

Abstract
Purpose
Photoreceptors can be prevented from undergoing apoptosis in response to constant light by the application of exogenous neuroprotective agents, including brain-derived neurotrophic factor (BDNF). BDNF, however, cannot exert its effect directly on photoreceptors because they do not express receptors for BDNF. It has been proposed that BDNF released from Müller cells provides a feed-forward loop, increasing ciliary neurotrophic factor (CNTF) and basic fibroblast growth factor (bFGF) production in Müller cells, which may enhance photoreceptor survival. The authors hypothesized that retinas with reduced BDNF levels in which the BDNF-mediated release of neuroprotective signals is dampened are more susceptible to light-induced photoreceptor degeneration.
Methods
Young adult BDNF+/+ and BDNF+/− littermates (B6.129-BDNFtm1-LT) were analyzed. Retinal neurotrophin and growth factor mRNA levels were determined by quantitative RT-PCR, photoreceptor function was assessed through electroretinography, and survival was documented in morphologic sections and in TUNEL assays. Oxidative stress was assayed by measuring glutathione peroxidase activity.
Results
baseline, BDNF+/− animals had significantly increased levels of glial-derived neurotrophic factor (GDNF) mRNA compared with their wild-type littermates. After light damage GDNF, CNTF, and BDNF mRNA levels dropped 14- to 16-fold in the BDNF+/+ mice but remained almost unchanged compared with baseline levels in the BDNF+/− mice. Preservation of neurotrophin levels in BDNF+/− mice correlated with photoreceptor cell survival, preservation of function, and reduced oxidative stress.
Conclusions
Contrary to the hypothesis, reducing BDNF levels resulted in photoreceptor protection against light damage. Survival was paralleled by a reduction in oxidative stress and the preservation of neurotrophin levels, suggesting that chronic reduction of BDNF in the retina provides a level of preconditioning against stress.
Retinal photoreceptors signal the absorption of light. Photoreceptor outer segments contain all the components of the signal transduction cascade for the absorption of light and the subsequent changes in rod outer segment membrane conductance, whereas the inner segments translate conductance changes to synaptic transmitter release to the postsynaptic neurons. Photoreceptors are stable but fragile cells.1 Like most neurons in the central nervous system (CNS), photoreceptors last the lifespan of an individual. What makes them fragile is their specialization, their absorption of light, and the energy requirements to carry out this task. First, outer segment membranes have a quick turnover such that, in rats for example, outer segments are replaced by shedding every 2 weeks.2 Resultant shed fragments must also be phagocytized efficiently by the retinal pigment epithelium to remove debris. Second, photoreceptors require a large amount of energy in the form of adenosine triphosphate (ATP) to maintain the structure and the ionic polarization of the outer segment.3,4 Thus, photoreceptors use three to four times more oxygen in the light-adapted state and six to eight times more oxygen in the dark-adapted state than other CNS neurons. Oxygen is supplied to the photoreceptors by the vasculature of the choroid. The choroidal circulation is fast to dissipate the heat generated by the photoreceptor metabolism.5 However, the choroidal blood flow shows little autoregulation in response to the photoreceptor oxygen requirements (reviewed by Yu and Cringle6). Therefore, the decrease in oxygen consumption that occurs when photoreceptors go from a dark- to a light-adapted state causes an increase in oxygen tension. Thus, the photoreceptors have to be equipped with mechanisms to deal with short periods of hypoxia and hyperoxia.
How do photoreceptors survive during these changes in oxygen tension during the day? Is it possible that photoreceptors create their own protective environment in response to light? Protective mechanisms could include antioxidants, growth factors or cytokines, or signaling pathways regulating antiapoptotic genes. Penn et al.7 showed that retinal levels of antioxidants such as ascorbic acid, vitamin E, and glutathione were higher in rats reared in bright light conditions than in rats reared in dim light conditions, possibly counteracting oxygen toxicity. Normal light conditions at a normal circadian rhythm (12 hours light, 12 hours dark) and, hence, normal physiological light levels are effective in upregulating basic fibroblast growth factor (bFGF) in rats during the day, the time of higher oxygen exposure.1 In addition, photoreceptors can be protected from light-induced cell death if the retina is exposed first to a period of bright light. This preconditioning stimulus results in significant increases in retinal bFGF and ciliary neurotrophic factor (CNTF).8 Thus, the retina is equipped with two mechanisms to protect its photoreceptors, a short-term mechanism that protects against brief exposures to bright light and a long-term mechanism that protects against circadian light exposure. Each mechanism seems to induce the production of neuroprotective agents such as bFGF, CNTF, and antioxidants.
Photoreceptors can also be prevented from undergoing apoptosis by the application of exogenous neuroprotective agents. Wenzel et al.9 have elegantly summarized all the components that have been tested. They range from blocking reactive oxygen species and nitric oxide production (e.g., Ranchon et al.,10 Ranchon et al.,11 Goureau et al.12) to blocking glucocorticoid receptor activity and, thus, blocking AP-1 activity13 injecting various neurotrophins and growth factors. Intravitreal injections of lens epithelium-derived growth factor (LEDGF),14 bFGF,15 pigment epithelium-derived factor (PEDF),16 CNTF (Axokine; Regeneron, Tarrytown, NY),16 brain-derived neurotrophic factor (BDNF), NT-4, and insulin-like growth factor II (IGF-II)15 all can prevent or reduce light-induced photoreceptor degeneration to some degree. However, the molecular targets and the means of action have not yet been determined for neurotrophins and growth factors.
Recently, Harada et al.17 have suggested a microglia–Müller cell interaction model involved in controlling light damage–mediated photoreceptor degeneration. The model is based on available data on the molecular targets of some of these neuroprotective agents and the known distribution pattern of their respective receptors within the retina. They have put this model to the test by examining gene expression and cytokine production in light-damaged retinas and activated retinal microglia. Some key features of this model are that, during light damage, activated microglia release glial-derived neurotrophic factor (GDNF) and CNTF, each of which modulates bFGF and BDNF production and release from Müller cells, and that BDNF participates in a feed-forward mechanism, increasing CNTF and bFGF production again in Müller cells, which together may enhance photoreceptor survival.
To test this model, we examined retinas in which the BDNF-mediated amplification of the microglia-derived neuroprotective signal might be dampened by analyzing photoreceptor survival and function in BDNF+/− mice, which express only 50% of BDNF mRNA and protein, after exposing them to damaging light. We predicted that these mice would be more susceptible to light damage because their normal neuroprotective mechanisms against brief exposures to very bright light might be impacted. However, we found the opposite to be true. After light damage, not only did BDNF+/− retinas contain more photoreceptors, exhibit fewer TUNEL-positive cells, and have higher ERG amplitudes, their levels of glutathione peroxidase activity, a marker of oxidative stress, were reduced. In addition, the light damage–induced decrease in mRNA levels of GDNF, CNTF, and BDNF observed in the BDNF+/+ retinas was abolished in the BDNF+/− retinas. Thus, it appears that the reduced levels of BDNF in the BDNF+/− retinas triggered compensatory mechanisms reminiscent of preconditioning against stress.
Materials and Methods
Animals
B6.129-BDNFtm1-LT mice were generously provided by Lino Tessarollo18 and were maintained as heterozygous breeding pairs to provide wild-type (BDNF+/−) and heterozygous (BDNF+/−) littermates. A background analysis produced by Charles River Laboratories Genetic Testing Services, testing the original breeders with a 110-marker panel that distinguished between C57BL/6 and 129, demonstrated that our colony ranged from 93.2% to 95.2% C57BL/6 and that it appeared to have been backcrossed to C57BL/6 to N4 or N5 and then intercrossed. Animals were housed in the MUSC Animal Care Facility under a 12-hour light/12-hour dark cycle with access to food and water ad libitum. Ambient light intensity at the eye level of the animals was 85 ± 18 lux. All experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the University Animal Care and Use Committee.
