

Abstract
Purpose
Axonal damage and loss of neurons correlate with permanent vision loss and neurologic disability in patients with optic neuritis and multiple sclerosis (MS). Current therapies involve immunomodulation, with limited effects on neuronal damage. The authors examined potential neuroprotective effects in optic neuritis by SRT647 and SRT501, two structurally and mechanistically distinct activators of SIRT1, an enzyme involved in cellular stress resistance and survival.
Methods
Experimental autoimmune encephalomyelitis (EAE), an animal model of MS, was induced by immunization with proteolipid protein peptide in SJL/J mice. Optic neuritis developed in two thirds of eyes with significant retinal ganglion cell (RGC) loss detected 14 days after immunization. RGCs were labeled in a retrograde fashion with fluorogold by injection into superior colliculi. Optic neuritis was detected by inflammatory cell infiltration of the optic nerve.
Results
Intravitreal injection of SIRT1 activators 0, 3, 7, and 11 days after immunization significantly attenuated RGC loss in a dose-dependent manner. This neuroprotective effect was blocked by sirtinol, a SIRT1 inhibitor. Treatment with either SIRT1 activator did not prevent EAE or optic nerve inflammation. A single dose of SRT501 on day 11 was sufficient to limit RGC loss and to preserve axon function.
Conclusions
SIRT1 activators provide an important potential therapy to prevent the neuronal damage that leads to permanent neurologic disability in optic neuritis and MS patients. Intravitreal administration of SIRT1 activators does not suppress inflammation in this model, suggesting that their neuroprotective effects will be additive or synergistic with current immunomodulatory therapies.
Multiple sclerosis (MS) is an autoimmune-mediated neurodegenerative disease with characteristic foci of inflammatory demyelination in the brain, spinal cord, and optic nerves.1 Recent studies have demonstrated not only that axonal damage and neuronal loss are significant pathologic components of MS and experimental autoimmune encephalomyelitis (EAE)2–4 but that this neuronal damage is thought to cause the permanent neurologic disability often seen in MS patients.4–9 Current treatments for MS involve immunomodulation, which can reduce the incidence of inflammatory relapses.1 However, existing therapies are often not fully effective, and limited evidence suggests that these therapies prevent the long-term neuronal damage and physical disability of MS patients.10,11 New therapies that prevent neurodegeneration through nonimmunomodulatory mechanisms have a tremendous potential to work synergistically with current MS therapies.
Optic neuritis is an inflammatory demyelinating condition of the optic nerve that often occurs in MS patients.1 Axonal loss develops in patients with optic neuritis and correlates with decreased visual function.12,13 In an observational prospective study, 27 of 38 patients with optic neuritis had significant retinal nerve fiber layer thinning, but this was not detected until 3 to 6 months after most (85%) patients initially sought care.14 Neuroprotective therapies initiated during the acute inflammatory phase, therefore, have tremendous potential to prevent significant subsequent neuronal loss in patients with optic neuritis and MS.
Significant retinal ganglion cell (RGC) loss has been demonstrated in several experimental models of optic neuritis, including relapsing/remitting EAE,15 chronic EAE,16 and a model of spontaneous isolated optic neuritis.17 In the relapsing/remitting model—the most common disease course of MS and optic neuritis in human patients—SJL/J mice develop a high incidence (greater than 60%) of optic neuritis beginning 9 to 11 days after immunization with proteolipid protein peptide (PLP). RGC loss occurs over the next several days with significant RGC loss detected by day 14, suggesting that neuronal loss occurs secondarily to inflammation-induced damage.15
SIRT1 is a member of a highly conserved gene family (sirtuins) encoding NAD+-dependent deacetylases,18–20 originally found to deacetylate histones leading to increased DNA stability and prolonged survival in yeast and higher organisms,21–23 including mammals.24 Members of the sirtuin family have since been found to function as deacetylases for numerous protein targets involved in many cellular pathways, including cellular stress responses, apoptosis, and axonal degeneration.25–27 The activity of sirtuins can be regulated by other compounds that can serve as activators (e.g., plant polyphenols) that increase SIRT1 activity by lowering the Km of its substrates or that can serve as inhibitors (e.g., sirtinol).25,27–30 Neuroprotection mediated by SIRT1 activation has been demonstrated in axotomized dorsal root ganglion neurons31 and in neurons containing the Huntingtin gene mutation responsible for neurodegeneration in Huntington disease.32 We hypothesized that SIRT1 activation is neuroprotective for RGCs during optic neuritis. To test this hypothesis, two distinct SIRT1 activators, SRT647 (nicotinamide riboside)33 and SRT501 (a proprietary formulation of resveratrol)34 were examined for their neuroprotective effects in EAE mice, and the effects on RGC survival were quantified.
METHODS
Mice
Six-week-old SJL/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Treatment of the animals, housed at animal facilities at the Thomas Jefferson University (Philadelphia, PA), adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
RGC Labeling
Retrograde labeling of RGCs was performed as described previously.15 Briefly, mice were anesthetized by intraperitoneal injection of 0.2 mL solution containing 10 mg/mL ketamine (Sigma, St. Louis, MO) and 1 mg/mL xylazine (Sigma). Holes were drilled through the skull above each superior colliculus through a midsagittal skin incision. Unless otherwise noted, 2.5 μL of 1.25% hydroxystilbamidine (Fluorogold; Molecular Probes, Eugene, OR) in PBS was injected stereotactically into each superior colliculus 1 week before EAE immunization (for axonal transport function studies, fluorogold injections were made 2 days before mice were killed).
Induction and Evaluation of EAE
EAE was induced as previously described.15 Briefly, mice were anesthetized with ketamine/xylazine and were injected subcutaneously at two sites on the back with 0.1 mL solution containing 0.5 mg/mL proteolipid protein peptide 139–151 (University of Pennsylvania Protein Chemistry Laboratory, Philadelphia, PA) emulsified in complete Freund adjuvant (CFA; Difco, Detroit, MI) containing 2.5 mg/mL Mycobacterium tuberculosis (Difco). Control mice were injected with equal volumes of PBS and CFA. All mice were injected with 200 ng pertussis toxin (List Biological, Campbell, CA) in 0.1 mL PBS intraperitoneally on day 0 (day of immunization) and again on day 2. Mice were scored daily for clinical signs of EAE using a 5-point scale, as described previously15,35: no disease = 0; partial tail paralysis = 0.5; tail paralysis or waddling gait = 1.0; partial tail paralysis and waddling gait = 1.5; tail paralysis and waddling gait = 2.0; partial limb paralysis = 2.5; paralysis of one limb = 3.0; paralysis of one limb and partial paralysis of another = 3.5; paralysis of two limbs = 4.0; moribund state = 4.5; death = 5.0.
