

Abstract
Insulin-like growth factor (IGF) signaling is a critical regulator of somatic growth during fetal and adult development, primarily through its stimulatory effects on cell proliferation and survival. IGF signaling is also required for development of the reproductive system, although its precise role in this regard remains unclear. We have hypothesized that IGF signaling is required for embryonic germline development, which requires the specification and proliferation of primordial germ cells (PGCs) in an extragonadal location, followed by directed migration to the genital ridges. We tested this hypothesis using loss-of-function studies in the zebrafish embryo, which possesses two functional copies of the Type-1 IGF receptor gene (igf1ra, igf1rb). Knockdown of IGF1Rb by morpholino oligonucleotides (MO) results in mismigration and elimination of primordial germ cells (PGCs), resulting in fewer PGCs colonizing the genital ridges. In contrast, knockdown of IGF1Ra has no effect on PGC migration or number despite inducing widespread somatic cell apoptosis. Ablation of both receptors, using combined MO injections or overexpression of a dominant-negative IGF1R, yields embryos with a PGC-deficient phenotype similar to IGF1Rb knockdown. TUNEL analyses revealed that mismigrated PGCs in IGF1Rb-deficient embryos are eliminated by apoptosis; overexpression of an antiapoptotic gene (Bcl2l) rescues ectopic PGCs from apoptosis but fails to rescue migration defects. Lastly, we show that suppression of IGF signaling leads to quantitative changes in the expression of genes encoding CXCL-family chemokine ligands and receptors involved in PGC migration. Collectively, these data suggest a novel role for IGF signaling in early germline development, potentially via cross-talk with chemokine signaling pathways.
Introduction
Insulin-like growth factor (IGF) signaling plays a central role in the regulation of animal growth and development (Barbieri et al., 2003; LeRoith et al., 2001; Wood et al., 2005a). In mammals, IGF-1 and IGF-2 ligands exert their effects by activating the Type-1 IGF receptor (IGF1R), triggering a variety of cellular responses including proliferation, migration, and cell survival (Butler et al., 1998; Yano et al., 1999). During postnatal growth, IGF signaling serves as the primary endocrine mediator of growth hormone (GH) signaling (LeRoith et al., 2001), but appears to function independently of GH during prenatal development (Wollmann, 2000).
The importance of IGF signaling to animal growth was clearly demonstrated by targeted deletion of genes encoding IGF ligands and receptors in mice (Baker et al., 1993; DeChiara et al., 1990; DeChiara et al., 1991; Liu et al., 1993). Mice lacking either IGF ligand survive to adulthood, but exhibit proportional growth retardation during fetal and postnatal life, while deletion of both ligands or IGF1R leads to severe fetal dwarfism and perinatal mortality. Importantly, these effects are consistent with growth defects in humans with mutations in IGF-related genes, confirming a conserved role for IGF signaling in regulating vertebrate growth (Abuzzahab et al., 2003; Nakae et al., 2001).
There is also evidence that IGF signaling is specifically required for both development and function of the reproductive system. Both IGF-1 and the IGF1R are abundantly expressed in reproductive tissues of adult vertebrates (Nuttinck et al., 2004; Ohtsuki et al., 2005; Perrot et al., 2000), and IGF-1 nullizygous mice exhibit significantly reduced fertility as adults (Baker et al., 1996; Liu et al., 1993). The precise cause of the fertility defects remains unclear; an analysis of testicular germ cell numbers in IGF-1 nullizygous mouse revealed a disproportionate reduction in the number of germ cells (relative to both organ and body size; Baker et al., 1996), suggesting specific defects in germ cell development in the absence of normal IGF signaling. To our knowledge however, a detailed analysis of germ cell development in IGF-deficient animals has not been performed.
In vitro studies indicate that IGF-1 can inhibit apoptosis of fetal mouse germ cells (Morita et al., 1999) and chicken primordial germ cells (Park and Han, 2000), but these findings have not been extended to an examination of IGF signaling and germline development in vivo. However, insulin-like signaling was shown to play a direct role in vivo in promoting germline stem cell renewal in Drosophila (LaFever and Drummond-Barbosa, 2005), suggesting that regulation of germline development by insulin-like signaling may be conserved throughout animal evolution.
The zebrafish is an excellent model for in vivo studies of vertebrate germline development. The transparency of zebrafish embryos permits direct visualization of PGC development, a coordinated sequence of events involving cell fate specification, proliferation, and directed migration. While a number of conserved signaling pathways required for PGC development (e.g., Cxcl-family chemokine signaling) have been identified in zebrafish (Ara et al., 2003; Doitsidou et al., 2002; Knaut et al., 2003; Molyneaux et al., 2003; Schier, 2003; Stebler et al., 2004), our understanding of this process remains largely incomplete. The well-described effects of IGF signaling on both cell proliferation and migration led us to hypothesize that IGF signaling regulates one or more facets of PGC development. In this study we used the zebrafish to test this hypothesis, using two strategies to suppress IGF signaling during the PGC migration period. We examined PGC development in zebrafish embryos after single or combined knockdown of IGF1Ra and IGF1Rb, using morpholino antisense oligonucleotides or a dominant-negative IGF1R. Our results indicate that in zebrafish embryos, IGF signaling through IGF1Rb is necessary to ensure correct migration of PGCs to the genital ridges, and that defective migration results in subsequent elimination of mismigrated PGCs by apoptosis. We also present evidence that the effects of IGF signaling on PGC development may be manifest through modifications of CXCL-family chemokine signaling.
Materials and Methods
Animals
Adult wild-type zebrafish were reared and maintained using standard methods. Embryos were generated from natural crosses, staged according to Kimmel et al. (1995), and reared as previously described (Wood et al., 2005b). All experiments were conducted in accordance with guidelines as established by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Chemicals and reagents
Standard chemicals and reagents were purchased from Fisher (Pittsburgh, PA) unless otherwise specified. RNA polymerases and DNase (RNase-free) were purchased from Promega (Madison, WI), and restriction endonucleases were purchased from New England BioLabs (Beverly, MA). Platinum® Taq DNA polymerase, Superscript II reverse transcriptase and oligonucleotide primers (for conventional and quantitative RT-PCR) were purchased from Invitrogen Life Technologies, Inc. (Carlsbad, CA). FailSafe™ PCR reaction buffers were purchased from EpiCentre (Madison, WI), and the In Situ Cell Death Detection Kit (TMR Red) was purchased from Roche Applied Science (New Jersey, NJ). Gene-specific morpholino-modified oligonucleotides (MO) were purchased from Gene Tools, LLC (Philomath, OR). Polyclonal antiserum raised against zebrafish Vasa protein was a generous gift from Holger Knaut and Christiane Nüsslein-Volhard (Max Planck Institute for Developmental Biology, Tübingen, Germany). Alexa Fluor® 488 secondary antibodies were purchased from Molecular Probes (Eugene, OR).
