer (NAG, Downers Grove, IL) with custom designed modules. Shaded surfaces are displayed at an isosurface value of 0.6σ (SD) from the mean.
Å−1. To evaluate the reliability of the reconstructions, we randomly split the original images into two half sets and calculated the phase differences or Fourier ring correlation coefficients as a function of spatial frequency between the two reconstructions obtained from the half sets. The effective resolution was based on the phase difference being <45° and the Fourier ring correlation coefficients being twice those of random noise (16). To evaluate the extent of icosahedral symmetry, we calculated a disagreement factor (see Fig. 2D) as a function of radius for two independent reconstructions of each capsid type. Graphics rendering was carried out by using explorer (NAG, Downers Grove, IL) with custom designed modules. Shaded surfaces are displayed at an isosurface value of 0.6σ (SD) from the mean.
Figure 2.
Comparison of the outer shells and internal cores of wild-type and protease-minus capsid reconstructions. One central, 4.67-Å thick section traversing twofold axes and Tb triplexes was extracted from wild-type (A) and protease-minus (B) capsids. (C) The densities at the same radius were averaged to show the density distribution as a function of radius for the wild-type (solid curve) and protease-minus (dotted curve) capsid reconstructions. The arrow indicates an additional density peak close to the capsid shell of protease-minus capsids. (D) Disagreement factor (defined as 2 |D1 − D2|/(D1 + D2), where D1 and D2 represents density values in two independent reconstructions, 1 and 2, of each capsid type) was calculated as wild-type (solid line) and protease-minus (dashed line) capsids. The regions enclosed by dashed boxes in A and B are enlarged and shown as contour plots of E and F, respectively. A twofold axis is indicated by dashed lines. Two radial distances from the center of the capsid (R = 450 Å and 635 Å) are marked by dot-dashed arcs in E. Arrowheads in F designate additional rod-like densities present in the protease-minus capsid.
RESULTS
Symmetry of the Scaffolding Protein Core.
Recombinant baculovirus generated capsids containing unprocessed scaffolding protein were made by coexpressing all of the capsid proteins except for the UL26 encoded protease. Because the protease is not expressed, its autoproteolytic products, VP21 and VP24, are not present and the scaffolding protein is in the uncleaved, higher Mr form (preVP22a) and not the processed form (VP22a) found in wild-type B capsids (Fig. 1A) (14, 15).
Figure 1.
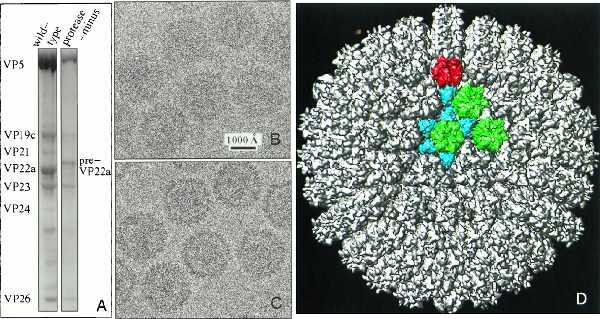
(A) SDS-gel of purified wild-type B capsids (left) and recombinant baculovirus-expressed protease-minus capsids (right). In the absence of an active protease, the major scaffolding protein is present in the unprocessed form, preVP22a, and the two autoproteolytic products of the protease, VP21 and VP24, are absent. (B and C) Focal pair images of ice-embedded protease-minus capsids at 1.1 μm (B) and 2.9 μm (C) underfocus respectively. (D) 3D reconstruction of protease-minus capsid as viewed along a threefold axis. Highlighted in color are morphological components found in an asymmetric unit including one penton (red), three types of hexon (green), and six types of triplex (aquamarine).