Intravitreal Injections
SRT647 (Sirtris Pharmaceuticals, Cambridge, MA) and sirtinol (Sigma) were diluted in PBS, and SRT501 (Sirtris) was diluted in 2% hydroxypropyl methylcellulose (HPMC) and 0.2% dioctyl sodium sulfosuccinate (DOSS; Wilson Laboratories, Mumbai, India). Intravitreal injections were given by methods described previously,36 with minor modifications. Mice were anesthetized with ketamine/xylazine, and eyes were visualized under a dissecting microscope. The conjunctiva was lifted with jeweler’s forceps and cut down to the sclera with Vannas scissors, and the incision was extended circumferentially around the corneal limbus. The sclera was then penetrated with a 30-gauge needle that passed into the vitreous just posterior to the lens. The tip of a 32- or 33-gauge blunt needle on a 10-μL Hamilton syringe (Hamilton, Reno, NV) containing 0.8 μL solution (vehicle or drug) was then introduced into the vitreous and slowly injected by an assistant while the syringe and needle were held in place under the microscope. Identical treatment was repeated in the contralateral eye of each mouse. After injection, ointment (Polysporin; Pfizer, New York, NY) was applied to each eye. The final drug concentration in the eye is estimated to be one sixth the concentration of the solution injected based on the volume injected and the average size of the vitreal space measured in the SJL/J mouse eyes. Concentrations given in the text and figures represent this estimated final dilution. Mice were randomly selected for each drug or placebo (vehicle) treatment group.
Quantification of RGC Numbers
RGC numbers were counted as described previously.15 Briefly, mice were killed, and each eye was removed and fixed in 4% paraformaldehyde in PBS. Dissected retinas were flat mounted on glass slides, viewed by fluorescence microscopy (Eclipse E600; Nikon, Tokyo, Japan), and photographed at 20× magnification in 12 standard fields: 1/6, 3/6, and 5/6 of the retinal radius from the center of the retina in each quadrant. Each photographed field measured 0.22 × 0.28 mm (0.062 mm2); thus, the total area counted per eye was 0.74 mm2. RGC numbers shown in each experiment represented the total number of RGCs counted per eye (RGCs/0.74 mm2). RGCs were counted by a masked investigator using image analysis software (Image-Pro Plus 5.0; Media Cybernetics, Silver Spring, MD).
Histopathologic Evaluation of Optic Nerves
After the mice were humanely killed, each optic nerve was removed, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 5-μm longitudinal sections. Nerves were stained with hematoxylin and eosin, and the presence of inflammatory cell infiltration was assessed by a masked investigator using a 4-point scale as in previous studies15,37: no infiltration = 0; mild cellular infiltration of the optic nerve or optic nerve sheath = 1; moderate infiltration = 2; severe infiltration = 3; massive infiltration = 4. Eyes with any level of optic nerve inflammation (score 1–4) were considered to have optic neuritis, whereas eyes with no detectable optic neuritis were considered not to have optic neuritis.
Electroretinographic Measurements
Whole eye electroretinographic (ERG) measurements were performed on 6-week-old C57BL/6 mice using previously described methods.38–40 In brief, mice were anesthetized with ketamine/xylazine after 12-hour dark adaptation, and eyes were dilated with one drop of 1% tropicamide (Alcon, Fort Worth, TX). Mice were placed on a 37°C platform with a reference electrode placed in the mouth. Contact lenses containing embedded platinum wires were placed on each eye as corneal electrodes. Differential amplifiers with bandwidths of 0.1 Hz to 1.0 kHz were used to record full-field ERGs. Mice were placed in a recording chamber, and ERGs were evoked with 10-ms flashes of 20 cd · s/m2 light generated through a Ganzfeld stimulator (Lace, Pisa, Italy). ERG measurements were performed on each eye before treatment and were repeated 2 weeks after intravitreal injection of vehicle, SRT647, SRT501, or sirtinol. Mean scotopic b-wave and total a-wave responses were compared among groups.
Statistical Analysis
RGC numbers, or optic nerve inflammation scores, were analyzed by one-way ANOVA followed by Bonferroni multiple comparison test using biostatistics software (GraphPad Prism 3.02; GraphPad Software, San Diego, CA). Data shown represent the mean ± SEM number of RGCs, as has been reported in previous studies quantifying RGC numbers.15–17 Clinical EAE scores were compared between treatment groups by ANOVA for repeated measures at each day after immunization with the biostatistics software (GraphPad Prism).
RESULTS
Prevention of Acute RGC Loss, but Not Inflammation by SRT647 Treatment from Time of EAE Induction
The ability of SRT647, an indirect activator of SIRT1, to attenuate RGC loss was examined. RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0), and mice were killed on day 14. As in previous studies,15 EAE eyes with optic neuritis had significantly fewer surviving RGCs at day 14 than eyes of control mice (Figs. 1A, 1B, 2A). Significant differences were found in total RGC numbers added across 12 standardized retinal fields, but not within any one region of the retina for all experiments. Eyes of EAE mice with optic neuritis treated with repeated intravitreal injections of 16.67 mM SRT647 on days 0, 3, 7, and 11 after immunization (n = 12) had significantly more RGCs (505.2 ± 35.8/eye) than eyes of placebo-treated mice with optic neuritis (268.4 ± 59.4/eye; n = 8) but significantly fewer RGCs than eyes of control mice (690.8 ± 80.9/eye; Fig. 2A). Optic neuritis eyes treated with 66.67 mM SRT647 (n = 8) had significantly greater RGC survival (709.9 ± 66.8) than in optic neuritis eyes treated with 16.67 mM SRT647, and equivalent RGC survival to control eyes (Figs. 1C, 2A). SRT647 treatment did not alter RGC survival in control eyes (769.8 ± 116.1; n = 8; Fig. 2A). EAE eyes that did not develop optic neuritis had no significant loss of RGCs compared with control eyes (data not shown), as found in previous studies.15
Figure 1.