In situ hybridization
Partial cDNA sequences corresponding to divergent regions of igf1ra and igf1rb were generated by RT-PCR using gene-specific primers as previously described (Wood et al., 2005b), and subcloned into the PCRII-TOPO™ vector. The gene-specific oligonucleotide primer sequences are as follows:
igf1ra (forward): 5'-GCCATCTTTCCTGGAGATCA-3'
igf1ra (reverse): 5'-AGACAAAGGGAGGAGGGAAA-3'
igf1rb (forward): 5'-CCCTCTAGAACCGTCTTCCA-3’
igf1rb (reverse): 5'-GATCCTGTCTGGCGGAAATA-3'.
Accuracies of amplified sequences were confirmed by automated sequencing using commercial primers. Template DNA for riboprobe synthesis was generated by restriction enzyme digestion, and digoxigenin-labeled ribonucleotide probes were generated by in vitro transcription. Whole-mount in situ hybridizations were performed as previously described (Wood et al., 2005b). Images were captured with a digital camera (MicroFire, Optronics®, Goleta, CA) mounted to a Nikon Eclipse TE-2000SE microscope (Melville, NY).
Messenger RNA synthesis
Capped synthetic messenger RNA was generated by in vitro transcription (mMESSAGE mMACHINE®, Ambion, Austin, TX) according to supplier’s instructions. Template plasmid DNA for zebrafish bcl2l mRNA was generously provided by Monte Westerfield (University of Oregon, Eugene, OR). The dominant-negative IGF1R:GFP fusion protein construct (dnIGF1R:GFP) has been previously described (Schlueter et al., 2007).
Embryo microinjections
Stock MO solutions were diluted in Danieau buffer and injected into fertilized embryos (~1 nl/embryo) at the one-cell stage, as previously described (Wood et al., 2005b). To ensure efficient knockdown, two non-overlapping MO sequences were used against the translational start site of each igf1r subtype; the specificity and efficacy of each of these MO sequences have been confirmed using multiple approaches (Schlueter et al., 2006). The MO against each receptor subtype were injected together (2.5 ng total MO per embryo); it was determined in pilot studies that these quantities of MO (i.e., 1.25 ng igf1ra-MO1 + 1.25 ng igf1ra-MO2) yielded reproducible effects (mild, proportional growth restriction) for each receptor subtype, whereas equivalent quantities of a control-MO cocktail (Control MO-a + Control MO-b) yielded embryos indistinguishable from non-injected embryos. For phenotypic analyses, embryos were raised as described above, fixed at desired stages of development in 4% buffered paraformaldehyde, dehydrated in methanol, and stored at –20ºC until analysis. For quantitative RT-PCR (qRT-PCR) analyses, injected embryos were snap-frozen in liquid N2 in preparation for RNA extraction.
For synthetic mRNA injections, stock solutions were diluted in RNase-free Danieau buffer, and either injected alone (dnIGF1R:GFP, Blc2l, GFP), or co-injected (Bcl2l) with gene-specific MO. Synthetic mRNA was microinjected at nominal concentrations of 750 pg/embryo (dnIGF1R:GFP) or 100 pg/embryo (Bcl2l); for controls, mRNA encoding GFP was injected at 750 pg/embryo.
Whole-mount immunostaining
Fluorescent immunostaining for PGCs was performed in whole embryos as previously described (Westerfield, 1995), using polyclonal antiserum against zebrafish Vasa protein, diluted 1:8000 in blocking buffer (PBS, containing 0.1% Tween 20, 1% DMSO, 2% goat serum). The secondary antibody (Alexa Fluor™ 488 goat anti-rabbit IgG) was diluted 1:1000 in blocking buffer. Primordial germ cells (Vasa-positive cells) were localized and enumerated by examining whole embryos under a fluorescent microscope. Negative controls were incubated in the absence of primary antibody to confirm the germ cell-specificity of the primary antibody.
Western immunoblot analysis
Western immunoblot analysis was used to confirm expression of the dnIGF1R:GFP fusion protein, using prim-5 stage zebrafish embryos. Yolk proteins were removed by puncturing the yolk sac and washing the embryos in PBS. Residual carcasses were then homogenized in SDS sample buffer (1% SDS, 0.1 M Tris-HCl, 10% glycerol, 0.02% bromophenol blue, 2% 2-mercaptoethanol), and the homogenates were electrophoretically fractionated in 10% SDS-PAGE gels under denaturing conditions. After transfer to Immobilon-P PVDF membranes, the blots were incubated for 4 h in blocking buffer (Tris-buffered saline containing 0.1% Tween-20 and 5% non-fat dry milk; TBS-TM), and then incubated overnight at 4ºC in TBS-TM containing rabbit antiserum against GFP (Torrey Pines Biolabs, Houston, TX). Primary antibody was detected using HRP-conjugated donkey anti-rabbit IgG (1:5000; Jackson ImmunoResearch Labs, West Grove, PA), and labeled proteins were visualized by the ECL detection system.
Whole-mount in situ cell death (TUNEL) analysis
Fixed embryos were rehydrated and permeabilized as described (Schlueter et al., 2006), and incubated for 1 h at 37ºC in TUNEL assay reagents. Negative controls were incubated in the absence of terminal transferase enzyme. Stained embryos were resuspended in glycerol:PBS (70:30) and examined by fluorescence microscopy. To specifically identify TUNEL-positive PGCs, we performed subsequent immunocytochemical staining (as above) before analysis by fluorescence microscopy.
Quantitative RT-PCR
Quantitative RT-PCR (qRT-PCR) was employed to determine if knockdown of IGF1Ra or IGF1Rb affected mRNA levels for genes encoding known germ cell guidance molecules. Efforts focused on mRNA encoding the chemokine ligand Cxcl12a (NP_840092) and its cognate receptor Cxcr4b (AAF17561), a conserved ligand-receptor pair whose functions are essential for directional PGC migration in vertebrate embryos (Doitsidou et al., 2002; Knaut et al., 2003; Molyneaux et al., 2003; Schier, 2003). Ornithine decarboxylase 1 (odc1; NM_131801) mRNA was used as the internal reference standard (Draper et al., 2001). The following primers were used (lower case letters indicate hairpin primer extensions; [FAM] indicates position of fluorescent label):
cxcl12a (forward): 5’-cgccCGTAGTAGTCGCTCTGATGG[FAM]G-3’
cxcl12a (reverse): 5’-TGGGACTGTGTTGACTGTGGAA-3’
cxcr4b (forward): 5’-cggcCTGGTTGCCTTACTGTGC[FAM]G
cxcr4b (reverse): 5’-CCATTTCTCCAGACCCTGTTCC-3’
odc1 (forward): 5’-gacaGCGGTGAACCTCCTTGGCTG[FAM]C-3’
odc1 (reverse): 5’-CGGTGCAAGCCGTCATAGTG-3’.