Figs. 1B and C show a corresponding specimen area in a focal pair of micrographs of protease-minus capsids. As visible in the out-of-focus micrograph (Fig. 1C), each capsid has an electron-dense internal core that represents the scaffolding protein. Images of wild-type B capsids look similar (20). We reconstructed the 3D structures of both B capsids and protease-minus capsids to a nominal resolution of 12 Å, using 355 and 385 particle images from close-to-focus micrographs, respectively. By comparing independent reconstructions of each capsid, we estimated the effective resolution of our reconstructions at 13–15 Å (20). The external structural features of the wild-type and protease-minus capsids are almost identical. Except for finer scale features, their overall structures and intersubunit contacts exhibit the same patterns as seen in earlier reconstructions of the wild-type A capsid (16), B capsid (2, 21, 22), and C capsid (23, 24). Fig. 1D shows the outer surface of the protease-minus capsid reconstruction viewed down a threefold axis. The colored region highlights the unique morphological components of the capsid shell, including one penton (red), three types of hexon (green), and six types of triplex (aquamarine). Wild-type and protease-minus capsids exhibit differences in the mass distributions of their cores, which can be seen in the comparison of a 4.67-Å thick central slice extracted from their reconstructions (Figs. 2 A and B). This is confirmed by radial density plots, which reveal substantial differences inside a radius of 450 Å (Fig. 2C). There is a layer of density from 340- to 450-Å radii in the protease-minus capsids, which is completely absent from the wild-type B capsids; whereas, the average density between 70- and 330-Å radii is lower in the protease-minus capsid than in the wild-type B capsid. Thus, it appears that there is a substantial radial translocation of protein mass as a result of maturational cleavage of the scaffolding proteins. In contrast, the two capsid types have identical density distributions for the region between 460- and 625-Å radii, indicating there is no radial density dislocation in the capsid shell upon proteolysis of the scaffolding protein. Independent reconstructions of each capsid type showed identical profiles, confirming that the radial density distribution differences between the cores of the protease-minus and B capsid is statistically significant and represents the result of scaffolding cleavage.
Because the procedure for 3D reconstruction is based on the assumption that the particle has icosahedral symmetry, we would expect a close agreement between two independent reconstructions of the same capsid in regions where the structure is indeed icosahedrally symmetrical. Therefore, we calculated the structural disagreement factor as a function of radius for two independent reconstructions of the wild-type B capsid (Fig. 2D). The structural features are clearly similar in the region of the capsid shell between 440- and 620-Å radii. However, the high disagreement factor inside 440-Å radius strongly suggests that there is no icosahedral symmetry in this region and thus the detailed structural features other than the radial density distribution are not reliable. Comparison of two protease-minus reconstructions yielded comparable results (Fig. 2D). To confirm this observation, we calculated the phase difference as a function of spatial frequency with cores or shells computationally extracted from the two independent reconstructions of each capsid type. Both capsid types showed similar patterns, with the phase differences between the extracted capsid shells remaining significantly <90° until 13-Å resolution, but those between the extracted cores reaching 90° at 200 Å (data not shown). Because similar patterns were obtained for both wild-type and protease-minus capsids, it is clear that the proteolytic status of the scaffold is not responsible for its lack of icosahedral symmetry.
Contacts Between Scaffold and Capsid Shell.
As can be seen from Figs. 2 A, B, and C, there is a sharp drop in mass density at the inner edge of the capsid shell, which implies that there is very little contact between the proteins of the core and the shell. However, the intact scaffolding protein is known to interact with VP5 (7) and these interactions are needed to form a sealed capsid (8, 9). Such interactions must occur presumably at the inner surface of the shell that forms the interface with the capsid interior. To look for continuity between the scaffold and shell, we computationally isolated the density between radii of 430 and 630 Å. Figs. 2 E and F show enlargements of the regions enclosed by the dashed boxes in Figs. 2 A and B. Although the capsid shells are remarkably similar in each case, additional densities can be seen extending inwards from the inner surface of the protease-minus capsid shell (arrowheads in Fig. 2F). Other internal densities also distribute differently between the two capsids. However, because these are unattached masses that are not in direct contact with the capsid floor, they do not represent sites of interaction between the capsid shell and the scaffold. To show the pattern of contacts, the colored region in Fig. 1D has been extracted from both the wild-type and the protease-minus capsid reconstructions and rotated to show a comparison between their side views (Fig. 3).
Figure 3.

Visualization of additional densities attached to the floor of protease-minus capsids. The region corresponding to the colored portion in Fig. 1D was extracted from the wild-type (A) and the protease-minus (B) capsid reconstructions and viewed from one side. The additional masses that are only present in protease-minus capsids are shown in white. For clarity, floating densities that do not make contacts with the capsid floor have been removed.