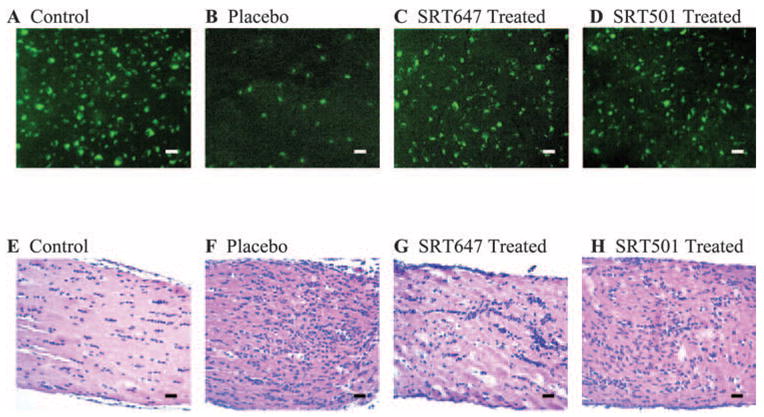
SIRT1 activators attenuated RGC loss during optic neuritis without preventing inflammatory cell infiltration of the optic nerve. RGCs were labeled in a retrograde fashion by injection of fluorogold into the superior colliculi 1 week before immunization. EAE mice were immunized with PLP, and control mice were sham immunized with PBS on day 0. Mice were killed on day 14, and retinas and optic nerves were isolated. (A) Numerous fluorescence-labeled RGCs are shown in a representative field of the retina from a control mouse eye. (B) Fewer RGCs are observed in an EAE mouse eye with optic neuritis treated with placebo. (C) Numerous RGCS are shown from an EAE mouse eye with optic neuritis treated with 66.67 mM SRT647 by intravitreal injections on days 0, 3, 7, and 11 after immunization. (D) Numerous RGCS are shown from an EAE mouse eye with optic neuritis treated with 100 μM SRT501 by intravitreal injections on days 0, 3, 7, and 11 after immunization. (E) Longitudinal section of the optic nerve from a control mouse stained with hematoxylin and eosin demonstrates the normal cellularity of the optic nerve. (F) Moderate to severe inflammatory cell infiltration shown in the optic nerve of an EAE mouse treated with placebo indicates acute optic neuritis. (G) Mild to moderate inflammation of the optic nerve and sheath observed in an EAE mouse eye treated with 66.67 mM SRT647. (H) Moderate inflammation of the optic nerve in an EAE mouse eye treated with 100 μM SRT501. Original magnification, 20×. Scale bars = 25 μm.
Figure 2.
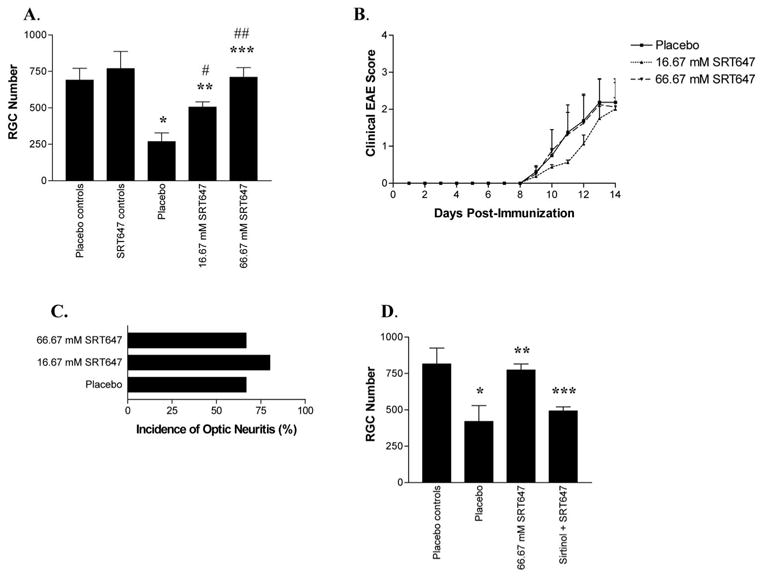
SRT647 attenuated RGC loss in a dose-dependent manner without reducing clinical EAE or inflammatory optic neuritis. (A) Control and EAE mice were treated with placebo or SRT647 on days 0, 3, 7, and 11 after immunization and were killed on day 14. Eyes from control mice treated with 16.67 mM SRT647 had no significant difference in RGC numbers compared with placebo-treated control eyes. EAE eyes with optic neuritis (placebo treated) had decreased RGC numbers (*P < 0.001) compared with controls. Optic neuritis eyes treated with 16.67 mM SRT647 had more RGCs than placebo treated optic neuritis eyes (**P < 0.01) but fewer RGCs than controls (#P < 0.05). SRT647 (66.67 mM) further attenuated RGC loss, with RGC numbers increased compared with placebo-treated optic neuritis eyes (***P < 0.001) and 16.67 mM SRT647–treated eyes (##P < 0.01) and no difference compared with control eyes. (B) Mice immunized with PLP were evaluated daily for clinical EAE. No significant difference in EAE score was found in EAE mice treated with placebo, 16.67 mM, or 66.67 mM SRT647. (C) Optic neuritis was detected by histologic evaluation of optic nerves on day 14 after immunization. No significant difference in incidence of optic neuritis was detected between EAE mice treated with placebo, 16.67 mM SRT647, or 66.67 mM SRT647. (D) The SIRT1 inhibitor sirtinol blocked the neuroprotective effects of SRT647. EAE optic neuritis eyes (placebo treated) had fewer RGCs than control eyes (*P < 0.05) and optic neuritis eyes treated with 66.67 mM SRT647 (**P < 0.05). Optic neuritis eyes treated with 66.67 mM SRT647 and 100 μM sirtinol had fewer RGCs than optic neuritis eyes treated with 66.67 mM SRT647 alone (***P < 0.001), and RGC numbers were not significantly different from those in placebo-treated optic neuritis eyes.
Intravitreal injection of SRT647 did not attenuate clinical EAE. EAE mice treated with placebo (n = 8) or either dose of SRT647 (n = 8 mice/dose) developed a similar time course of clinical EAE, beginning 9 days after immunization and progressing through day 14, when mice were killed (Fig. 2B). SRT647 also did not attenuate inflammatory infiltration of the optic nerve, as demonstrated by inflammatory cell infiltration of the optic nerve sheath and the parenchyma present in placebo-treated and SRT647-treated optic nerves (Figs. 1F, 1G). The incidence of optic neuritis was not significantly different between placebo (8 of 12 eyes examined), 16.67 mM SRT647–treated (12 of 15 eyes), or 66.67 mM SRT647–treated (8 of 12 eyes) eyes (Fig. 2C), and the degree of inflammation did not differ between placebo (1.13 ± 0.35 average inflammation score; n = 8 eyes), 16.67 mM SRT647–treated (1.25 ± 0.45, n = 12), or 66.67 mM SRT647–treated (1.25 ± 0.46, n = 8) eyes.