Total RNA was isolated from control-MO-injected and MO-injected embryos (Prim-5 stage) using Trizol reagent, and residual genomic DNA was removed by digestion with RNase-free DNase. Each sample consisted of mRNA isolated from a pool of ten embryos, to ensure sufficient quantities of mRNA for analysis. First-strand cDNA was generated by reverse-transcription as described (Wood et al., 2005b), using 500 ng of DNA-free RNA. Real-time PCR amplifications were performed in a Cepheid SmartCycler II (Fisher Scientific) for 45 cycles under the following conditions: 95ºC for 150 s, 95ºC for 10 s; 60ºC for 10 s; 72ºC for 10 s. The identity of PCR products amplified with FAM-labeled primers was confirmed by electrophoretic and melting point analysis; quantification of relative mRNA levels for each target gene in treatment and control groups was determined as previously described (Pfaffl, 2001).
Data analysis
Values are presented as means ± standard error of the mean (SEM). Mean PGC numbers in treatment and control groups were compared statistically using one-way analysis of variance (ANOVA), followed by the Tukey's post-hoc test for multiple comparisons. Target gene (cxcl12a, cxcr4b) mRNA levels are presented as relative (percentage) values normalized to odc (100%); mean relative changes in mRNA levels were statistically compared among treatment and control groups by paired Student’s t-test. In all statistical tests, mean values were considered significantly different when P < 0.05.
Results
Both igf1ra and igf1rb are ubiquitously expressed in zebrafish embryos
Although it was previously reported that mRNAs encoding both IGF1Ra and IGF1Rb are ubiquitously expressed in zebrafish embryos, as determined by whole-mount in situ hybridization (Maures et al., 2002), we sought to re-examine mRNA expression in greater detail, specifically in PGCs and the genital ridges (Supplemental Fig. 1). As previously reported, both genes exhibit ubiquitous mRNA expression in zebrafish embryos (Supplemental Fig. 1A–D). To examine the mRNA expression of igf1ra and igf1rb specifically within the PGCs and the genital ridges, we used fluorescence immunohistochemistry (Supplemental Fig. 1E–H) with an antibody raised against zebrafish Vasa protein (Knaut et al., 2000) to visualize PGCs in whole embryos directly following in situ hybridization labeling of mRNA for either IGF1Ra or IGF1Rb.
The double-labeling approach confirmed that both PGCs and the genital ridge regions express mRNA for both IGF1Ra and IGF1Rb (Supplemental Fig. 1G–H), but we were unable to detect any relative differences in expression of the receptor subtypes in either PGCs or the genital ridges.
Morpholino knockdown of igf1rb reduces total PGC number
To test the hypothesis that PGC development requires signaling through IGF1R, we first counted the total number of PGCs in equivalent-stage embryos after separate or combined knockdown of IGF1Ra and IGF1Rb with gene-specific morpholino antisense oligonucleotides (MO). The prim-5 stage of development was chosen for analysis because in normal untreated (wild-type) embryos, virtually all PGCs have completed their migration to the genital ridges by this stage. The efficacy and specificity of the MO sequences have recently been confirmed in zebrafish using multiple approaches (Schlueter et al., 2006). We identified and quantified PGCs by fluorescence immunostaining of Vasa protein (Fig. 1) as described above.
Figure 1.
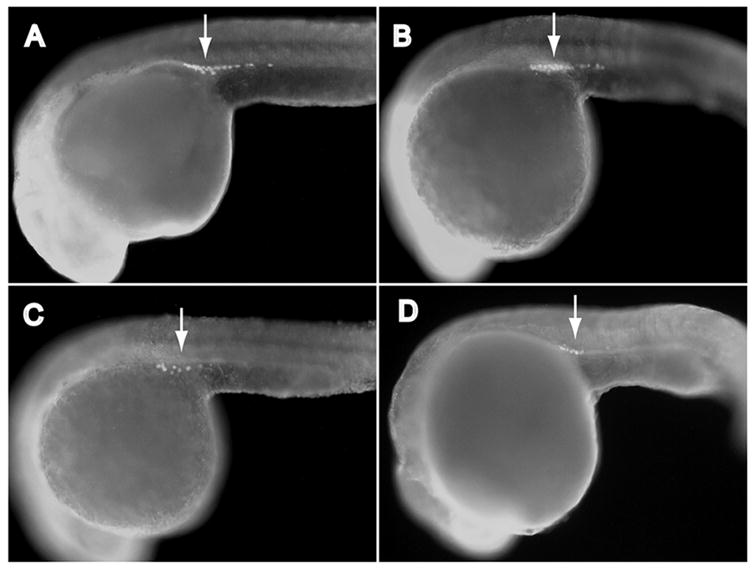
Igf1rb is required to establish normal primordial germ cell (PGC) numbers at the prim-5 stage. Immunohistochemical identification of PGCs (Vasa-positive cells, arrows) in prim-5 stage zebrafish embryos, after ablation of Type-1 IGF receptors (igf1ra, igf1rb): (A) control (non-targeting) morpholinos; (B) igf1ra-MO; (C) igf1rb-MO; (D) igf1ra-MO + igf1rb-MO. Magnification 200x. Mean PGC numbers of treatment groups are summarized in Table 1.
Control-MO-injected embryos (Fig. 1A) developed similarly to non-injected (wild-type) embryos, and at the prim-5 stage no significant differences were detected in the mean number of PGCs among Control-MO-injected and wild-type embryos (Table 1). Knockdown of IGF1Ra by antisense MO injection resulted in slightly delayed development, as described previously (Schlueter et al., 2006), but had no detectable effects on the mean number of PGCs, relative to control-MO-injected or wild-type embryos, when compared at the prim-5 stage (Fig. 1B, Table 1). Knockdown of IGF1Rb by MO injection similarly induced slightly delayed development, but also induced a significant reduction in the total number of visible PGCs at the prim-5 stage (Fig. 1C, Table 1). Simultaneous MO-mediated knockdown of both IGF1Ra and IGF1Rb, using combined igf1ra-MO + igf1rb-MO injections, yielded embryos with PGC numbers similar to those observed in IGF1Rb-deficient embryos (Fig. 1D, Table 1), suggesting no additive effects of double receptor knockdown beyond those observed after IGF1Rb knockdown.