Light Damage
At 3 months of age, animals were moved to the light treatment room and were dark adapted overnight. At 12:00 noon, each animal’s pupils were dilated with a drop of phenylephrine HCl (2.5%) and atropine sulfate (1%). Light damage was induced by exposing the animals to 15,000 lux of white light for 1.5 hours, provided by four 54-W fluorescent bulbs (T5HO F54W/865; General Electric, Fairfield, CT) suspended approximately 40 cm above the cages.19 After light exposure, animals were returned to complete darkness for 10 days and then underwent electrophysiological, histologic, and molecular analyses. The term light damage will be used here to connote the combined effects of the intense light exposure and the damage executed during those 10 days in complete darkness.
ERG Recordings
Electroretinographic (ERG) recordings were performed as described.20,21 Animals were dark adapted overnight and anesthetized with xylazine and ketamine (20 and 80 mg/kg, respectively), and each pupil was dilated with a drop of phenylephrine HCl (2.5%) and atropine sulfate (1%). Body temperature was stabilized with a DC-powered heating pad and held at 37°C. A needle ground electrode was placed in the tail, and a needle reference electrode was placed in the forehead. ERG responses were measured with contact lenses containing a gold-ring electrode22 held in place by a drop of methyl cellulose. Electroretinograms were recorded with a visual electrodiagnostic testing system (EPIC-2000; LKC Technologies, Inc., Gaithersburg, MD) with a strobe-flash stimulus (Grass Technologies, West Warwick, RI). ERG responses were recorded at a gain of 2 k using a notch filter at 60 Hz, bandpass filtered between 0.1 and 1500 Hz, and digitized online at 1 kHz with 12-bit accuracy.
Stimuli to isolate rod responses consisted of 10-μsec single flashes at a fixed intensity (2.48 photopic cd · s/m2 at the dome’s inner surface, as calibrated by the manufacturer) under scotopic conditions. Single-flash responses were averaged two to four times with an interstimulus interval (ISI) ranging from 15 seconds to 2 minutes, depending on the light intensity used.
For all ERG recordings, a-wave amplitude was measured from baseline to the a-wave trough, b-wave amplitude was measured from the a-wave trough or baseline to the peak of b-wave, and implicit time was measured from the onset of stimulus to the a-wave trough or b-wave peak.
Anatomy
After the ERG recordings, the animals were humanely killed, and their eyes were removed while maintaining their dorsoventral orientation with the superior rectus muscle and optic nerve as landmarks. Eyes for semithin sections and light microscopy were removed, postfixed in 4% paraformaldehyde and 2% glutaraldehyde, and bisected dorsal to ventral through the optic nerve. Each half was embedded in an epoxy resin (EPON and Araldite; Electron Microscopy Sciences, Hatfield, PA) mixture, and sections were cut at 1 μm through the horizontal meridian and stained with toluidine blue. Photoreceptor layers were counted in the central (superior and inferior, within 350 μm of the optic nerve head) and peripheral (superior and inferior, within 350 μm of the ciliary body) retina. Three measurements were made per field and were averaged to provide a single value for each area, and the four area values were averaged to give a value for the retina as described previously.23 One of the heterozygous samples was damaged during processing and could not be analyzed.
TUNEL Labeling
TUNEL labeling was performed according to our published protocol24 according to the protocol provided by the manufacturer (Roche Diagnostics, Indianapolis, IN). In short, eyes were fixed in 2% paraformaldehyde for 2 hours at 4°C followed by dehydration and paraffin embedding, as described. TUNEL (TdT-mediated dUTP nick-end) labeling was performed on 7-μm paraffin sections, and the DNA strand breaks were labeled with fluorescein for visualization. Slides were examined with a fluorescein isothiocyanate (FITC) filter on a microscope (Carl Zeiss, Oberkochen, Germany) equipped with fluorescence.
Quantitative RT-PCR
All chemicals used in this study were at least molecular biology grade material and were purchased from Fisher Scientific (Pittsburgh, PA), unless otherwise noted. Animals were humanely killed by cervical dislocation, and retinas were isolated and stored (RNAlater; Ambion, Austin, TX) at −20°C. Retinas from two animals per genotype per time point were pooled, and total RNA was isolated (Trizol; Ambion), followed by a clean-up using RNA marker kit mini-columns (RNA Easy; Qiagen, Valencia, CA). RNA quality was examined by gel electrophoresis and spectrophotometry, as described previously.25 Real-time PCR was performed, also as described previously.25 RNA (2 μg each) was used to generate first-strand cDNA in reverse-transcription reactions (Invitrogen, Carlsbad, CA). PCR amplifications were conducted with the use of a PCR kit (QuantiTect Syber Green; Qiagen) with 0.01 U/μL enzyme (AmpErase UNG; Applied Biosystems, Foster City, CA) to prevent carryover contamination (see Table 1 for primer sequences, gene accession number, and expected length). Real-time PCR was performed in triplicate in a sequence-detection system (GeneAmp 5700; Applied Biosystems) under the following cycling conditions: 50°C for 2 minutes, 94°C for 15 minutes, 40 cycles of 94°C for 15 seconds, and 58°C for 1 minute. Quantitative values were obtained using the cycle number (Ct value), which is inversely proportional to the amount of a specific mRNA species in the tissue sample. Relative gene expression levels were calculated using the equation y = (1 + AE)Δ ΔCt, where AE is the amplification efficiency of the target gene (set at 1.0 for all calculations), and ΔΔCt is the difference between the mean experimental and control ΔCt values. The ΔCt value is the difference between the Ct value for a retina-associated gene and the β-actin internal reference control gene.26
Table 1.
QRT-PCR Primers
| Name | Gene Accession No. | Sequence (5′-3′ Forward) | Sequence (3′-5′ Reverse) | Size (bp) |
|---|---|---|---|---|
| β-actin | NM_007393 | GCT ACA GCT TCA CCA CCA CA | TCT CCA GGG AGG AAG AGG AT | 123 |
| BDNF | NM_007540 | TGG CTG ACA CTT TTG ACC AC | CAA AGG CAC TTG ACT GCT GA | 131 |
| GDNF | NM_010275 | TGG GCT ATG AAA CCA AGG AG | CAA CAT GCC TGG CCT ACT TT | 142 |
| CNTF | NM_053007 | ATG ACT GAG GCA GAG CGA CT | AGG CAG AAA CTT GGA GCG TA | 157 |
| FGF-2 | NM_008006 | AAG CGG CTC TAC TGC AAG AA | TGG CAC ACA CTC CCT TGA TA | 151 |
| NGF | NM_013609 | ATG GGG GAG TTC TCA GTG TG | GCA CCC ACT CTC AAC AGG AT | 180 |
Enzymatic Assays
Enzymatic activity was measured using appropriate fluorescent substrates provided in the enzyme activity assay kit in retina homogenates (glutathione peroxidase; Calbiochem, San Diego, CA). Retinas were dissected out, kept in lysis buffer at 40°C, sonicated, and centrifuged at 20,000g for 15 minutes. The supernatant was assayed for protein content by spectrophotometric measurement of the absorbance at 340 nm and was expressed as change in relative fluorescent units (RFUs) per hour per milligram of protein. Protein content was measured according to the Bradford Folins reagent method (Bio-Rad Laboratories, Hercules, CA). Results are presented as percentage change in activity, taking BDNF+/+ mice raised in cyclic light as 100%.
Statistical Analysis
For all experiments, data were expressed as mean ± SEM. ERG data were analyzed using repeated-measures analysis of variance (ANOVA; StatView; SAS Institute, Cary, NC). After ANOVA, individual data pairs were compared using Student’s t-test analysis, accepting a significance level of P < 0.05. Morphologic and biochemical data were analyzed with the standard Student’s t-test, also accepting a significance level of P < 0.05.