SRT647 Neuroprotection Blocked by SIRT1 Inhibition
To confirm that SIRT1 activity mediated the observed neuroprotective effects of SRT647, SIRT1 activity was blocked with a known inhibitor, sirtinol, at a concentration previously shown to be effective.30,32 RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0), and mice were killed on day 14. In EAE eyes with optic neuritis, treatment with 66.67 mM SRT647 on days 0, 3, 7, and 11 (n = 5) attenuated the RGC loss observed in placebo-treated (n = 5) eyes (773.4 ± 41.6 vs. 420.2 ± 109.3 RGCs/eye), but RGC survival in eyes treated with both 66.67 mM SRT647 and 100 μM sirtinol was significantly lower (492.6 ± 28.1; n = 5) than in control eyes (815.6 ± 108.6, n = 12) or optic neuritis eyes treated with SRT647 alone (773.4 ± 41.6; Fig. 2D), suggesting a role of SIRT1 in the effect of SRT647.
Prevention of Acute RGC Loss, but Not Inflammation, by SRT501 Treatment from Time of EAE Induction
Neuroprotective effects of SRT501, a direct SIRT1 activator with an EC50 value of approximately 50 μM (Milne J, et al., manuscript submitted), were examined during optic neuritis. RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0), and mice were killed on day 14. Intravitreal injection of 6 μM SRT501 on days 0, 3, 7, and 11 after immunization (n = 10 eyes) led to a trend of increased RGC survival (451.8 ± 43.0 RGCs vs. 384.9 ± 31.2 RGCs in placebo-treated optic neuritis eyes; n = 14) that was not statistically significant (Fig. 3A). Injection of 13 μM SRT501 (n = 15) resulted in a significant increase in RGC numbers (585.5 ± 51.0) that was not statistically different from numbers in controls (685.6 ± 35.8; n = 10), despite the trend toward fewer RGC numbers. Optic neuritis eyes treated with 100 μM SRT501 (n = 9) had even greater RGC survival (644.0 ± 49.2), with RGC numbers equivalent to control eyes (Figs. 1D, 3A). Intravitreal injection of the highest dose of SRT501 into control eyes (n = 6) did not reduce RGC survival (675.3 ± 55.8; Fig. 3A).
FIGURE 3.
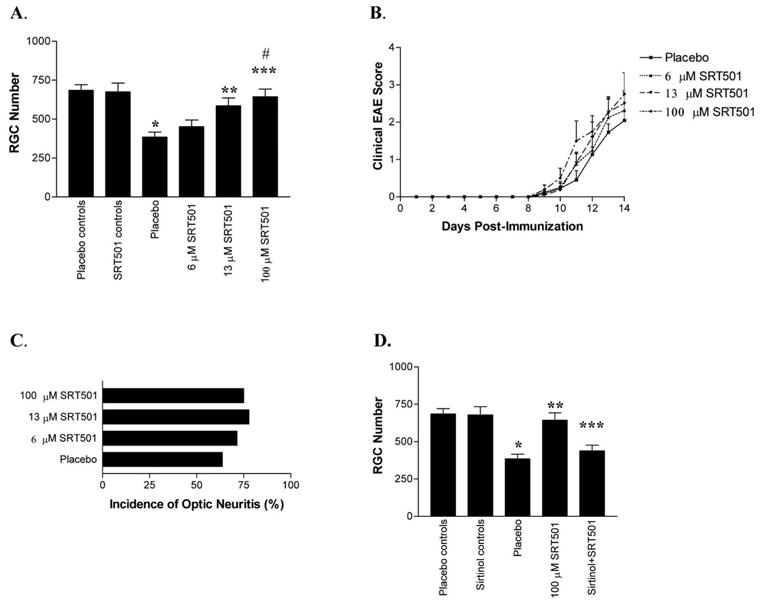
SRT501 attenuated RGC loss in a dose-dependent manner without reducing clinical EAE or inflammatory optic neuritis. (A) Control and EAE mice were treated with placebo or SRT501 on days 0, 3, 7, and 11 and were killed on day 14. Eyes from control mice treated with 100 μM SRT501 had no significant difference in RGC number from placebo-treated control eyes. EAE optic neuritis eyes had decreased RGC numbers (*P < 0.001) compared with controls. A small increase in RGC numbers in optic neuritis eyes treated with 6 μM SRT501 versus placebo was not significant. EAE optic neuritis eyes treated with 13 μM SRT501 had more RGCs than placebo (**P < 0.01). SRT501 (100 μM) further attenuated RGC loss, with RGC numbers increased compared with placebo (***P < 0.001) and with 6 μM SRT501–treated eyes (#P < 0.01) but no difference compared with control eyes. A trend toward increased RGC numbers in 100 μM SRT501 compared with 3 μM SRT501-treated eyes was not significant. (B) Mice immunized with PLP were evaluated daily for clinical EAE. No significant difference in EAE score was found between EAE mice treated with placebo or those treated with SRT501. (C) No significant difference in incidence of histologic optic neuritis was detected between EAE mice treated with placebo or SRT501. (D) The SIRT1 inhibitor sirtinol blocked the neuroprotective effects of SRT501. Sirtinol (100 μM) in control eyes did not significantly change RGC numbers compared with placebo-treated control eyes. EAE optic neuritis eyes had fewer RGCs than control eyes (*P < 0.001) and optic neuritis eyes treated with 100 μM SRT501 (**P < 0.001). Optic neuritis eyes treated with both 100 μM SRT501 and 100 μM sirtinol had fewer RGCs than optic neuritis eyes treated with 100 μM SRT501 alone (***P < 0.05), and RGC numbers were not significantly different from those of placebo-treated optic neuritis eyes.
As with SRT647, repeated intravitreal injections of SRT501 did not attenuate the development of clinical EAE. Mice treated with placebo (n = 11), 6 μM (n = 8), 13 μM (n = 11), or 100 μM (n = 8) SRT501 all developed similar clinical disease scores beginning at day 9 and progressing through day 14 after immunization (Fig. 3B). SRT501-treated eyes developed typical inflammatory infiltration of the optic nerve (Fig. 1H). No significant difference in the percentage of eyes developing optic neuritis was detected between EAE mice treated by intravitreal injection with placebo (14 of 22 eyes examined), 6 μM (10 of 14 eyes), 13 μM (17 of 22 eyes), or 100 μM SRT501 (9 of 12 eyes; Fig. 3C). The degree of optic nerve inflammation also did not vary significantly in eyes treated with placebo (1.14 ± 0.36 average inflammation score; n = 14 eyes), 6 μM SRT501 (1.40 ± 0.70; n = 10), 13 μM SRT501 (1.53 ± 0.64; n = 15), or 100 μM SRT501 (1.55 ± 0.73; n = 8).