Because only material in close contact with the capsid shell is icosahedrally ordered, only those densities extending <40Å inward from the capsid floor are shown in Fig. 3. Unattached densities were removed for clarity. The densities on the capsid floors (between radii of 460 and 490 Å) are shown in purple. The additional masses existing only inside the protease-minus capsid are shown in white (Fig. 3B). These masses form several clusters of attached, rod-like densities extending inwards in the protease-minus capsid (Fig. 3B). To reveal more clearly their pattern of contacts with the capsid floor, we superimposed the additional masses from the protease-minus capsid on the wild-type B capsid structure (Fig. 4A). In this display, only those densities within a radial distance of ≈10 Å from the capsid floor are shown for clarity (Fig. 4A). The most prominent attached masses are located beneath the positions of four of the triplexes surrounding the C hexon (Fig. 4A). The enlarged stereo view of this region reveals that all the points of contact are situated at junctions between the floor domains of adjacent hexon subunits (Fig. 4B). Two schematic drawings, shown as viewed from inside (Fig. 4C) and from outside (Fig. 4D) the capsid, illustrate the locations of these contacts. Extensive contacts (large filled circles) are formed at two of three quasi-equivalent positions underneath each of the triplexes Tb, Tc, and Te. In addition, a smaller white density also can be seen attached at the third quasi-equivalent position under these triplexes (indicated by small filled circles). A different pattern of contacts is evident beneath the triplex Tf, which occupies the icosahedral threefold position. However, only one small white density is present under Td, and no additional densities were detected under the pentonal triplex Ta (Fig. 4).
Figure 4.
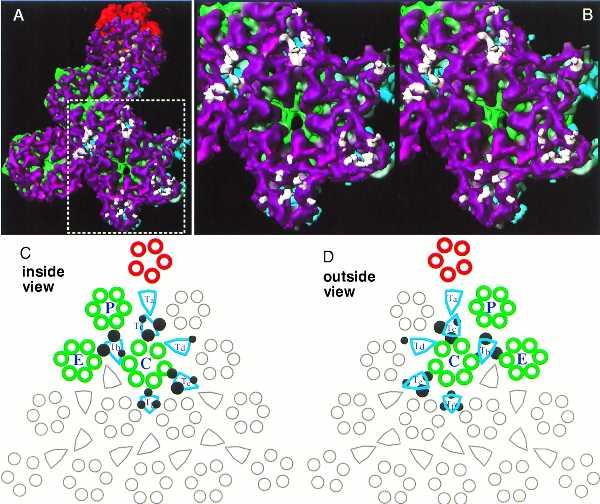
Contact points of scaffolding proteins on the capsid floor. The additional rod-like densities from the protease-minus capsid are super-imposed on the capsid shell of the wild-type B-capsid (A). For clarity, only those densities that are within a radial distance of ≈10 Å from the shell are shown. Panel B shows a stereo pair of an enlarged region surrounding the C hexon (indicated by the dotted rectangle in A). The schematic diagrams of one triangular face of the HSV capsid depict the points of contact of the additional mass densities on the floor as viewed from inside (C) and outside (D) of the capsid. All the morphological components in a triangular face are shown, and those components in one of the three asymmetric units are highlighted in color. Each asymmetric unit includes one-fifth of the penton (red), one each of P and C hexons (green), one-half an E hexon (green), one each of the triplexes Ta, Tb, Tc, Td, and Te, and one-third of the triplex Tf (aquamarine). Filled dark circles designate the points of contact of the scaffolding and the capsid shell. Larger circles represent more extensive contacts, and smaller circles indicate less extensive ones.
DISCUSSION
The mechanism by which the scaffold influences capsid morphogenesis in herpes viruses is not well understood. In the absence of scaffolding proteins, the outer capsid proteins can self-assemble to form aberrant structures. If all three essential shell proteins are present (VP5, VP19C, and VP23), these structures have the appearance of partial and incomplete shells (14, 15, 25). If just VP5 and VP19C are present, they form densely staining spherical particles (26), which are markedly smaller (900 Å) than intact capsids (1,250 Å). Both forms contain recognizable capsomeres and have local sixfold symmetry (15, 25, 26). A similar situation has been described for a number of double-stranded DNA phages. In P22 for example, although the absence of scaffolding protein does not prevent assembly of normal-sized capsids, it greatly reduces its rate. In addition, aberrant spiral forms and smaller than normal capsids are formed (27). These smaller capsids are icosahedral but have T = 4 symmetry rather than the T = 7 symmetry of the full sized forms (28). Thus, it is a common property of HSV and P22 that scaffolding proteins are not needed for the assembly of the outer shell proteins, which appear to have an intrinsic propensity to adopt icosahedral symmetry, but rather are involved in defining the curvature and ultimate size of the capsid shell. The lack of icosahedral symmetry in the scaffolds of wild-type B and protease-minus capsids described here, and in in vitro-assembled procapsids (2), further suggests that the scaffold may not need to adopt a unique organization to fulfill its function, provided that its overall dimensions are within the tolerance necessary to ensure the correct shell conformation.