Table 1.
Mean number of PGCs in zebrafish embryos after targeted knockdown of Type-1 IGF receptors using morpholino oligonucleotides (MO), or a dominant-negative IGF1R:GFP fusion protein (dnIGF1R:GFP). Sample size indicated in parentheses. N.D., not determined.
| Stage of Development | ||
|---|---|---|
| Treatment Group | Prim-5 | 18-somite (18S) |
| Wild-type (non-injected) | 32.6 ± 0.9 (10)a | N.D. |
| Control MO-a/MO-b | 33.9 ± 1.1 (21)a | 34.6 ± 3.4 (10) |
| igf1ra-MO | 33.1 ± 1.8 (9)a | 30.6 ± 0.5 (8) |
| igf1rb-MO | 18.2 ± 2.2 (18)b | 32.9 ± 3.2 (10) |
| igf1ra-MO + igf1rb-MO | 19.3 ± 2.8 (8)b | 32.7 ± 1.82 (15) |
| igf1rb-MO + bcl2l* | 28.8 ± 1.5 (20)a | N.D. |
| dnIGF1R:GFP | 16.0 ± 3.1 (9)b | 34.4 ± 2.6 (8) |
| GFP | 35.9 ± 1.7 (20)a | N.D. |
Superscript letters denote significant differences among same-stage embryos (ANOVA, Tukey’s, P < 0.05). No significant differences were detected among means of 18S embryo groups.
mRNA encoding zebrafish Bcl2l.
Overexpression of dominant-negative IGF1R reduces PGC number
To confirm the MO knockdown results using an alternative approach to suppress IGF signaling, we injected embryos with synthetic mRNA encoding a dominant-negative IGF1R:GFP fusion protein (dnIGF1R:GFP; Schlueter et al., 2007). Overexpression of dnIGF1R:GFP yielded GFP-positive embryos (Fig. 2A) with a developmental delay phenotype similar to that observed in igf1r-MO-injected embryos, confirming functionality of the dnIGF1R:GFP fusion protein. Overexpression of dnIGF1R:GFP (Fig. 2C) also resulted in a significant reduction in mean PGC number in embryos at the prim-5 stage, relative to control and wild-type embryos (Table 1). The mean number of visible PGCs in embryos overexpressing dnIGF1R:GFP was statistically similar to the mean values observed in both igf1rb-MO-injected embryos and igf1ra-MO + igf1rb-MO-injected embryos (ANOVA, Tukey’s, P > 0.05; Table 1). Injection of mRNA encoding GFP alone had no significant effects on mean PGC number relative to wild-type or Control-MO-injected embryos.
Figure 2.
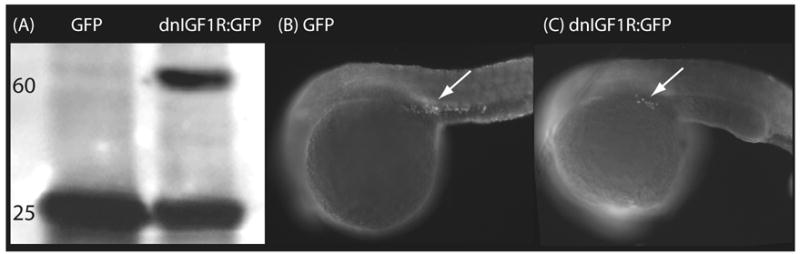
(A) Western immunoblot analysis of zebrafish lysates demonstrating overexpression of dominant-negative IGF1R:GFP fusion protein (dnIGF1R:GFP). First lane, lysates from zebrafish embryos injected with synthetic mRNA encoding GFP only; second lane, lysates from zebrafish embryos injected with synthetic mRNA encoding dnIGF1R:GFP. Upper band in dnIGF1R:GFP corresponds to the predicted molecular weight (~60 kDa) of dnIGF1R:GFP after denaturation in reducing conditions. (B) Vasa immunostaining of PGCs (arrows) in zebrafish embryo overexpressing GFP only (control); (C) Vasa immunostaining of PGCs in zebrafish embryo overexpressing dnIGF1R:GFP fusion protein. Magnification (B–C), 200x. Mean PGC numbers in B and C are summarized in Table 1.
Suppression of IGF signaling does not disrupt early PGC proliferation
IGF signaling stimulates mitotic proliferation in a diversity of cell types (Onagbesan et al., 1994; Otteson et al., 2002; Pozios et al., 2001), and zebrafish PGCs undergo 2–3 mitotic divisions prior to initiating migration towards the genital ridges (Braat et al., 1999). To determine whether the PGC deficiency in igf1rb-MO-injected embryos is due to defects in PGC proliferation, we enumerated PGCs in igf1r-MO-injected embryos at an earlier stage of development (18S). As shown (Table 1), the mean number of PGCs in 18S igf1r-MO-injected embryos (igf1ra-MO, igf1rb-MO, igf1ra-MO + igf1rb-MO), and embryos overexpressing dnIGF1R:GFP, were not significantly different from PGC numbers in 18S control-MO-injected embryos, that were likewise similar to PGC numbers in control-MO-injected embryos at the prim-5 stage. These results suggest that PGCs initially proliferated normally in all treatment groups, but subsequently declined in number between 18 and 24 hpf, specifically after targeted knockdown of IGF1Rb. Notably, this time interval corresponds directly to the terminal stages of PGC migration to the genital ridges (Weidinger et al., 1999).
Knockdown of IGF1Rb disrupts PGC migration
In the course of enumerating PGCs in 18S embryos, we noticed that more Vasa-positive cells in igf1rb-MO-injected embryos were found in “ectopic” positions (i.e., distant from the genital ridges) relative to other treatment groups. Because cell migration is a universal feature of PGC development in metazoans (Molyneaux and Wylie, 2004), we hypothesized that the PGC deficiency in igf1rb-MO-injected embryos may be a secondary consequence of defective PGC migration. We therefore counted the number of ectopic PGCs in 20-somite (20S) stage MO-injected embryos; we specifically chose this stage of development to facilitate discrimination between mismigrated PGCs and normally migrating PGCs that had not yet reached the genital ridges. We defined ectopic PGCs as Vasa-positive cells found in cranial, dorsal, and caudal regions of the embryo proper, and in regions of the yolk cell distal to the genital ridges (Fig. 3A–C). Vasa-positive cells found along the yolk cell extension, or in regions of the yolk cell directly adjacent to the genital ridges were considered to be normal PGCs in the terminal stages of migration toward the genital ridges. Consistent with our hypothesis, we found a significantly increased mean number of ectopic PGCs in 20S igf1rb-MO-injected embryos (6.2 ± 0.8; n=12) relative to 20S control MO-injected embryos (0.7 ± 0.2; n=27, ANOVA, P < 0.001; Fig. 3D). Notably, ectopic PGCs in igf1rb-MO-injected embryos appeared to be distributed in a random fashion, suggesting their displacement by an active, but unguided, mechanism. The mean number of ectopic PGCs in 20S igf1ra-MO-injected embryos (0.5 ± 0.2; n=10) was not significantly different from 20S control MO-injected embryos (P = 0.6).