SRT501 Neuroprotection Blocked by SIRT1 Inhibition
To confirm that SIRT1 activity mediated the observed neuroprotective effects of SRT501, SIRT1 activity was blocked with sirtinol. RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0), and mice were killed on day 14. Intravitreal injection of 100 μM sirtinol into control eyes on days 0, 3, 7, and 11 (n = 4) did not alter RGC survival (679.5 ± 55.0 RGCs vs. 685.6 ± 35.8 RGCs in placebo controls; n = 10; Fig. 3D). In EAE eyes with optic neuritis, 100 μM SRT501 (n = 9) attenuated the RGC loss (644.0 ± 49.2) observed in placebo-treated eyes (384.9 ± 31.2, n = 14), but eyes treated with 100 μM SRT501 and 100 μM sirtinol (n = 5) had significantly lower RGC survival (438.4 ± 38.9) than control eyes or optic neuritis eyes treated with SRT501 alone (Fig. 3D).
Long-term Reduction of RGC Loss by SRT501 Treatment from Time of EAE Induction
To determine whether SRT501 merely delays the loss of RGCs rather than providing longstanding neuroprotection, effects of SRT501 on RGC survival were examined 2 weeks after remission of EAE and acute optic neuritis. RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0). Mice were treated with 100 μM SRT501 on days 0, 3, 7, and 11 after immunization and were killed on day 30. Eyes from EAE mice treated with SRT501 (n = 16) had significantly more RGCs (563.1 ± 35.9) than eyes from placebo-treated EAE mice (431.2 ± 48.3; n = 12; Fig. 4B), though RGC numbers were lower than in control eyes (788.3 ± 96.8; n = 10). Clinical EAE was not affected by SRT501 treatment because placebo-treated (n = 6) and SRT501-treated (n = 8) mice developed relapsing EAE, similar to previous reports of relapsing EAE,15 with the first clinical episode peaking by day 14, remission of clinical symptoms several days later, and the beginning of a second relapse of disease starting at day 30 (Fig. 4C).
Figure 4.
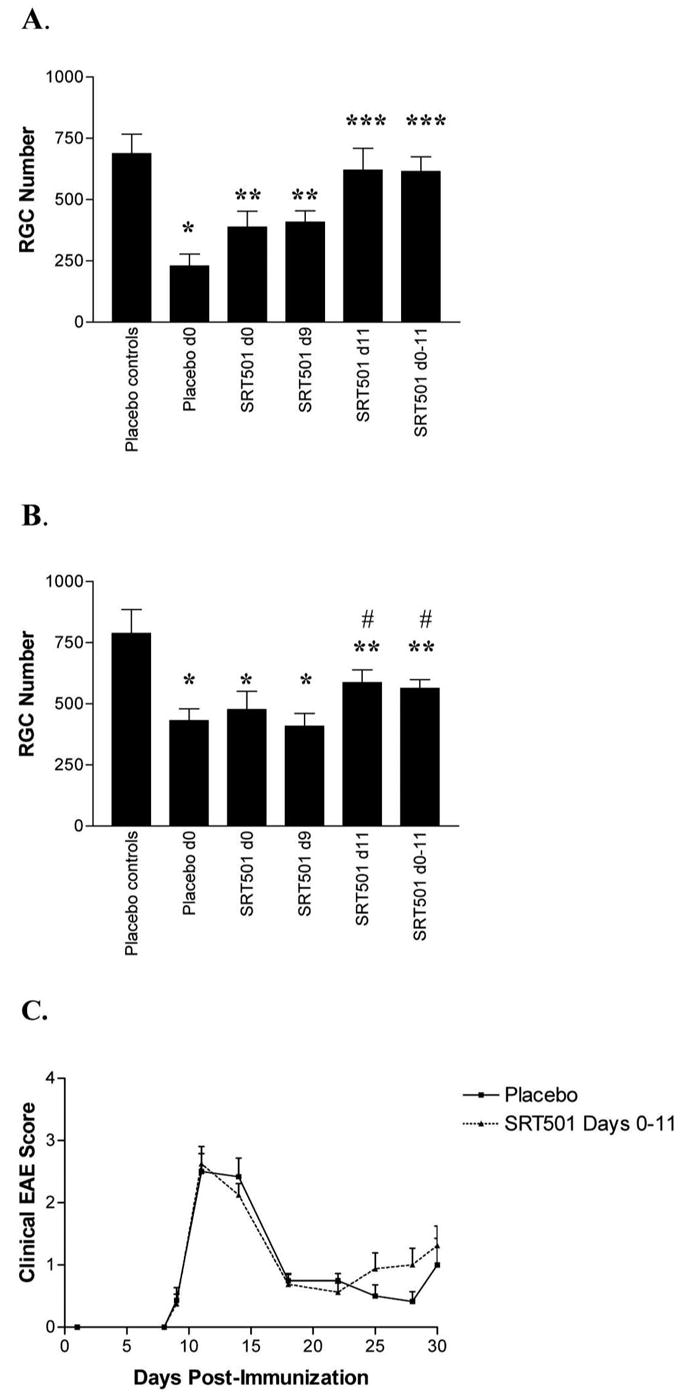
Single-dose SRT501 provided neuroprotection in acute optic neuritis and longer term neuroprotection at 30 days. (A) Fourteen days after immunization, RGC loss was detected in optic neuritis eyes treated with placebo compared with controls (*P < 0.001). SRT501 (100 μM) given on either day 0 or day 9 after immunization led to a trend of increased RGC numbers compared with placebo treated optic neuritis eyes that was not significant, and eyes still had fewer RGCs than controls (**P < 0.05). After a single dose of 100 μM SRT501 on day 11, or multiple doses at days 0, 3, 7, and 11, RGC numbers were increased compared with placebo treated optic neuritis eyes (***P < 0.01) and were equivalent to controls. (B) Thirty days after immunization, significant RGC loss is present in eyes from EAE mice treated with placebo compared with controls, and similar RGC loss occurs in eyes from EAE mice treated with 100 μM SRT501 on day 0 or day 9 (*P < 0.05). EAE eyes treated with 100 μM SRT501 on day 11 alone, or with multiple treatments on days 0, 3, 7, and 11, had more RGCs than placebo treated eyes (**P < 0.05), although RGCs were reduced compared with controls (#P < 0.05). (C) Mice immunized with PLP were evaluated for 30 days for clinical EAE. No significant difference in EAE score was found between EAE mice treated with placebo or 100 μM SRT501 on days 0, 3, 7, and 11.