Results
BDNF Status Affects mRNA Expression of Neurotrophins and Growth Factors
Harada et al.17 have suggested a model whereby in light damage, activated microglia invade the degenerating outer nuclear layer and release nerve growth factor (NGF), GDNF, and CNTF, which in turn affect bFGF, BDNF, and CNTF production and release from Müller cells. BDNF increases CNTF and bFGF production again in Müller cells, amplifying the microglia-mediated signal. Müller cell–derived factors are considered to control photoreceptor degeneration or neuroprotection, with NGF regulating apoptosis and GDNF and BDNF regulating survival. The model might predict that the neuroprotection provided by BDNF is affected in retinas with reduced BDNF levels, leading to faster photoreceptor degeneration in the presence of damaging light.
Quantitative RT-PCR measurements were performed on whole retina samples, testing basal levels (Figs. 1A, 1B) in BDNF+/+ and BDNF+/− mice. As expected, at baseline, BDNF mRNA levels were reduced to 50% in the heterozygote animals compared with their wild-type littermates (P = 0.005). CNTF and bFGF levels were unchanged (P > 0.1); however, NGF mRNA levels were slightly but significantly reduced (P = 0.0004), and GDNF mRNA levels were twice the levels found in wild-type littermates (P = 0.03).
Figure 1.
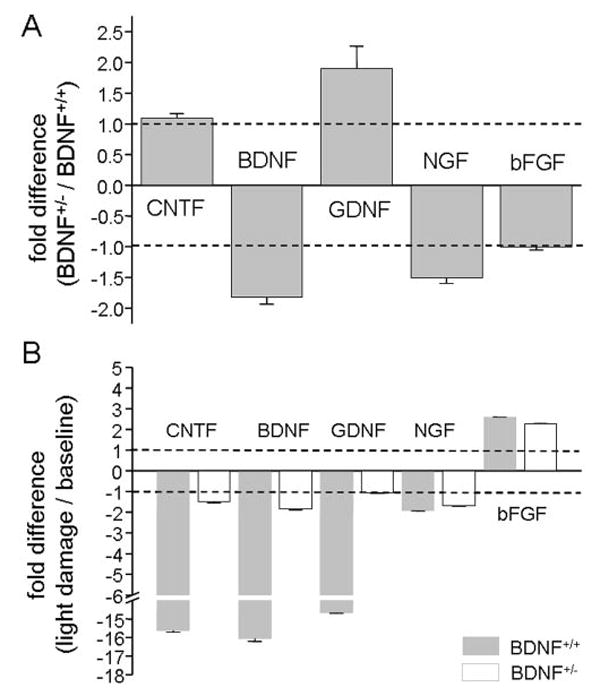
Quantitative RT-PCR measurements performed on whole retina samples from BDNF+/+ and BDNF+/− mice. Measurements were performed for BDNF, CNTF, GDNF, NGF, and bFGF on samples isolated from mice (A) exposed to cyclic light and (B) after light damage.
After light damage, GDNF, CNTF, and BDNF mRNA levels dropped significantly (14- to 16-fold) in the BDNF+/+ mice (P < 0.001). In the BDNF+/− mice, CNTF and GDNF remained unchanged compared with baseline levels (P > 0.3), whereas BDNF levels dropped slightly (P = 0.04). NGF and bFGF levels changed independently of genotype; NGF levels dropped twofold (BDNF+/+, P = 0.002; BDNF+/−, P < 0.001), but bFGF levels were increased twofold to threefold (BDNF+/+, P < 0.001; BDNF+/−, P = NS). Overall, all relative changes of neurotrophin mRNA levels from baseline to light damage were significantly larger in BDNF+/+ than in BDNF+/− mice (CNTF, P = 0.001; BDNF, P = 0.02; GDNF, P = 0.02; NGF, P = 0.008; bFGF, P = 0.03).
Neurotrophin Status Affects Susceptibility of Photoreceptors to Light Damage: Physiology
The lack of a large decrease in some neurotrophins in BDNF+/− mice suggests that the heterozygous mice might be less susceptible to light damage than their wild-type littermates. Dark-adapted, single-flash ERG responses of BDNF+/+ and BDNF+/− mice were compared at increasing flash intensities before and after intense light exposure in adult mice (3 months old) to determine whether neurotrophin status had any effects on the susceptibility of photoreceptors to light damage.
Analysis of scotopic, single-flash ERG responses before light damage revealed that, overall, a-wave amplitudes were slightly but significantly diminished in BDNF+/− mice compared with BDNF+/+ mice (P < 0.001), an interaction that was amplitude specific (P = 0.004; Figs. 2, 3; Table 2 for further information on F parameters and values). In particular, maximum a-wave responses were approximately 20% smaller in the BDNF+/− mice (BDNF+/+, –191 ± 4.9 μV; BDNF+/−, –154 ± 4.3 μV; P = 0.003). Similarly, b-wave amplitudes were reduced in the BDNF+/− animals at baseline (P = 0.013), also revealing an amplitude by genotype interaction (P < 0.001), and maximum b-wave responses were reduced by 28% (BDNF+/+, 567 ± 25.4 μV; BDNF+/−, 428 ± 32 μV; P = 0.02). When ERG responses to the two lowest light intensities were excluded in the analysis, the genotype-by-amplitude effect was rendered null for a-waves and b-waves (a-wave: F [3,27] = 2.56; P = 0.08; b-wave: F [3,27] = 1.16; P = 0.34). Implicit times, on the other hand, were identical between the two genotypes, irrespective of ERG component (a-wave implicit times, P = 0.81; b-wave implicit times [genotype], P = 0.69).
Figure 2.
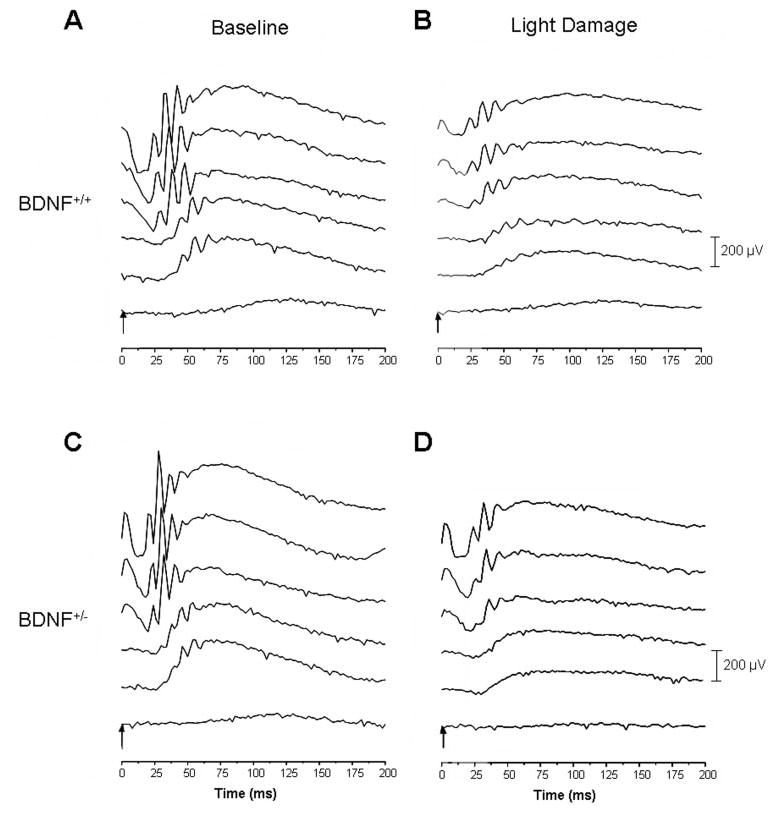
Families of ERGs elicited from BDNF+/+ (A, C) and BDNF+/− (B, D) mice before (A, B) and after (C, D) light damage, at increasing light intensities (40, 30, 20, 10, 6, and 0 log units of attenuation). At higher light intensities, rod ERG amplitudes were approximately 25% smaller in BDNF+/− mice than in their age-matched wild-type littermates. Light damage resulted in a large reduction of a- and b-wave amplitudes in the BDNF+/+ mouse retina (maximum amplitudes were reduced by approximately 40% and 60%, respectively) but had a less deleterious effect on the ERG elicited from the BDNF+/− retina (maximum amplitudes were reduced by only approximately 13% and 40%, respectively). For complete data analysis, see Figure 3, Table 2, and Results section.