Figure 3.
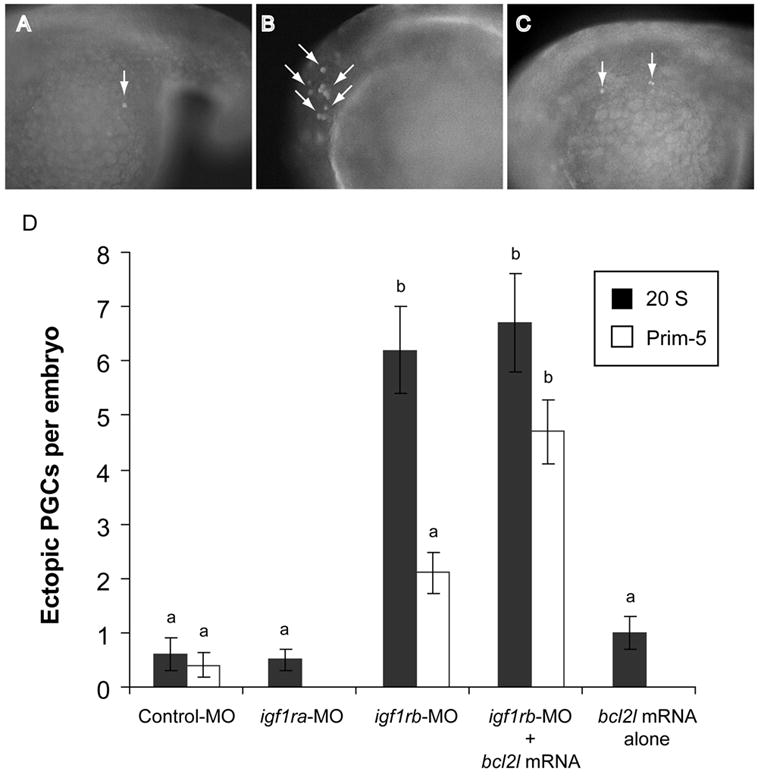
IGF1Rb is required for normal PGC migration. Ectopic PGCs (arrows) were detected rarely in Control-MO-injected embryos (A), whereas they were frequently observed, in both somatic tissues (B) and throughout the yolk cell (C) of igf1rb-MO-injected embryos. (D) Mean numbers of ectopic PGCs in zebrafish embryos after morpholino injections, and/or over-expression of an anti-apoptotic Bcl2-like protein (Bcl2l). Data represent means ± SEM, with sample size indicated in parentheses. Superscript letters denote significant differences between groups (ANOVA, Tukey’s, P < 0.05). Magnification (A–C), 200x.
Ectopic PGCs are selectively eliminated by apoptosis
Signaling through the IGF1R is a potent anti-apoptotic signal in many cell types, including germ cells (Byrne et al., 2002; Delaney et al., 1999; Loir, 1999). We therefore reasoned that the decline in mean PGC number in igf1rb-MO-injected embryos may be due to the elimination of PGCs by apoptosis. We therefore examined igf1ra-MO- and igf1rb-MO-injected embryos by TUNEL analysis for evidence of apoptotic DNA fragmentation.
We first examined whole embryos, and consistent with the known anti-apoptotic functions of IGF signaling, TUNEL analyses revealed increased DNA fragmentation throughout MO-injected embryos after knockdown of either receptor subtype, in both 18S (Fig. 4A–C) and prim-5 (Fig. 4D–F) embryos. TUNEL-positive cells in both igf1ra-MO- and igf1rb-MO-injected embryos were most abundant in anterior neural tissues and dorsal regions of the trunk, with greater intensities observed in 18S versus prim-5 stage embryos. Overall, DNA fragmentation was most intense in igf1ra-MO-injected embryos (Fig. 4B, E); although DNA fragmentation was visibly increased in igf1rb-MO-injected embryos (Fig. 4C, F) relative to control MO-injected embryos, the intensity of DNA fragmentation in igf1rb-MO-injected embryos was noticeably less than that in igf1ra-MO-injected embryos. When considered in the context of the PGC-deficient phenotype, the difference in relative intensities of DNA fragmentation among igf1ra-MO- and igf1rb-MO-injected embryos suggests that IGF1Rb may play a more selective role in promoting PGC survival, whereas IGF1Ra is predominantly involved in somatic cell survival.
Figure 4.
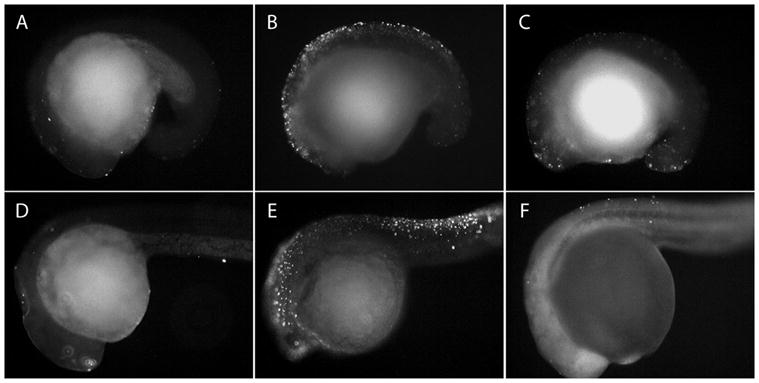
Targeted knockdown of either IGF1R subtype results in increased apoptosis. In situ cell death (TUNEL) analyses of zebrafish embryos after injection with (A, D) control morpholinos; (B, E) IGF1Ra antisense morpholinos (igf1ra-MO); (C, F) IGF1Rb antisense morpholinos (igf1rb-MO). Embryos in upper panels are 18-somite stage embryos; embryos in lower panels are prim-5 stage embryos. Relative to control-MO-injected embryos (A, D), TUNEL-positive cells are more abundant in both igf1ra-MO- and igf1rb-MO-injected embryos, at both stages of development. Magnification 100x.