Attenuation of RGC Loss by Single Dose of SRT501 after Onset of Optic Nerve Inflammation
Single-dose administrations of SRT501 (100 μM) were evaluated to determine potential therapeutic windows for treating optic neuritis. RGCs of SJL/J mice were labeled in a retrograde fashion with fluorogold. EAE was induced 1 week later by immunization with PLP (day 0), and mice were killed on day 14 or day 30. A single dose of SRT501 given on day 0 (the day of immunization) showed a trend toward increased RGC survival (388.1 ± 64.0 RGCs; n = 10 eyes) compared with placebo-treated optic neuritis eyes on day 14 (229.1 ± 49.1; n = 7) that was not significant (Fig. 4A) and that had no effect on RGC survival in EAE eyes at day 30 (Fig. 4B). SRT501 treatment on day 9 after immunization also resulted in a nonsignificant increase in RGC survival in optic neuritis eyes on day 14 (407.8 ± 46.5; n = 10; Fig. 4A) and had no effect at day 30 (Fig. 4B). Treatment with a single dose of SRT501 on day 11 resulted in significant attenuation of RGC loss in optic neuritis eyes on day 14, with RGC survival (620.4 ± 88.8; n = 7) equivalent to that in control eyes (686.9 ± 79.9, n = 10) and optic neuritis eyes treated with repeated SRT501 injections on days 0, 3, 7, and 11 (614.2 ± 60.5; n = 10; Fig. 4A). Treatment on day 11 also resulted in significant long-term attenuation of RGC loss at day 30 (586.0 ± 52.6; n = 16), with similar efficacy as seen with repeated injections (563.1 ± 35.9; n = 16; Fig. 4B).
Axon Preservation and Maintained Retrograde Transport Function with SRT501 Neuroprotection
In previous experiments, retrograde labeling of RGCs was performed 1 week before induction of EAE to ensure that labeling was complete well before optic nerve inflammation and axonal damage were induced. To determine whether RGC axons remained intact and maintained functional retrograde transport after SRT501 treatment, nonlabeled mice were immunized on day 0, and fluorogold injections for retrograde labeling were not given until 2 days before mice were killed. On day 11, eyes from control mice were treated with placebo (vehicle) by intravitreal injection, and EAE mouse eyes were treated with placebo or 100 μM SRT501. RGC labeling was detectable 2 days after injection of fluorogold, with similar RGC numbers detected in control eyes as in previous experiments (Fig. 5). Mice killed on day 14, after fluorogold injection on day 12, had significantly lower RGC numbers in placebo-treated optic neuritis eyes (377.0 ± 35.0; n = 7) than in controls (599.3 ± 84.4, n = 4), and SRT501 significantly attenuated this RGC loss (669.6 ± 64.6; n = 8; Fig. 5A). Similar RGC loss and neuroprotection by SRT501 was detected in optic neuritis eyes on day 16 (614.3 ± 41.9 RGCs in control eyes [n = 6] vs. 350.7 ± 44.8 in placebo-treated eyes [n = 6] vs. 598.1 ± 26.9 in SRT501-treated eyes [n = 7]), after fluorogold injection on day 14 (Fig. 5B).
Figure 5.
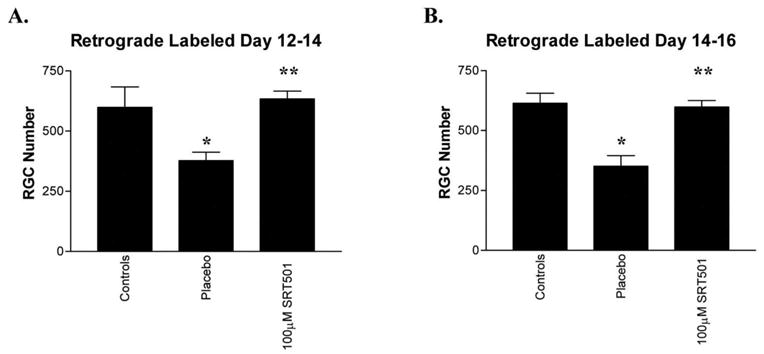
Retrograde axonal transport function was maintained in SRT501 protected RGCs. EAE mice were immunized with PLP in CFA and control mice sham-immunized with PBS in CFA on day 0 without previous fluorogold labeling. (A) Fluorogold was injected into the superior colliculi on day 12 and mice were killed on day 14. Significant RGC loss was detected in placebo-treated EAE eyes that developed optic neuritis compared with control eyes (*P < 0.05). A single intravitreal administration of 100 μM SRT501 on day 11 significantly attenuated this RGC loss (**P < 0.01). (B) Fluorogold was injected into the superior colliculi on day 14, and mice were killed on day 16. Placebo-treated EAE eyes that developed optic neuritis had significant RGC loss compared with control eyes (*P < 0.001) that was attenuated by a single intravitreal administration of 100 μM SRT501 on day 11 (**P < 0.001).
Assessment of Toxicity of SIRT1 Activators to Photoreceptors or Retinal Function
Although SJL/J mice are known to undergo photoreceptor degeneration at an early age, before EAE induction, previous studies have shown this process does not alter quantification of RGCs in control and EAE mice.15 However, this precludes the use of visual or electrophysiologic functional assessment. Despite this limitation, SRT647, SRT501, or sirtinol treatment did not appear to be toxic to retinal cells because no change in RGC numbers was found in control eyes after treatment (Figs. 2A, 3A, 3D), and no change in retinal thickness was observed between treated and untreated eyes.
To determine whether treatment is toxic to photoreceptors and electrophysiologic function, full-field flash ERG measurements were performed on untreated, wild-type, 6-week-old C57BL/6 mice. Intravitreal injection of vehicle, 66.67 mM SRT647, 100 μM SRT501, or 100 μM sirtinol was given in each eye, and ERGs were repeated 2 weeks later. No significant difference in amax responses were found among eyes treated with vehicle (314.8 ± 93.7 μV), SRT647 (319.2 ± 88.2 μV), SRT501 (328.0 ± 15.0 μV), or sirtinol (348.0 ± 60.5 μV). Similarly, no significant difference in scotopic bmax responses were found among eyes treated with vehicle (169.2 ± 56.6 μV), SRT647 (168.0 ± 53.3 μV), SRT501 (174.8 ± 9.2 μV), or sirtinol (214.4 ± 43.9 μV; n = 5 eyes/treatment group). Correspondingly, no difference in retinal thickness was observed among treatment groups by histology (data not shown).