Figure 3.
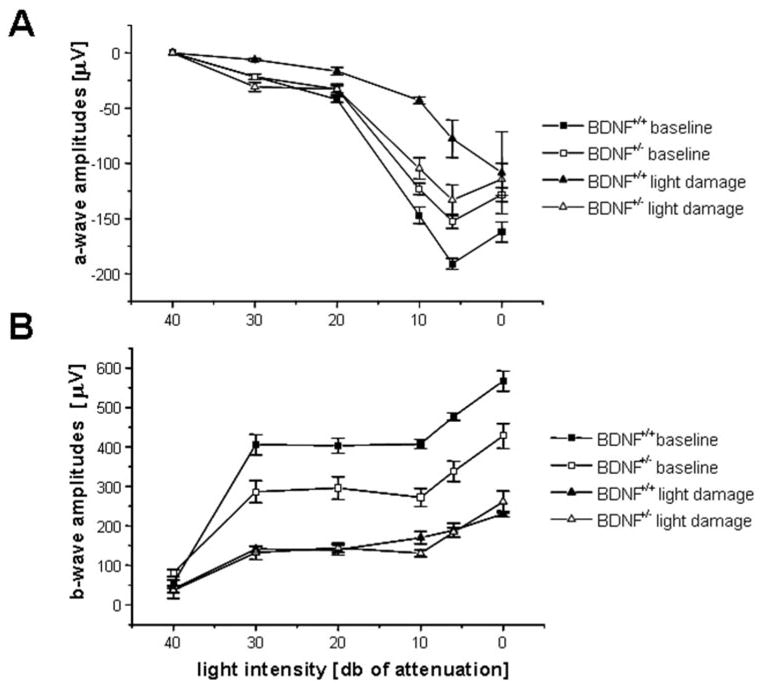
ERG amplitudes were determined for a-waves (A) and b-waves (B) at six light intensities before (closed symbols) and after (open symbols) light damage. Data are plotted as mean ± SEM. For statistical analysis, see Table 2 and Results section.
Table 2.
Statistical Analysis of ERG Components Using Repeated-Measure ANOVA
| ERG Component | Treatment | Analysis | F Parameters | F Value | P |
|---|---|---|---|---|---|
| a-Wave amplitudes | Baseline | Genotype | 1,9 | 23.60 | <0.001 |
| a-Wave amplitudes | Baseline | Genotype * amplitude | 4,36 | 4.76 | 0.004 |
| a-Wave kinetics | Baseline | Genotype | 1,9 | 0.06 | 0.81 |
| b-Wave amplitudes | Baseline | Genotype | 1,9 | 9.67 | 0.013 |
| b-Wave amplitudes | Baseline | Genotype * amplitude | 5,45 | 12.17 | <0.0001 |
| b-Wave kinetics | Baseline | Genotype | 1,9 | 0.17 | 0.69 |
| a-Wave amplitudes | Light damage | Genotype | 1,9 | 10.48 | 0.01 |
| a-Wave amplitudes | Light damage | Genotype * amplitude | 4,36 | 2.69 | 0.046 |
| a-Wave kinetics | Light damage | Genotype | 1,9 | 6.22 | 0.03 |
| a-Wave kinetics | Light damage | Genotype * kinetics | 4,36 | 5.41 | 0.002 |
| b-Wave amplitudes | Light damage | Genotype | 1,9 | 1.23 | 0.3 |
| b-Wave kinetics | Light damage | Genotype | 1,9 | 6.81 | 0.03 |
| b-Wave kinetics | Light damage | Genotype * kinetics | 5,45 | 6.01 | 0.0002 |
Animals were reexamined using ERG recordings after light damage. A significant genotype-dependent effect was observed with respect to the reduction in a-wave amplitudes after light damage (P = 0.01), an effect that showed an interaction between genotype and amplitude (P = 0.046). In wild-type mice, the maximum a-wave amplitudes were reduced to approximately 60% of baseline levels (−77.8 ± 17.0 μV; P < 0.001), whereas in BDNF+/− mice, maximum a-wave amplitudes were reduced by only approximately 13% of baseline levels (−114.4 ± 14.5 μV, P = 0.40). On the other hand, no genotype-specific effects were observed for the b-wave amplitudes (P = 0.3). b-Wave amplitudes were reduced to approximately 40% of baseline levels in BDNF+/+ mice (230.3 ± 7.6 μV; P < 0.001) and to approximately 60% of baseline levels in BDNF+/− mice (261 ± 28 μV; P = 0.002). Reduced susceptibility to light damage in the BDNF+/+ animals was reflected by the lack of change in the implicit time in the two ERG components compared with their wild-type littermates. Although the implicit time at maximum light intensity increased in the BDNF+/+ mice (a-wave: 19 ± 2.7 seconds, P = 0.07; b-wave: 116 ± 10.1 seconds, P = 0.008), it remained unchanged in the BDNF+/− animals (a-wave: 12.7 ± 0.4 seconds, P = 1; b-wave: 79.7 ± 6.5 seconds, P = 0.3). Thus, for a- and b-wave kinetics, a genotype (a-wave, P = 0.03; b-wave, P = 0.002) and genotype-by-kinetics interaction was observed (a-wave, P = 0.03; b-wave, P < 0.001).
Neurotrophin Status Affects Susceptibility of Photoreceptors to Light Damage: Structure
Rod photoreceptor survival was examined in plastic sections of control and light-damaged BDNF+/+ and BDNF+/− mice. A representative section from the central retina for each genotype is shown in Figures 4A–4D. At 3 months of age, both genotypes had similar numbers of rows of photoreceptors in the retina when averaged for the four quadrants (Figs. 4A, 4B; 5). The BDNF+/+ retina contained an average of 9.81 ± 0.06 rows, and the BDNF+/− retina contained 8.88 ± 0.38 rows of photoreceptors (P = 0.13). However, after light damage, the average number of rows of photoreceptors decreased by approximately 61% in the wild-type mice, to 4.81 ± 0.67 (P = 0.007), but by less than 10% in the BDNF+/− mice, to 8.07 ± 0.41 (P = 0.26; genotype: F [1,8] = 21.5; P = 0.002; Figs. 4C, 4D; 5). The effect is attributed to the prevention of cell loss in the central retina of the BDNF+/− mice (genotype: F [1,18] = 50.3; P < 0.001), whereas both genotypes showed a small but comparable loss of photoreceptors in the periphery that did not reach statistical significance (genotype: F [1,18] = 4.21; P = 0.06; Fig. 5). The numbers of rows of photoreceptors after light damage paralleled the preservation of photoreceptor function determined in the BDNF+/+ and BDNF+/− mice. This lack of photoreceptor cell loss was reflected in the absence of TUNEL-labeled cells in the BDNF+/− compared with the BDNF+/+ retinas after light damage (Figs. 4E, 4F).
Figure 4.