To examine this in more detail, we used combined immunostaining (anti-Vasa, Fig. 5A, D) and TUNEL (Fig. 5B, E) to look specifically for TUNEL-positive PGCs in MO-injected embryos. We again examined 20S-stage embryos, to facilitate detection of PGCs before their degradation and elimination, which appears to occur by the prim-5 stage (based on mean PGC numbers, Table 1). As shown in Fig. 5A–C, TUNEL-positive PGCs were readily identifiable in igf1rb-MO-injected embryos; notably however, TUNEL-positive PGCs were restricted exclusively to ectopic positions. We did not detect TUNEL-positive PGCs in the genital ridges of any embryo treatment group (e.g., Fig. 5D–F). This observation supports the hypothesis that ectopic PGCs are selectively eliminated by apoptosis, whereas PGCs that correctly migrate to the genital ridges are resistant to death by apoptosis. Thus, more total PGCs are eliminated in igf1rb-MO-injected embryos (relative to other treatment groups), because they contain comparatively more ectopic PGCs, ultimately leading to fewer total PGCs at the prim-5 stage (Table 1).
Figure 5.
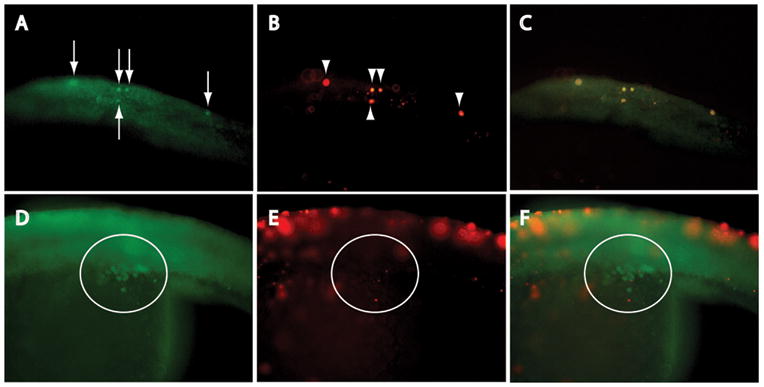
Ectopic PGCs undergo apoptosis. (A) Vasa immunostaining, and (B) TUNEL analysis of ectopic PGCs in dorsal trunk of igf1rb-MO-injected embryo; (C; merged image) colocalization of Vasa-positive (arrows) and TUNEL-positive (arrowheads) cells confirms DNA fragmentation in ectopic PGCs. (D) Vasa-positive, but (E) TUNEL-negative PGCs in genital ridge (circled) of igf1rb-MO-injected embryo; images merged in (F). An absence of DNA fragmentation in genital ridge PGCs confirms survival of the subpopulation of PGCs that successfully migrate to the genital ridge. Anterior is to the left in all images; magnification 200x.
Overexpression of Bcl2l rescues ectopic PGCs in IGF1Rb-deficient embryos
We next analyzed rescue of PGCs by overexpression of an anti-apoptotic Bcl2 protein family member (Bcl2l; Chen et al., 2001; Her et al., 2006), to confirm further that PGCs are eliminated by apoptosis. Co-injection of synthetic mRNA encoding zebrafish Bcl2l significantly rescued mean total PGC numbers in igf1rb-MO-injected embryos, when examined at the prim-5 stage (Table 1), supporting our interpretation that the PGC deficiency in these embryos is due to the elimination of PGCs by apoptosis. The mean number of PGCs in embryos co-injected with bcl2l mRNA and igf1rb-MO was statistically similar to the mean number of PGCs in both control MO-injected and wild-type embryos, and significantly greater than the mean number of PGCs in embryos after injection with igf1rb-MO, igf1ra-MO + igf1rb-MO, and overexpression of dnIGF1R:GFP. These data confirm a statistically significant rescue of PGC numbers by Bcl2l overexpression.
To determine whether Bcl2l overexpression rescues ectopic PGCs specifically, we quantified the number of ectopic PGCs in both 20S and prim-5 embryos, with or without Bcl2l overexpression. As reported above (Fig. 3D), a significantly greater mean number of ectopic PGCs was detected in 20S igf1rb-MO-injected embryos (6.2 ± 0.8, n = 11), relative to 20S control-MO-injected embryos (0.6 ± 0.3, n = 18). Similarly, we detected a significantly increased mean number of ectopic PGCs in 20S igf1rb-MO/bcl2l mRNA co-injected embryos (6.7 ± 0.9; n = 12; ANOVA, P < 0.001), relative to control-MO-injected embryos (Fig. 3D). Injection of bcl2l mRNA alone had no effects on the mean number of ectopic PGCs in 20S embryos (1.0 ± 0.3; n = 15; ANOVA, P > 0.05) relative to control MO-injected embryos (Fig. 3D) indicating that Bcl2l overexpression does not affect PGC migration.
At the prim-5 stage, we detected significantly fewer ectopic PGCs in igf1rb-MO-injected embryos (2.1 ± 0.38; n = 10; ANOVA, P < 0.001), relative to 20S igf1rb-MO-injected embryos (Fig. 3D), supporting our hypothesis that the decline in total PGC number in igf1rb-MO-injected embryos is due to the elimination of ectopic PGCs by apoptosis. Accordingly, we detected a significantly greater mean number of ectopic PGCs in prim-5 igf1rb-MO/bcl2l mRNA co-injected embryos (4.7 ± 0.6; n = 10), relative to both prim-5 igf1rb-MO-injected (ANOVA, P < 0.05) and prim-5 control MO-injected embryos (ANOVA, P < 0.001). These data confirm that overexpression of Bcl2l promotes the survival of ectopic PGCs that would otherwise be eliminated by apoptosis. Combined TUNEL analysis and Vasa immunostaining of igf1rb-MO/bcl2l mRNA co-injected embryos confirmed a reduction in the overall number of TUNEL-positive cells, and an absence of DNA fragmentation in ectopic PGCs (Fig. 6).
Figure 6.
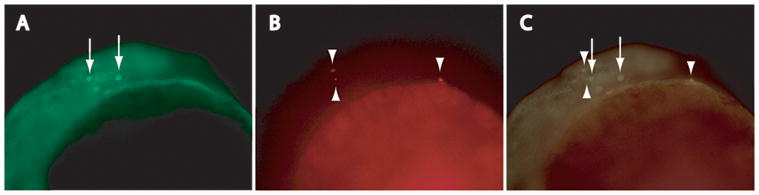
Ectopic PGCs are rescued from apoptosis by overexpression of Bcl2l. (A) Vasa positive cells (arrows), and (B) rare TUNEL-positive cells (arrowheads) in dorsal trunk of igf1rb-MO/bcl2l mRNA co-injected embryo; (C; merged image) lack of colocalized Vasa and TUNEL signals (arrows, arrowheads) confirms absence of DNA fragmentation in ectopic PGCs (compare with Fig. 6A–C). Magnification 200x.