DISCUSSION
SRT647 and SRT501 are neuroprotective for RGCs during acute optic neuritis, as shown by the fact that each SIRT1 activator attenuates RGC loss in a dose-dependent manner. SRT647 and SRT501 are chemically distinct from each other and represent direct and indirect activators of SIRT1. As such, the findings reported with both agents in this animal model help confirm the role of SIRT1 activation as important for neuroprotection in CNS demyelinating disease. SRT501 also provides significantly longer term neuroprotection for RGCs at day 30, suggesting that the increased RGC survival is not merely delayed at day 14 but can have long-lasting neuroprotective effects. This level of neuroprotection was seen with multiple- and single-dose drug administration, demonstrating that treatment at day 11, after optic nerve inflammation begins,15 provides effective neuroprotection. This is important because it suggests that treatment may be effective when patients are expected to present with clinical symptoms. No change in RGC number was found in control eyes treated with SRT647 or SRT501, demonstrating that these compounds are not toxic to RGCs. No systemic adverse effects were observed, further suggesting that these compounds are well tolerated.
Importantly, the fact that SRT501-treated eyes with optic neuritis also demonstrated no reduction in RGC numbers when retrograde labeling was performed after optic neuritis began (2 days before mice were killed) suggests that SRT501 neuroprotection supports RGC survival and maintains intact RGC axons with functional axonal transport. These results suggest that active optic nerve inflammation does not interfere with axonal transport in this model. Unfortunately, because of photoreceptor receptor degeneration in SJL/J mice, visual function could not be assessed in these mice, though we have previously shown that the RGCs are not affected in control eyes.15 However, treatment of C57BL/6 mice with SRT647 or SRT501 did not alter ERG responses, demonstrating that, at least in this strain, the compounds are not toxic to photoreceptors or overall retinal function.
The mechanism of neuroprotection by SRT647 and SRT501 likely involves SIRT1 deacetylase activity. This is suggested by the fact that two structurally and mechanistically distinct SIRT1 activators had significant neuroprotective effects and is confirmed by the finding that sirtinol, a known SIRT1 inhibitor, blocked the RGC protection. SIRT1 is expressed in virtually all cell types and localizes to cell nuclei,41 raising a concern that the use of a SIRT1 inhibitor may have significant toxicity. Fortunately, at the concentration used, sirtinol did not have any detectable retinal toxicity, similar to the SIRT1 activators.
Specific substrates of SIRT1 mediating RGC survival are not yet known. Future examination of specific substrates will be useful to identify focused downstream targets for potential therapeutic intervention. The sirtuin family of deacetylases, including SIRT1, is known to act on proteins involved in apoptosis, such as p5342,43 and Ku70,44 and on structural proteins, including α-tubulin,45 alteration of which may lead to axonal degeneration. Apoptosis has been shown previously to mediate RGC loss in a chronic EAE optic neuritis model16 and in a spontaneous optic neuritis model,17 and we have found similar apoptotic cell death in relapsing EAE optic neuritis (unpublished observations, 2006), making apoptotic proteins likely candidates for the downstream targets of SIRT1-mediated RGC neuroprotection. Alternatively, SIRT1 regulation of cellular stress responses may mediate neuroprotection because mitochondrial oxidative stress is known to induce neuronal damage in experimental optic neuritis.46
Interestingly, SRT501 exerts significant neuroprotective effects at day 11, but not at day 9, despite the fact that some optic nerve inflammation begins by day 9.15 This suggests that the molecular pathways underlying RGC loss in this model are not yet activated or present for SIRT1 to act on until a couple of days after inflammation develops.
Optic neuritis and other MS lesions are characterized by inflammation and demyelination.1,2 Current therapies include immunosuppression with steroids and immunomodulation with interferon beta and glatiramer acetate.1 These treatments target the inflammatory component but have limited efficacy in preventing relapse or progression of disease. In addition, evidence is limited to suggest immunomodulatory therapies prevent neuronal damage and long-term disability in MS patients.10,11 Although recently described immunomodulatory therapy with natalizumab alone or in combination with interferon beta did reduce the progression of disability in MS patients,47,48 effects were not complete, and adverse effects might have limited the use of this combination therapy. In EAE rats, glatiramer acetate had neuroprotective effects for RGCs when given before optic neuritis developed, but no effect was found when treatment was started at the onset of disease.49 Neuroprotective therapies that work through nonimmunomodulatory mechanisms may have tremendous clinical benefits. The neuroprotective effects of SRT647 and SRT501 occurred without suppression of the inflammatory infiltration of the optic nerve because incidence of optic neuritis did not differ between placebo and treatment groups and was similar to the previously reported incidence.15 In addition, localized treatment with intravitreal injections did not reduce spinal cord inflammation, as evidenced by the lack of effect on clinical EAE score. The ability of SIRT1 to prevent RGC loss without reducing inflammation suggested that the therapeutic effects of SIRT1 activators had a strong potential to be additive or synergistic to disease-modifying effects of current immunomodulatory therapies. Future studies will evaluate the effects of immunomodulatory therapies in optic neuritis in combination with SIRT1 activators. Synergistic effects of two mechanistically distinct immunomodulatory therapies have been shown in EAE,50 demonstrating the usefulness of studying combination therapies in this MS model.
The potent neuroprotective effects of SIRT1 activators suggest an important potential therapy for optic neuritis and probably for other diseases leading to RGC death, such as ischemic, toxic, and traumatic optic neuropathies and glaucoma. SIRT1 activators did not reduce inflammation; hence, they may be neuroprotective in these noninflammatory-mediated optic nerve diseases. Neuroprotective effects of SIRT1 activation have been demonstrated in other noninflammatory neurologic disease models. SIRT1 activity reduces neuronal death induced by the Huntington gene mutation32 and prevents degeneration of axotomized dorsal root ganglion neurons.31 The current route of administration of SRT501 and SRT647, by intravitreal injection, may be useful for optic nerve insults that have a limited time course and treatment period, such as optic neuritis and ischemic optic neuropathy, though less invasive routes would be preferred. For chronic RGC loss in diseases such as glaucoma, topical or oral formulations with ocular penetration would be ideal; such formulations are being developed.