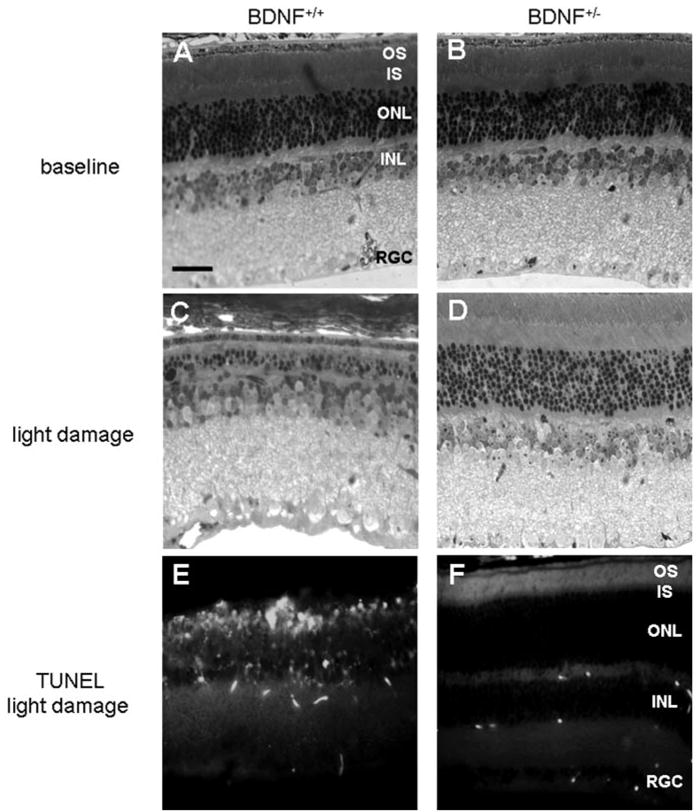
Comparison of BDNF+/+ (left) and BDNF+/− (right) mouse retinas. Photographs of sections were taken in the central retina for comparison. Sections stained with toluidine blue revealed that the 10-day light damage regimen (C, D) resulted in the loss of approximately 50% of the cells in the outer nuclear layer of the BDNF+/+ retina but did not affect the BDNF+/− retina compared with their respective controls (A, B). TUNEL-labeling confirmed positive cells in BDNF+/+, but not BDNF+/−, retinas after light damage (E, F)
Figure 5.
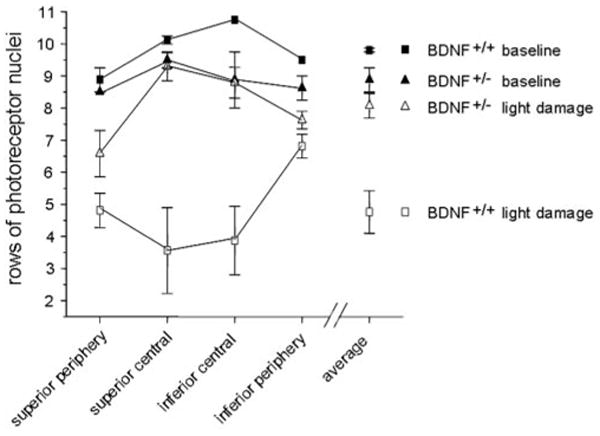
Rows photoreceptor cells in BDNF+/+ and BDNF+/− mice before and after light damage. Four different measurements were taken across the retina, counting rows of photoreceptor nuclei in the superior periphery, superior central, inferior central, and inferior periphery and determining the overall average per retina. Little change was observed after light damage in the BDNF+/− mouse retinas, but there was severe loss of photoreceptors in the BDNF+/+ retina, especially in the central regions. Data are plotted as mean ± SEM.
BDNF Status Affects Oxidative Stress in Response to Light Damage
To establish the presence of oxidative stress during the progression of photoreceptor degeneration after intense light exposure, the enzymatic activity of glutathione peroxidase (GPx) was measured in BDNF+/+ and BDNF+/− mice before and after light damage. As shown in Figure 6, GPx activity was elevated in BDNF+/+ mouse retinas after light damage (P = 0.016) but not in the retinas of BDNF+/− mice. At baseline, no genotypic difference could be detected (P = 0.15).
Figure 6.
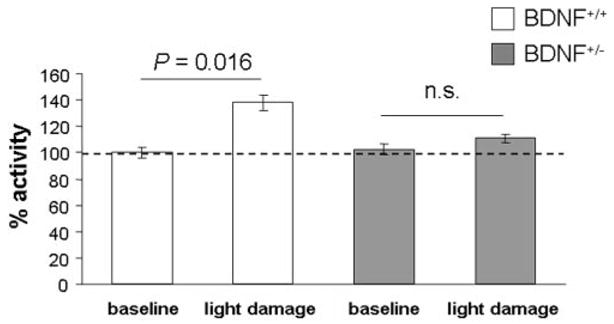
GPx activity in BDNF+/+ and BDNF+/− mice before and after light damage. GPx activity levels were indistinguishable in the two sets of mice when raised in cyclic light but showed a genotype-specific change after light damage. Data are plotted as mean ± SEM.
Discussion
Microglia–Müller Glial Cell Network in Light-Induced Photoreceptor Degeneration
The experiments described here were designed to further understand the role of BDNF in photoreceptor neuroprotection, in particular the light-induced degeneration. LaVail and colleagues (Cao et al.)16 have demonstrated that photoreceptor degeneration in response to constant bright light (~150 ft-c; ~1600 lux) can be ameliorated by exogenous BDNF, even though the photoreceptors do not express trkB receptors. The proposed pathway includes trkB-mediated release of additional neuroprotective factors by the Müller glial cells (e.g., see Wahlin et al.27 and Rohrer et al.28). Based on elegant experiments examining growth factor and neurotrophic factor production and release by activated microglial cells from control and constant light-exposed retinas (800–1300 lux) and microglia-conditioned medium on trophic factor expression in cultured Müller glial cells, Harada et al.17 put forward a model suggesting that microglia–Müller glial cell interactions mediated the generation of neuroprotective signals during prolonged light exposure. The proposed cascade is triggered by the invasion of activated microglia into the degenerating outer nuclear layer, which then release NGF, GDNF, and CNTF. Müller glial cells in return would increase their production and secretion of bFGF, BDNF, and CNTF. BDNF is envisioned as an amplification signal because it increases CNTF and bFGF production in an auto-crine fashion (i.e., it increases CNTF and bFGF production through the stimulation of trkB receptors on Müller cells). It is unclear whether this network is operational under normal cyclic light, but, for the sake of this set of experiments, we assumed that it is. Thus, based on the proposed connections, the model makes certain predictions (Fig. 7). First, it predicts that under cyclic light conditions, BDNF-mediated CNTF and bFGF production and release might be dampened. Second, it predicts that the amplification of the neuroprotection signal provided by BDNF is reduced in BDNF+/− mice, speeding up light-induced photoreceptor degeneration.
Figure 7.
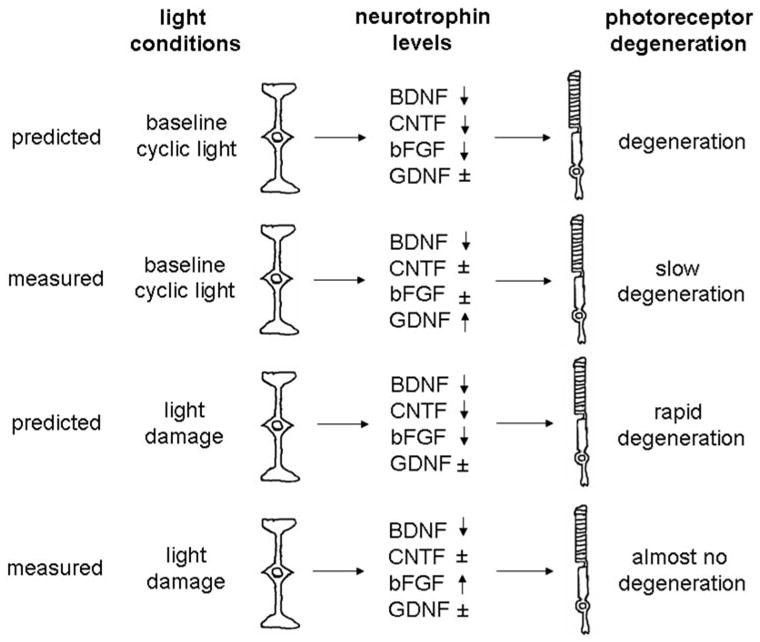
Summary of predicted and measured results based on the microglia–Müller glial network in light-degenerated retinas.17 Microglial cells have been reported to constitutively release NGF, CNTF, and GDNF. Microglia-derived GDNF and CNTF are expected to increase BDNF and bFGF production in Müller cells, with BDNF amplifying neurotrophin production by increasing CNTF and bFGF production in Müller cells, possibly providing a rescue signal. Microglia-derived NGF is expected to reduce bFGF production in Müller cells, possibly resulting in photoreceptor apoptosis. During excessive light stimulation, both arms of the network are expected to be activated. Only the results from the BDNF+/− mice are depicted in control (cyclic light) and experimental (light damage) conditions. Thus, for each condition, the predicted/measured net result of growth factor/neurotrophin production is indicated as is the expected effect on photoreceptor degeneration. Based on the reduced levels of BDNF, it was predicted that the BDNF-dependent CNTF and bFGF production would be reduced (↓) and the GDNF levels would be unaffected (±) to varying degrees under cyclic and constant light conditions, causing degeneration and rapid degeneration, respectively. However, we found the opposite, namely that in the presence of reduced BDNF levels, GDNF levels increased (↑) under cyclic light levels, concomitant with slow basal photoreceptor degeneration. Under light exposure conditions, the expected drop in bFGF and CNTF expression did not occur, resulting in slow to almost no photoreceptor degeneration.