Increased expression of cxcr12a in IGF1Rb-deficient embryos
In efforts to elucidate the mechanism by which loss of IGF1Rb leads to PGC mismigration, we sought to determine whether knockdown of IGF1Rb leads to detectable changes in the expression of genes encoding a chemokine ligand/receptor pair (Cxcl12a/Cxcr4b) that plays a critical role in guiding germ cell migration in vertebrates (Knaut et al., 2003). Embryos in all treatment groups were examined during the PGC migratory period (20S). Targeted knockdown of IGF1Rb had no detectable effect on cxcr4b mRNA levels (111.3 ± 19.5, n=4, P=0.48; Fig. 7) as determined by qRT-PCR analysis, but resulted in a significant increase in cxcl12a mRNA levels (152.3 ± 13.9; n=4, P<0.01). Conversely, knockdown of IGF1Ra had no detectable effect on relative cxcl12a mRNA levels (84.8 ± 5.0%; n=4, P=0.11), though did lead to a detectable reduction in cxcr4b mRNA levels (34.8 ± 2.6%; n=4, P=0.001).
Figure 7.
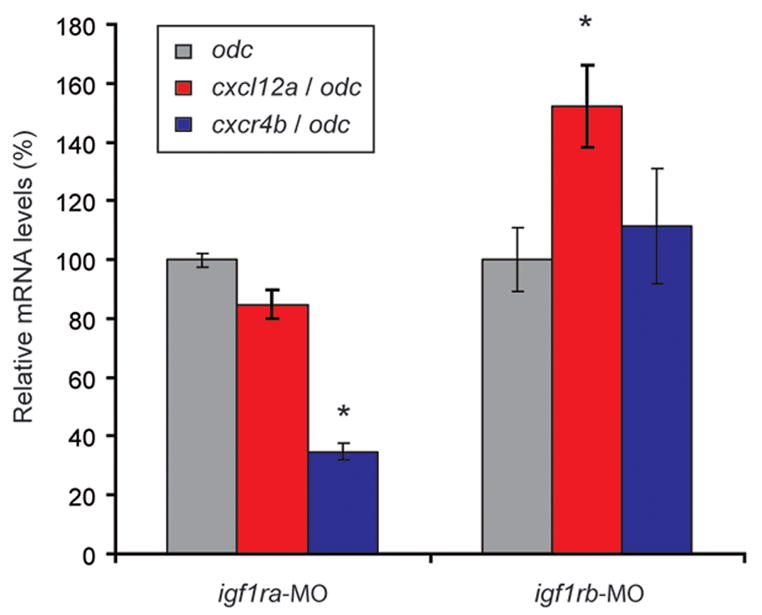
Relative mRNA levels of cxcl12a and cxcr4b in zebrafish embryos injected with morpholino oligonucleotides targeting IGF1Ra (igf1ra-MO) and IGF1Rb (igf1rb-MO). Values are normalized relative to odc mRNA levels (100%), and presented as means ± SEM (n=4). *denotes statistical difference from Control-MO-injected embryos (paired Students t-test, P < 0.05).
Discussion
In this study we employed an in vivo loss-of-function approach to investigate the role of IGF signaling in primordial germ cell development, using the embryonic zebrafish as a model. Our findings suggest novel functions for IGF signaling in PGC development, and provide further support for recent suggestions of divergent evolution of IGF1R signaling following duplication of the ancestral igf1r gene (Schlueter et al., 2006). Specifically, we have shown that suppression of IGF signaling through one (IGF1Rb) of the two duplicate IGF receptors in zebrafish results in defective migration and apoptosis of PGCs, resulting in fewer numbers of PGCs colonizing the genital ridges. Suppressing expression of the other receptor duplicate (IGF1Ra) results in widespread somatic cell death, but has no observable effects on PGC development.
While IGF signaling is a known survival factor for multiple cell types, we believe the PGC apoptosis observed after suppression of IGF1Rb is a secondary consequence of misdirected PGC migration. This interpretation is based upon the observations that PGC apoptosis does not occur until the terminal stages of PGC migration, and among this cell population, appeared to be restricted to ectopic (mismigrated) cells. Furthermore in 18S embryos, significant somatic cell apoptosis could be observed after suppression of both IGF1Ra and IGF1Rb, whereas normal numbers of PGCs were observed in embryos of both groups at this stage. This suggests that in IGF1Rb-deficient embryos, normal numbers of PGCs are initially specified but subsequently exhibit a differential susceptibility to apoptosis, corresponding to their stage of development and/or position in the embryo. Only upon mismigration were PGCs observed to degenerate, which is not wholly unexpected; the elimination of mismigrated PGCs has been well-documented in other vertebrates (Anderson et al., 1999; Molyneaux and Wylie, 2004), possibly as a mechanism to avert the development of extragonadal germ cell tumors (Donovan and de Miguel, 2003).
There are three developmental defects that could lead to PGC migratory defects, and one or more of these could explain the mismigration phenotype observed in IGF1Rb-deficient embryos: (1) An intrinsic loss of cell motility, leaving PGCs unable to respond to extrinsic migratory cues; (2) an intrinsic failure to correctly interpret extrinsic cues; and (3) defective development or death of IGF-dependent somatic cells that provide guidance cues to migratory PGCs. We believe the first possibility can be ruled out, based upon the random distribution of ectopic PGCs in IGF1Rb-deficient embryos. For example, we detected ectopic PGCs in cranial regions of some IGF1Rb-deficient embryos, but in caudal or ventral regions of others, none of which corresponds to the normal pathways followed by migratory PGCs (Weidinger et al., 1999; Weidinger et al., 2002). This random distribution of PGCs in IGF1Rb-deficient embryos suggests that these cells retain intrinsic migratory behaviors, but fail to either receive or correctly interpret extrinsic migratory cues. While this may rule out intrinsic cell-motility defects, it remains unclear whether PGCs cell-autonomously require IGF signaling through IGF1Rb to correctly interpret extrinsic directional cues, or whether the observed defects stem from defective development of IGF-dependent somatic cells upon which PGCs rely for directional cues. Resolving this issue is complicated by the spatially and temporally ubiquitous patterns of expression of both IGF1Ra and IGF1Rb during zebrafish embryogenesis (Maures et al., 2002). On-going studies seek to develop methods to cell-autonomously suppress IGF1R function specifically within PGCs, while maintaining normal receptor function in somatic cells.