SIRT1 activation also has great potential for preventing neuronal loss throughout the central nervous system in patients with MS. Clinical trials that include patients with optic neuritis have helped shape the current clinical practice patterns for use of steroids and immunomodulators in MS.51–53 Development of SIRT1 activators with good systemic bioavailability will likely lead to new MS therapies that prevent long-term disability. Future studies will examine the intraocular penetration and potential neuroprotection of systemic SIRT1-activating drugs.
Overall, the current studies demonstrate that SIRT1 activation prevents RGC loss in optic neuritis through SIRT1 family enzymatic activity. SIRT1 activators are well tolerated without RGC toxicity and provide neuroprotection even in the presence of active inflammation. SRT501 and other SIRT1 activators have important therapeutic potential for preventing permanent neuronal loss and disability from optic neuritis and MS and may have a therapeutic role in preventing vision loss in other optic nerve diseases.
Acknowledgments
The authors thank Jean Bennett and Albert Maguire for assistance with techniques for intravitreal injections and Elsa Aglow and Mahasweta Dutt for expert technical assistance.
Supported by National Institutes of Health Grant EY015098, a Career Development Award from Research to Prevent Blindness, grants from Sirtris Pharmaceuticals, and unrestricted funding from the Paul and Evanina Mackall Foundation Trust and the F. M. Kirby Foundation.
Footnotes
Disclosure: K.S. Shindler, None; E. Ventura, None; T.S. Rex, None; P. Elliott, Sirtris Phamaceuticals (I, E); A. Rostami, Sirtris Phamaceuticals (F)
References
- 1.Arnold AC. Evolving management of optic neuritis and multiple sclerosis. Am J Ophthalmol. 2005;139:1101–1108. doi: 10.1016/j.ajo.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 2.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 3.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 4.Kornek B, Storch MK, Weissert R, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 6.Wujek JR, Bjartmar C, Richer E, et al. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- 7.Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 8.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;102:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 9.Davie CA, Barker GJ, Webb S, et al. Persistent functional deficit in multiple sclerosis and autosomal dominant ataxia associated with axon loss. Brain. 1995;118:1583–1592. doi: 10.1093/brain/118.6.1583. [DOI] [PubMed] [Google Scholar]
- 10.Hickman SJ, Kapoor R, Jones SJ, Altmann DR, Plant GT, Miller DH. Corticosteroids do not prevent optic nerve atrophy following optic neuritis. J Neurol Neurosurg Psychiatry. 2003;74:1139–1141. doi: 10.1136/jnnp.74.8.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parry A, Corkill R, Blamire AM, et al. β-Interferon treatment does not always slow the progression of axonal injury in multiple sclerosis. J Neurol. 2003;250:171–178. doi: 10.1007/s00415-003-0965-8. [DOI] [PubMed] [Google Scholar]
- 12.Fisher JB, Jacobs DA, Markowitz CE, et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology. 2006;113:324–332. doi: 10.1016/j.ophtha.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 13.Trip SA, Schlottmann PG, Jones SJ, et al. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol. 2005;58:383–391. doi: 10.1002/ana.20575. [DOI] [PubMed] [Google Scholar]
- 14.Costello F, Coupland S, Hodge W, et al. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol. 2006;59:963–969. doi: 10.1002/ana.20851. [DOI] [PubMed] [Google Scholar]
- 15.Shindler KS, Guan Y, Ventura E, Bennett J, Rostami A. Retinal ganglion cell loss induced by acute optic neuritis in a relapsing model of multiple sclerosis. Multiple Sclerosis. 2006;12:526–532. doi: 10.1177/1352458506070629. [DOI] [PubMed] [Google Scholar]
- 16.Meyer R, Weissert R, Diem R, et al. Acute neuronal apoptosis in a rat model of multiple sclerosis. J Neurosci. 2001;21:6214–6220. doi: 10.1523/JNEUROSCI.21-16-06214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan Y, Shindler KS, Tabuela P, Rostami A. Retinal ganglion cell damage induced by spontaneous autoimmune optic neuritis in MOG-specific TCR transgenic mice. J Neuroimmunol. 2006;178:40–48. doi: 10.1016/j.jneuroim.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 19.Landry J, Sutton A, Tafrov ST, et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith JS, Brachmann CB, Celic I, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tissenbaum HA, Guarente L. Increased dosage of a Sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 23.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porcu M, Chiarugi A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol Sci. 2005;26:94–103. doi: 10.1016/j.tips.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 27.Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 28.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Denu JM. The Sir2 family of protein deacetylases. Curr Opin Chem Biol. 2005;9:1–10. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 31.Araki T, Sasaki Y, Millbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 32.Parker JA, Arango M, Abderrahmane S, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 33.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Priess-Handler independent route to NAD+ in funji and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 34.Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gran B, Zhang GX, Yu S, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 36.Liang F-Q, Anand V, Maguire AM, Bennett J. Intraocular delivery of recombinant virus. In: Rakoczy PE, editor. Methods in Molecular Medicine: Vision Research Protocols. Totowa, NJ: Humana Press; 2000. pp. 125–139. [DOI] [PubMed] [Google Scholar]
- 37.Shao H, Huang Z, Sun SL, Kaplan HJ, Sun D. Myelin/oligodendrocyte glycoprotein-specific T-cells induce severe optic neuritis in the C57BL/6 mouse. Invest Ophthalmol Vis Sci. 2004;45:4060–4065. doi: 10.1167/iovs.04-0554. [DOI] [PubMed] [Google Scholar]
- 38.Rex TS, Allocca M, Domenici L, et al. Systemic but not intraocular EPO gene transfer protects the retina from light- and genetic-induced degeneration. Mol Therapy. 2004;10:855–861. doi: 10.1016/j.ymthe.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 39.Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr UV-and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 43.Vaziri H, Dessain SK, Ng-Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 44.Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 45.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 46.Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Suppression of mitochondrial oxidative stress provides long-term neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2007;48:681–691. doi: 10.1167/iovs.06-0553. [DOI] [PubMed] [Google Scholar]
- 47.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 48.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 49.Maier K, Kuhnert AV, Taheri N, et al. Effects of glatiramer acetate and interferon-β on neurodegeneration in a model of multiple sclerosis. Am J Pathol. 2006;169:1353–1364. doi: 10.2353/ajpath.2006.060159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuve O, Youssef S, Weber MS, et al. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116:1037–1044. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 2003;121:944–949. doi: 10.1001/archopht.121.7.944. [DOI] [PubMed] [Google Scholar]
- 52.Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis: CHAMPS Study Group. N Engl J Med. 2000;343:898–904. doi: 10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 53.Beck RW, Cleary PA, Anderson MM, Jr, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581–588. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]