However, here we found that under cyclic light conditions, CNTF and bFGF levels were unchanged, whereas GDNF mRNA levels were increased twofold over the levels found in age-matched BDNF+/+ littermates. After light damage, GDNF, CNTF, and BDNF mRNA dropped significantly in the presence of normal levels of BDNF (BDNF+/+ mice) but, paradoxically, remained almost unchanged compared with cyclic light levels in the animals with reduced levels of BDNF (BDNF+/− mice). Thus, the second prediction was also not met because BDNF+/− mice were less susceptible to light damage than their wild-type littermates, as determined by histologic cell counts, TUNEL assay, and electrophysiological recordings. Overall, the reduction in oxidative stress and improved photoreceptor survival indicated that compensatory mechanisms reminiscent of preconditioning against stress were triggered in the BDNF+/− retinas.
Are there obvious reasons for the differences in predicted and actual results? First, we started with the premise that neurotrophins and growth factors are expressed predominantly by Müller glial cells and invading microglial cells (e.g., Valter et al.29 and Frasson et al.30) and, thus, that changes in expression levels should be measurable in whole retina samples. Although retinal ganglion cells (RGCs) express some of those neurotrophic factors, it was argued that expression levels should not change in these cells because there was no evidence that light damage affects RGCs early in the progression of photoreceptor degeneration. In addition, Müller glial cells outnumber RGCs by a factor of 6,31 yet the possibility that changes in gene expression in the RGCs might have masked the changes occurring in the Müller glia and the invading microglial cells should not be excluded. Testing this possibility was beyond the scope of this project. Second, Harada et al.17 examined neurotrophin levels in microglial and Müller glial cells isolated from a 35-day-old rat and cultured for multiple days. Neurotrophin levels under these conditions might differ from the expression profiles of cells within an existing cellular network. Third, the model was established based on juvenile rat retina instead of young adult mouse, which was used here. And finally, fourth, the light paradigms used here and those used by Harada et al.17 differed. With albino rats, Harada et al.17 were able to use the continuous light exposure model (24-hour light exposure at approximately 800–1300 lux from postnatal day [P] 22-P35), whereas we used approximately 15-fold brighter light (approximately 15,000 lux) for a shorter period (1.5 hours) followed by 10 days of darkness to allow for the execution of cell death because the BDNF heterozygous mice were on a B6 background (B6.129-BDNFtm1-LT) and were resistant to continuous light exposure. However, as shown by Hao et al.,32 the mechanisms differed whereby continuous light exposure and intense light exposure triggered cell death. Both mechanisms required the presence of bleachable rhodopsin; however, intense bright-light damage was independent of the presence of transducin, whereas continuous low-light damage required transducin. Thus, it is plausible that the glial network outlined by Harada et al.17 is not activated in intense bright light. Unfortunately, none of the growth factors or neurotrophins involved in the glial network have been tested in rescue experiments in the bright-light damage model (summarized in Wenzel et al.9), the results of which might enable greater understanding.
Finally, the possibility that the small reduction in photoreceptor activity could blunt the sensitivity of the photoreceptors to light damage must be considered. Bright light triggers apoptosis of photoreceptor cells through a mechanism requiring activation of rhodopsin, and the kinetics by which bleached rhodopsin is regenerated dictates cell sensitivity to light-induced photoreceptor degeneration (reviewed in Hao et al.32). BDNF+/− and BDNF+/+ mice are approximately 94% C57BL/6 and carry the methionine variant in position 450 of the Rpe65 protein. Rpe65 is required for the synthesis of 11-cis retinal, the chromophore for all pigments in the retina,33 and the Leu450Met variation was shown recently to cosegregate with lower sensitivity to light damage.34 Thus, a difference in regeneration kinetics caused by the Rpe65 variant can be excluded. Therefore, the only obvious remaining difference for the purpose of this argument is the reduction in photoreceptor activity in the BDNF+/− mice. Unfortunately, to our knowledge, no studies have titrated the amount of rhodopsin present in the retina with sensitivity to light damage, a study that could be performed in the Rho−/− mouse.
BDNF and TrkB in Photoreceptor Signaling and Development
To strengthen our understanding of the role of trkB in rod development and function, we previously analyzed a juvenile trkB−/− mouse28 and juvenile mice that carried two hypomorphic trkB alleles (trkBfbz/fbz) expressing roughly 25% of the normal trkB,35 which allowed us to correlate functional and biochemical retinal phenotypes with levels of trkB expression in these two independently created trkB transgenic lines. The main findings from this study were that retinal rod function and morphology were unaffected in juvenile trkB+/− or trkB+/fbz mice (2–3 weeks of age) that expressed 50% or approximately 63% of wild-type trkB levels, respectively. However, rhodopsin content and outer segment length were significantly reduced in trkB−/− and trkBfbz/fbz mice, resulting in reduced a-wave (which reflects rod function) and b-wave (which reflects the rod-mediated rod bipolar cell response) amplitudes and slower kinetics. In follow-up experiments, it was determined that the reduction in b-wave response was caused by a presynaptic defect rather than a failure of the postsynaptic bipolar cells.36 Although the specific mechanisms still must be identified, it can be concluded that trkB-positive cells specifically control the developmental maturation of mouse rod photoreceptors and the maturation and function of the rod presynaptic terminal. Here we found evidence that reducing BDNF, one of the two ligands for the trkB receptors, resulted in a small but significant reduction in maximum a- and b-wave amplitudes in adult animals (3 months of age; Figs. 2, 3) without affecting the kinetics of the responses (Table 2). The current results confirm and extend our results on juvenile animals, demonstrating that long-term BDNF-deprivation does not impair photoreceptor survival but does reduce the maximum rod light response. However, long-term BDNF deprivation did not impact the function of the rod presynaptic terminal as it did in juvenile mice. Interestingly, a BDNF-dependent effect on synaptic transmission or bipolar cell responsiveness was revealed after light damage because the maximum b-wave amplitudes were reduced by 40% compared with the more modest drop of approximately 13% in the maximum a-wave amplitudes. This effect, however, differs from the results reported for the trk-Bfbz/fbz mice because b-wave kinetics in the BDNF+/− mice after light damage was unaffected.