Despite the fact that the bulk of the scaffold does not exhibit icosahedral symmetry, the specificity of the interaction between the C-terminus of preVP22a and VP5 (7, 29) implies that these two proteins interact in a consistent manner and that their contacts should adopt the symmetry of the participating VP5 molecules. Proteolysis of preVP22a, which removes the C-terminal 25 aa, almost certainly breaks the connection between the scaffold and the capsid shell. However, this action does not result in the major conformational changes to the shell that usually accompany bacteriophage maturation. Consequently, differences in the nature of the scaffold, shell interactions are less likely to be obscured by gross changes in the overall structure. The C-terminal 25 aa of the scaffolding protein do not appear to be retained in the capsid following cleavage (A. Davison, personal communications). Therefore, it seems very likely that the rod-like densities present only in the protease-minus capsid correspond to ordered domains of uncleaved scaffolding proteins in contact with VP5.
This interpretation is supported by studies on other capsid preparations. We generated reconstructions of an independent preparation of recombinant, protease-minus capsids that was made without using detergent (this was done because the interaction between VP5 and preVP22a has been reported as detergent sensitive; ref. 7), of capsids purified from cells infected at nonpermissive temperature with the protease mutant, ts1201 (11), and of recombinant B capsids made using all six capsid protein genes including that for the protease (data not shown). In the first two cases, in which the scaffolding protein is in the uncleaved form, additional density continuous with the capsid shell is present at similar locations to those described above; whereas, in the protease-plus recombinant capsid, the inner floor closely resembles that of the wild-type B capsid. Such close correspondence between the cleavage status of the scaffolding protein and the presence or absence of the additional masses strongly supports the interpretation we present here that they are formed by the C-termini of preVP22a.
The localized symmetry enforced on the scaffold through its contact with the shell might not be expected to involve much more of the scaffolding protein than the 25 aa necessary for its interaction with VP5. The rod-like densities in the protease-minus capsid extend radially for ≈40 Å before merging into the region of disordered density. This corresponds, approximately, to the length occupied by 20–30 aa in an α-helical conformation. The mass contained in the rods is difficult to determine because the Fresnel fringe effect, which is seen at the inner edge of the capsid shell (see Figs. 2 A and B), probably makes the rod-like densities appear thinner than their actual size. It is also possible that a partial occupancy of these positions by preVP22a molecules has rendered these densities smaller. Despite this, the total mass making up each of the rod-like densities accounts for >25 aa, suggesting that the interaction of the scaffolding protein with VP5 may either involve a larger domain than previously thought or that more than one preVP22a molecule participates in each contact. It has been reported that the 25-aa terminal region of preVP22a binds to VP5 more efficiently when present as a parallel homodimer (30). If this is also true in the capsid, each rod could represent the combined C-termini of a preVP22a dimer. We observe three well defined pairs of rod-like densities for each asymmetric unit. Assuming each rod to represent the C termini of a preVP22a dimer, there would be 12 molecules for each asymmetric unit, which leads to a total of 720-scaffolding protein molecules in each capsid. The nature of the contacts formed underneath Tf is obscured by the threefold-averaging imposed at this position, but if we assume the existence of a pair of dimers at this location, the total number of scaffolding proteins would be 800. This would account for only two-thirds of the ≈1,200 copies of VP22a in B capsids estimated from biochemical studies (4). It is possible that the third, smaller density under Tb, Tc, and Te and the one density under Td are the result of binding by additional scaffolding proteins but at a lower occupancy rate. However, Oien et al. (29) have reported recently that functional scaffolds can contain both full length (preVP22a) and C-terminally truncated (VP22a) scaffolding proteins. Because the latter are known not to interact with VP5 and cannot support capsid assembly (31), it is clear that the incorporation of scaffolding proteins into a capsid does not entirely depend on their ability to interact with VP5. Thus, it is not unreasonable that the B capsid could contain more scaffolding protein molecules than are involved directly in binding to the capsid shell.
The separation between the members of each major pair of connections is ≈37 Å. The crystal structure of the human cytomegalovirus protease domain recently has been determined to atomic resolution (32–34). It exists as a dimer with two independent active sites located on opposite faces, with the two catalytic, nucleophile Ser 132 residues spaced 36.5 Å apart. The close correspondence between important structural elements in the human cytomegalovirus protease and conserved domains in the amino acid sequences of a number of herpes viruses strongly suggests that a similar spacing also may exist in the HSV protease. Such a close correlation between the spacing of the protease active sites and their likely targets in the uncleaved scaffold is provocative, but as the protease is not present in these capsids, it is not yet possible to draw any firm conclusions regarding its possible organization in the scaffold.
It is not clear why two of the three quasi-equivalent positions under each triplex appear to be favored as points of contact with the scaffold. Presumably this result reflects local factors that may be important during the assembly process but either no longer pertain to the capsids analyzed here or are not apparent at this resolution. It is interesting that the points of contact underlie the positions of the triplexes, which are heterotrimers comprising two copies of VP23 and one copy of VP19c (4, 16), because the triplexes also form nonsymmetrical connections, typically interacting strongly with only two of the three surrounding capsomeres (16, 20). As a consequence, VP5 molecules form visible connections to triplexes in certain quasi-equivalent locations but not in others. Although there is no evidence that the triplexes themselves are involved in the interaction between the scaffold and shell, the pattern of contacts between VP5 and triplexes appears to mirror the situation we observe here in which contacts exist between the scaffold and some, but not all, of the available quasi-equivalent locations on the interior of the capsid shell. The interaction of the scaffold and capsid shell seems to provide another example of the breakdown of quasi-equivalence at a local level. As the study of the capsid proceeds to higher resolution, it will be interesting to observe how such deviations from geometrical conformity relate to the biological role of the capsid.
Acknowledgments
This research was supported by NIH (AI38469, RR02250, and LM07093); National Science Foundation (BIR-9413229); and the Human Frontier Science Program (RG-537/96).
ABBREVIATIONS
- HSV-1
herpes simplex virus-1
- 3D
three dimensional
References
- 1.Newcomb W W, Homa F L, Thomsen D R, Booy F P, Trus B L, Steven A C, Spencer J V, Brown J C. J Mol Biol. 1996;263:432–446. doi: 10.1006/jmbi.1996.0587. [DOI] [PubMed] [Google Scholar]
- 2.Trus B L, Booy F P, Newcomb W W, Brown J C, Homa F L, Thomsen D R, Steven A C. J Mol Biol. 1996;263:447–462. doi: 10.1016/s0022-2836(96)80018-0. [DOI] [PubMed] [Google Scholar]
- 3.Rixon F J. Semin Virol. 1993;4:135–144. [Google Scholar]
- 4.Newcomb W W, Trus B L, Booy F P, Steven A C, Wall J S, Brown J C. J Mol Biol. 1993;232:499–511. doi: 10.1006/jmbi.1993.1406. [DOI] [PubMed] [Google Scholar]
- 5.Liu F, Roizman B. J Virol. 1991;65:5149–5156. doi: 10.1128/jvi.65.10.5149-5156.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preston V G, Rixon F J, McDougall I M, McGregor M, Al-Kobaisi M F. Virology. 1992;186:87–98. doi: 10.1016/0042-6822(92)90063-u. [DOI] [PubMed] [Google Scholar]
- 7.Hong Z, Beaudet-Miller M, Durkin J, R, Z, Kwong D. J Virol. 1996;70:533–540. doi: 10.1128/jvi.70.1.533-540.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennard J, Rixon F J, McDougall I M, Tatman J D, Preston V G. J Gen Virol. 1995;76:1611–1621. doi: 10.1099/0022-1317-76-7-1611. [DOI] [PubMed] [Google Scholar]
- 9.Matusick-Kumar L, Newcomb W W, Brown J C, McCann P J, III, Hurlburt W, Weinheimer S P, Gao M. J Virol. 1995;69:4347–4356. doi: 10.1128/jvi.69.7.4347-4356.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Addison C, Rixon F J, Preston V G. J Gen Virol. 1990;71:2377–2384. doi: 10.1099/0022-1317-71-10-2377. [DOI] [PubMed] [Google Scholar]