Reduction in BDNF Mimics Preconditioning
BDNF+/− mice exhibited less oxidative stress in response to constant light exposure and, concomitantly, showed improved photoreceptor survival and a more normal cytokine profile (Figs. 2, 4). These findings suggest that compensatory mechanisms similar to those occurring in preconditioning against stress were triggered in the BDNF+/− retinas. Preconditioning is typically referred to as the phenomenon whereby brief (or subthreshold) noxious episodes render the tissue or organism resistant to subsequent noxious exposure. Liu et al.8 showed that preconditioning Sprague–Dawley rats with bright light increased their resistance to subsequent light damage. Preexposure led to a prolonged increase in bFGF and CNTF, each of which is part of the neuroprotective network of cytokines also proposed by the microglia–Müller cell hypothesis. However, here we did not find an increase in bFGF or CNTF, but we did find an increase in another Müller cell–derived cytokine, GDNF, at baseline. GDNF promotes photoreceptor cell survival in the RCS rat,37 the rhodopsin 334ter rat,38 and the rd1 mouse,30,39 and it promotes cell survival after retinal ischemia.40 Exogenous GDNF, however, has not yet been tested as a therapeutic agent in the mouse light damage models. In the rat retina, GFRá-2, the receptor for GDNF, was increased in photoreceptors exposed to light damage, though no changes were noted in GDNF mRNA levels.41
Effects Produced by Reduction in BDNF Could Be Interpreted as Confounding/Compensatory Effects
Results in BDNF+/− mice, suggesting that they are less susceptible to oxidative stress, were clearly unexpected. Gene knockout mice have been used since the tools became available to test the involvement of genes in specific aspects of development, function, and disease. However, the issue of confounding effects when using knockout animals—particularly the presence of hitchhiking genes, which are flanking DNA regions from the donor strain and modifier genes—was recognized early.42–45 It was argued that if two independently created lines generated the same results, the experimental outcome should not be attributed to contamination by residual donor genes but rather to the gene knockout. However, this does not address the issue of compensatory effects, a concern that was quickly raised.46,47 Systemic compensatory effects could be avoided by using cell type–specific gene targeting (e.g., see Kos48) or viral vector–mediated expression of silencing RNA (e.g., see Aigner49). Even under those “controlled” conditions, local compensatory effects could not be excluded.
Could compensatory effects have developed in BDNF+/− mice over time? Conover and Yancopoulos50 have demonstrated, using combinatorial neurotrophin receptor and neurotrophin deletions, the presence of compensatory actions between neurotrophins, including the switching in neurotrophin dependence for some neuronal populations during central nervous system development. No other studies involve BDNF+/− mice that document compensatory effects, though the topic has been raised when discussing unexpected results (e.g., see Bender51). Clearly, in future experiments, changes in protein expression of all neurotrophin receptors and their ligands must be studied.
Compensatory responses have been reported in the retina of the antioxidant enzyme glutathione peroxidase-1 knockout mouse (GPx-1−/−) investigated by Gosbell et al.52 Based on the role of GPx as an antioxidant, it was expected that GPx-1−/− mice would be more susceptible to light damage. However, as in our study, the contrary was observed; photoreceptors in the knockout mice better withstood photo-oxidative damage than their wild-type littermates. Retinas of GPx-1−/− mice raised in cyclic light had increased mRNA levels of oxidative stress markers, suggesting that this constant, increased oxidative stress preconditions the retina against further insult. The exact mechanism, however, still must be determined. As in our study, retinas of the GPx-1−/− mice exhibited elevated levels of oxidative stress before light damage, whereas in the BDNF+/− mice, elevated levels of GDNF were observed at baseline before light exposure. Thus, pathway-specific compensatory mechanisms were triggered in the two different models in cycle light that might have preconditioned the retinas against the damaging effects of constant light.
In summary, contrary to expectation, the retinas of BDNF+/− mice, which have reduced levels of a neurotrophin known to be protective for photoreceptors, were not more sensitive to light-induced photoreceptor degeneration than the retinas of their wild-type counterparts. Similarly, the expected changes in cytokine levels, predicted by the Harada et al.17 model, did not occur in our model. After light damage, photoreceptor survival and function were significantly higher in retinas from BDNF+/− mice than from their wild-type littermates. In addition, lower levels of glutathione peroxidase activity, a marker of oxidative stress, were found in BDNF+/− retinas after light damage than in wild-type littermate retinas. Photoreceptor survival was paralleled by a lack of cytokine loss induced by the constant light exposure in wild-type animals. One of the neuroprotective mechanisms of the retina to deal with fluctuations in light exposure and oxygen consumption during a normal light-dark cycle is to modulate the levels of neuroprotective agents such as cytokines.1,7 Thus, it appears that in BDNF+/− retinas, the reduction in BDNF leads to an adaptive response in this neuroprotective pathway, including the upregulation of GDNF under cyclic light conditions, and a blunted response after light damage.
Acknowledgments
The authors thank Lino Tessarollo for providing the original breeding pairs for the BDNF colony, Lotta Granholm for helpful discussions, Joseph Vallone for technical assistance, and Luanna Bartholomew for critical review.
Supported by National Institutes of Health Grants EY-13520 and AG-23630; Vision Core Grant EY-14793; and an unrestricted grant to Medical University of South Carolina from Research to Prevent Blindness, Inc. BR is a Research to Prevent Blindness Olga Keith Weiss Scholar.
Footnotes
Disclosure: R.B. Wilson, None; K. Kunchithapautham, None; B. Rohrer, None
References
- 1.Stone J, Maslim J, Valter-Kocsi K, et al. Mechanisms of photoreceptor death and survival in mammalian retina. Prog Retin Eye Res. 1999;18:689–735. doi: 10.1016/s1350-9462(98)00032-9. [DOI] [PubMed] [Google Scholar]
- 2.LaVail MM. Outer segment disc shedding and phagocytosis in the outer retina. Trans Ophthalmol Soc UK. 1983;103(pt 4):397–404. [PubMed] [Google Scholar]
- 3.Ames A, 3rd, Li YY, Heher EC, Kimble CR. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J Neurosci. 1992;12:840–853. doi: 10.1523/JNEUROSCI.12-03-00840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demontis GC, Longoni B, Gargini C, Cervetto L. The energetic cost of photoreception in retinal rods of mammals. Arch Ital Biol. 1997;135:95–109. [PubMed] [Google Scholar]
- 5.Bill A, Sperber G, Ujiie K. Physiology of the choroidal vascular bed. Int Ophthalmol. 1983;6:101–107. doi: 10.1007/BF00127638. [DOI] [PubMed] [Google Scholar]
- 6.Yu DY, Cringle SJ. Retinal degeneration and local oxygen metabolism. Exp Eye Res. 2005;80:745–751. doi: 10.1016/j.exer.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Penn JS, Naash MI, Anderson RE. Effect of light history on retinal antioxidants and light damage susceptibility in the rat. Exp Eye Res. 1987;44:779–788. doi: 10.1016/s0014-4835(87)80041-6. [DOI] [PubMed] [Google Scholar]
- 8.Liu C, Peng M, Laties AM, Wen R. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344. doi: 10.1523/JNEUROSCI.18-04-01337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Ranchon I, Gorrand JM, Cluzel J, Droy-Lefaix MT, Doly M. Functional protection of photoreceptors from light-induced damage by dimethylthiourea and Ginkgo biloba extract. Invest Ophthalmol Vis Sci. 1999;40:1191–1199. [PubMed] [Google Scholar]
- 11.Ranchon I, Chen S, Alvarez K, Anderson RE. Systemic administration of phenyl-N-tert-butylnitrone protects the retina from light damage. Invest Ophthalmol Vis Sci. 2001;42:1375–1379. [PubMed] [Google Scholar]