- 11.Preston V G, Coates J A V, Rixon F J. J Virol. 1983;45:1056–1064. doi: 10.1128/jvi.45.3.1056-1064.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao M, Matsusick-Kumar L, Hurlburt W, DiTusa S F, Newcomb W W, Brown J C, McCann P J, III, Deckman I, Colonno R J. J Virol. 1994;68:3702–3712. doi: 10.1128/jvi.68.6.3702-3712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen D R, Newcomb W W, Brown J C, Homa F L. J Virol. 1995;69:3690–3703. doi: 10.1128/jvi.69.6.3690-3703.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tatman J D, Preston V G, Nicholson P, Elliott R M, Rixon F J. J Gen Virol. 1994;75:1101–1113. doi: 10.1099/0022-1317-75-5-1101. [DOI] [PubMed] [Google Scholar]
- 15.Thomsen D R, Roof L L, Homa F L. J Virol. 1994;68:2442–2457. doi: 10.1128/jvi.68.4.2442-2457.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Z H, Prasad B V V, Jakana J, Rixon F, Chiu W. J Mol Biol. 1994;242:458–469. doi: 10.1006/jmbi.1994.1594. [DOI] [PubMed] [Google Scholar]
- 17.Sherman M B, Brink J, Chiu W. Micron. 1996;27:129–139. doi: 10.1016/0968-4328(96)00018-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z H, Hardt S, Wang B, Sherman M B, Jakana J, Chiu W. J Struct Biol. 1996;116:216–222. doi: 10.1006/jsbi.1996.0033. [DOI] [PubMed] [Google Scholar]
- 19.Hardt S, Wang B, Schmid M F. J Struct Biol. 1996;116:68–70. doi: 10.1006/jsbi.1996.0012. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Z H, Chiu W, Haskell K, Spears H, Jakana J, Rixon F J, Scott L R. Biophys J. 1998;74:576–588. doi: 10.1016/S0006-3495(98)77816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker T S, Newcomb W W, Booy F P, Brown J C, Steven A C. J Virol. 1990;64:563–573. doi: 10.1128/jvi.64.2.563-573.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z H, He J, Jakana J, Tatman J, Rixon F, Chiu W. Nat Struct Biol. 1995;2:1026–1030. doi: 10.1038/nsb1195-1026. [DOI] [PubMed] [Google Scholar]
- 23.Schrag J D, Prasad B V V, Rixon F J, Chiu W. Cell. 1989;56:651–660. doi: 10.1016/0092-8674(89)90587-4. [DOI] [PubMed] [Google Scholar]
- 24.Booy F P, Newcomb W W, Trus B L, Brown J C, Baker T S, Steven A C. Cell. 1991;64:1007–1015. doi: 10.1016/0092-8674(91)90324-r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desai P, Watkins S C, Person S. J Virol. 1994;68:5365–5374. doi: 10.1128/jvi.68.9.5365-5374.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rixon F J, Addison C, McGregor A, Macnab S J, Nicholson P, Preston V G, Tatman J D. J Gen Virol. 1996;77:2251–2260. doi: 10.1099/0022-1317-77-9-2251. [DOI] [PubMed] [Google Scholar]
- 27.Earnshaw W, King J. J Mol Biol. 1978;126:721–747. doi: 10.1016/0022-2836(78)90017-7. [DOI] [PubMed] [Google Scholar]
- 28.Thuman-Commike P A, Greene B, Malinski J A, King J, Chiu W. Biophys J. 1998;74:559–568. doi: 10.1016/S0006-3495(98)77814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oien N L, Thomsen D R, Wathen M W, Newcomb W W, Brown J C, Homa F L. J Virol. 1997;71:1281–1291. doi: 10.1128/jvi.71.2.1281-1291.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelletier A, Do F, Brisebois J J, Lagace L, Cordingley M G. J Virol. 1997;71:5197–5208. doi: 10.1128/jvi.71.7.5197-5208.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preston V G, Al-Kobaisi M F, McDougall I M, Rixon F J. J Gen Virol. 1994;75:2355–2666. doi: 10.1099/0022-1317-75-9-2355. [DOI] [PubMed] [Google Scholar]
- 32.Qiu X, Culp J S, DeLilla A G, Hellmig B, Hoog S S, Janson C A, Smith W W, Abdel-Meguid S S. Nature (London) 1996;383:275–279. doi: 10.1038/383275a0. [DOI] [PubMed] [Google Scholar]
- 33.Shieh H-S, Kurumbail R G, Stevens A M, Stegeman R A, Sturman E J, Pak J Y, Wittwer A J, Palmier M O, Wiegand R C, Holwerda B C, Stallings W C. Nature (London) 1996;383:279–282. doi: 10.1038/383279a0. [DOI] [PubMed] [Google Scholar]
- 34.Tong L, Qian C, Massariol M-J, Bonneau P R, Cordingley M G, Lagace L. Nature (London) 1996;383:272–275. doi: 10.1038/383272a0. [DOI] [PubMed] [Google Scholar]