- 12.Goureau O, Jeanny JC, Becquet F, Hartmann MP, Courtois Y. Protection against light-induced retinal degeneration by an inhibitor of NO synthase. Neuroreport. 1993;5:233–236. doi: 10.1097/00001756-199312000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001;21:53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Machida S, Chaudhry P, Shinohara T, et al. Lens epithelium-derived growth factor promotes photoreceptor survival in light damaged and RCS rats. Invest Ophthalmol Vis Sci. 2001;42:1087–1095. [PubMed] [Google Scholar]
- 15.LaVail MM, Yasumura D, Matthes MT, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- 16.Cao W, Li F, Steinberg RH, LaVail MM. Development of normal and injury-induced gene expression of aFGF, bFGF, CNTF, BDNF, GFAP and IGF-I in the rat retina. Exp Eye Res. 2001;72:591–604. doi: 10.1006/exer.2001.0990. [DOI] [PubMed] [Google Scholar]
- 17.Harada T, Harada C, Kohsaka S, et al. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002;22:9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liebl DJ, Tessarollo L, Palko ME, Parada LF. Absence of sensory neurons before target innervation in brain-derived neurotrophic factor-, neurotrophin 3-, and TrkC-deficient embryonic mice. J Neurosci. 1997;17:9113–9121. doi: 10.1523/JNEUROSCI.17-23-09113.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenzel A, Grimm C, Samardzija M, Reme CE. The genetic modifier Rpe65Leu(450): effect on light damage susceptibility in c-Fos-deficient mice. Invest Ophthalmol Vis Sci. 2003;44:2798–2802. doi: 10.1167/iovs.02-1134. [DOI] [PubMed] [Google Scholar]
- 20.Gresh J, Goletz PW, Crouch RK, Rohrer B. Structure-function analysis of rods and cones in juvenile, adult, and aged C57bl/6 and Balb/c mice. Vis Neurosci. 2003;20:211–220. doi: 10.1017/s0952523803202108. [DOI] [PubMed] [Google Scholar]
- 21.Richards A, Emondi AA, Rohrer B. Long-term ERG analysis in the partially light damaged mouse retina reveals regressive and compensatory changes. Vis Neurosci. 2006;23:91–97. doi: 10.1017/S0952523806231080. [DOI] [PubMed] [Google Scholar]
- 22.Bayer AU, Cook P, Brodie SE, Maag KP, Mittag T. Evaluation of different recording parameters to establish a standard for flash electroretinography in rodents. Vis Res. 2001;41:2173–2185. doi: 10.1016/s0042-6989(01)00103-1. [DOI] [PubMed] [Google Scholar]
- 23.Rohrer B, Matthes MT, LaVail MM, Reichardt LF. Lack of p75 receptor does not protect photoreceptors from light-induced cell death. Exp Eye Res. 2003;76:125–129. doi: 10.1016/s0014-4835(02)00258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006;83:380–389. doi: 10.1016/j.exer.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Rohrer B, Pinto FR, Hulse KE, Lohr HR, Zhang L, Almeida JS. Multidestructive pathways triggered in photoreceptor cell death of the rd mouse as determined through gene expression profiling. J Biol Chem. 2004;279:41903–41910. doi: 10.1074/jbc.M405085200. [DOI] [PubMed] [Google Scholar]
- 26.Mitas M, Mikhitarian K, Walters C, et al. Quantitative real-time RT-PCR detection of breast cancer micrometastasis using a multi-gene marker panel. Int J Cancer. 2001;93:162–171. doi: 10.1002/ijc.1312. [DOI] [PubMed] [Google Scholar]
- 27.Wahlin KJ, Campochiaro PA, Zack DJ, Adler R. Neurotrophic factors cause activation of intracellular signaling pathways in Müller cells and other cells of the inner retina, but not photoreceptors. Invest Ophthalmol Vis Sci. 2000;41:927–936. [PubMed] [Google Scholar]
- 28.Rohrer B, Korenbrot JI, LaVail MM, Reichardt LF, Xu B. Role of neurotrophin receptor TrkB in the maturation of rod photoreceptors and establishment of synaptic transmission to the inner retina. J Neurosci. 1999;19:8919–8930. doi: 10.1523/JNEUROSCI.19-20-08919.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valter K, Bisti S, Gargini C, et al. Time course of neurotrophic factor upregulation and retinal protection against light-induced damage after optic nerve section. Invest Ophthalmol Vis Sci. 2005;46:1748–1754. doi: 10.1167/iovs.04-0657. [DOI] [PubMed] [Google Scholar]
- 30.Frasson M, Picaud S, Leveillard T, et al. Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci. 1999;40:2724–2734. [PubMed] [Google Scholar]
- 31.Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hao W, Wenzel A, Obin MS, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002;32:254–260. doi: 10.1038/ng984. [DOI] [PubMed] [Google Scholar]
- 33.Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 34.Danciger M, Lyon J, Worrill D, et al. New retinal light damage QTL in mice with the light-sensitive RPE65 LEU variant. Mamm Genome. 2004;15:277–283. doi: 10.1007/s00335-003-2336-2. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer B. Gene dosage effect of the TrkB receptor on rod physiology and biochemistry in juvenile mouse retina. Mol Vis. 2001;7:288–296. [PubMed] [Google Scholar]
- 36.Rohrer B, Blanco R, Marc RE, et al. Functionally intact glutamate-mediated signaling in bipolar cells of the TRKB knockout mouse retina. Vis Neurosci. 2004;21:703–713. doi: 10.1017/S0952523804045055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawrence JM, Keegan DJ, Muir EM, et al. Transplantation of Schwann cell line clones secreting GDNF or BDNF into the retinas of dystrophic Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2004;45:267–274. doi: 10.1167/iovs.03-0093. [DOI] [PubMed] [Google Scholar]
- 38.McGee Sanftner LH, Abel H, Hauswirth WW, Flannery JG. Glial cell line derived neurotrophic factor delays photoreceptor degeneration in a transgenic rat model of retinitis pigmentosa. Mol Ther. 2001;4:622–629. doi: 10.1006/mthe.2001.0498. [DOI] [PubMed] [Google Scholar]
- 39.Andrieu-Soler C, Aubert-Pouessel A, Doat M, et al. Intravitreous injection of PLGA microspheres encapsulating GDNF promotes the survival of photoreceptors in the rd1/rd1 mouse. Mol Vis. 2005;11:1002–1011. [PubMed] [Google Scholar]
- 40.Wu WC, Lai CC, Chen SL, et al. GDNF gene therapy attenuates retinal ischemic injuries in rats. Mol Vis. 2004;10:93–102. [PubMed] [Google Scholar]
- 41.Jomary C, Darrow RM, Wong P, Organisciak DT, Jones SE. Expression of neurturin, glial cell line-derived neurotrophic factor, and their receptor components in light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2004;45:1240–1246. doi: 10.1167/iovs.03-1122. [DOI] [PubMed] [Google Scholar]
- 42.Lathe R. Mice, gene targeting and behaviour: more than just genetic background. Trends Neurosci. 1996;19:183–186. doi: 10.1016/s0166-2236(96)20022-0. [DOI] [PubMed] [Google Scholar]
- 43.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 44.Crusio WE. Gene-targeting studies: new methods, old problems. Trends Neurosci. 1996;19:186–187. doi: 10.1016/s0166-2236(96)20023-2. discussion 188–189. [DOI] [PubMed] [Google Scholar]
- 45.Crawley JN. Unusual behavioral phenotypes of inbred mouse strains. Trends Neurosci. 1996;19:181–182. doi: 10.1016/s0166-2236(96)20021-9. discussion 188–189. [DOI] [PubMed] [Google Scholar]
- 46.Nelson RJ, Young KA. Behavior in mice with targeted disruption of single genes. Neurosci Biobehav Rev. 1998;22:453–462. doi: 10.1016/s0149-7634(97)00053-5. [DOI] [PubMed] [Google Scholar]
- 47.Lowenstein PR, Castro MG. Genetic engineering within the adult brain: implications for molecular approaches to behavioral neuroscience. Physiol Behav. 2001;73:833–839. doi: 10.1016/s0031-9384(01)00520-0. [DOI] [PubMed] [Google Scholar]
- 48.Kos CH. Cre/loxP system for generating tissue-specific knockout mouse models. Nutr Rev. 2004;62:243–246. doi: 10.1301/nr2004.jun243-246. [DOI] [PubMed] [Google Scholar]
- 49.Aigner A. Gene silencing through RNA interference (RNAi) in vivo: strategies based on the direct application of siRNAs. J Biotechnol. 2006;124:12–25. doi: 10.1016/j.jbiotec.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- 51.Bender R, Heimrich B, Meyer M, Frotscher M. Hippocampal mossy fiber sprouting is not impaired in brain-derived neurotrophic factor-deficient mice. Exp Brain Res. 1998;120:399–402. doi: 10.1007/s002210050413. [DOI] [PubMed] [Google Scholar]
- 52.Gosbell AD, Stefanovic N, Scurr LL, et al. Retinal light damage: structural and functional effects of the antioxidant glutathione peroxidase-1. Invest Ophthalmol Vis Sci. 2006;47:2613–2622. doi: 10.1167/iovs.05-0962. [DOI] [PubMed] [Google Scholar]