Our findings do contribute to recent progress in identifying signaling pathways regulating vertebrate PGC development. For example, signaling by the chemokine CXCL12 through the G-protein-coupled receptor CXCR4 is a highly conserved mechanism regulating directional migration of PGCs in mice, chick, and zebrafish (Ara et al., 2003; Doitsidou et al., 2002; Knaut et al., 2003; Molyneaux et al., 2003; Schier, 2003; Stebler et al., 2004), while stem cell factor, leukemia-inhibitory factor, and IGF-1 have all been shown to function as survival factors for PGCs in vitro (Morita et al., 1999; Park and Han, 2000; Pesce et al., 1993). Our study provides novel in vivo evidence that IGF signaling is also required to promote correct PGC migration, in a manner that is distinct from its documented effects on PGC survival, and more general effects on cell proliferation. Our findings also provide a potential explanation for the germ cell-deficient phenotype observed in mice after targeted ablation of IGF-1 (Baker et al., 1996).
The mechanism by which IGF promotes germ cell migration remains unclear, and warrants further investigation. A potential mechanism was suggested in a recent study with migratory breast cancer cells, which provided evidence for intracellular cross-talk between IGF1R and CXCR4 (Akekawatchai et al., 2005). Although demonstrating cross-talk at the protein level is a difficult prospect in vivo, we chose to examine mRNA levels of cxcl12a and cxcr4b after targeted knockdown of IGF1Ra and IGF1Rb. In IGF1Rb-deficient embryos, we failed to detect any significant changes in cxr4b mRNA levels, but observed a significant increase in cxcl12a mRNA levels. Conversely in IGF1Ra-deficient embryos, there were no significant changes in cxcl12a mRNA levels, though we did observe a significant reduction in cxcr4b mRNA levels. The significance of these changes in gene expression are not presently clear; while disruptions to endogenous cxcl12a expression has been shown to result in PGC mismigration (Knaut et al., 2003), the observed increase in cxcl12a expression in IGF1Rb-deficient embryos could alternatively be explained as a compensatory response by the embryo to mismigration and/or elimination of PGCs. Likewise, the decline in cxcr4b expression in IGF1Ra-deficient embryos could be a secondary effect associated with the demise of cxcr4b-expressing somatic cells, as cell death was particularly intense in IGF1Ra-deficient embryos (Chong et al., 2001).
Overall, our data are consistent with numerous published studies implicating a central role for phosphatidylinositol 3-kinase (PI3K)-Akt signaling during cell migration. Both IGF1R and CXCR4 are known activators of the PI3K-Akt pathway, and in zebrafish embryos, pharmacological suppression of PI3K signaling results in PGC migratory defects (Dumstrei et al., 2004). The specific signal transduction pathways utilized by zebrafish IGF1Ra and IGF1Rb have not been investigated in detail, although it was shown that MO-mediated suppression of both IGF1Ra and IGF1Rb in zebrafish embryos results in reduced Akt phosphorylation (using whole embryos lysates) (Schlueter et al., 2006). Thus, while both receptors appear to activate the PI3K-Akt pathway in zebrafish embryos, cell-specific responses have yet to be investigated in detail. It is possible that functional evolution of the receptor duplicates has occurred at the level of intracellular signal transduction in a cell-specific manner.
The sequence identity between the zebrafish receptor duplicates is relatively high (~70%), and both are highly similar to IGF1R orthologs in other vertebrates (Wood et al., 2005a). However, differences within selected regions of the mature zebrafish IGF1R peptides could account for the observed functional differences. For example, the IGF1Ra peptide contains a contiguous sequence of 15 amino acids in the cytoplasmic domain (directly adjacent to the tyrosine kinase domain) that is not present in IGF1Rb (Maures et al., 2002). While this region does not encode any known functional domains, its presence could presumably influence the activation of downstream signaling pathways, leading to differential cellular responses.
In summary we have shown that IGF signaling, through one of two duplicate IGF receptors, is required for successful migration of PGCs to the genital ridges during embryogenesis. As a consequence of suppressed IGF1Rb signaling, misdirected PGCs are eliminated by apoptosis, resulting in reduced numbers of PGCs colonizing the genital ridges. These findings provide new information about the embryonic functions of IGF signaling in vertebrates, and contribute to a growing body of evidence of the importance of insulin-like signaling for germline development in metazoans.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Calum Macrae, Eric Stone, Amy Doherty and staff members of the Cardiovascular Research Center at Massachusetts General Hospital for their assistance with zebrafish husbandry. We are grateful to Dr. Christian Waeber (Center for Neuroscience, Massachusetts General Hospital) for access to microscopy facilities, funded through NIH P30 NS 045776. This research was supported by Vincent Memorial Research Funds (AWW), and NSF Grant IBN#0110864 (CD). The thoughtful comments of Drs. Jonathan Tilly, Yuichi Niikura, Kaisa Selesniemi, James Pru, Monte Westerfield, Gang Peng and two anonymous reviewers greatly improved the quality of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W, Klammt J, Kratzsch J, Osgood D, Pfaffle R, Raile K, Seidel B, Smith RJ, Chernausek SD. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349:2211–22. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR. Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem. 2005;280:39701–8. doi: 10.1074/jbc.M509829200. [DOI] [PubMed] [Google Scholar]
- Anderson R, Fassler R, Georges-Labouesse E, Hynes RO, Bader BL, Kreidberg JA, Schaible K, Heasman J, Wylie C. Mouse primordial germ cells lacking beta1 integrins enter the germline but fail to migrate normally to the gonads. Development. 1999;126:1655–64. doi: 10.1242/dev.126.8.1655. [DOI] [PubMed] [Google Scholar]
- Ara T, Nakamura Y, Egawa T, Sugiyama T, Abe K, Kishimoto T, Matsui Y, Nagasawa T. Impaired colonization of the gonads by primordial germ cells in mice lacking a chemokine, stromal cell-derived factor-1 (SDF-1) Proc Natl Acad Sci U S A. 2003;100:5319–23. doi: 10.1073/pnas.0730719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J, Hardy MP, Zhou J, Bondy C, Lupu F, Bellve AR, Efstratiadis A. Effects of an IGF1 gene null mutation on mouse reproduction. Mol Endocrinol. 1996;10:903–18. doi: 10.1210/mend.10.7.8813730. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Barbieri M, Bonafe M, Franceschi C, Paolisso G. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am J Physiol Endocrinol Metab. 2003;285:E1064–71. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- Braat AK, Speksnijder JE, Zivkovic D. Germ line development in fishes. International Journal of Developmental Biology. 1999;43:745–760. [PubMed] [Google Scholar]
- Butler AA, Yakar S, Gewolb IH, Karas M, Okubo Y, LeRoith D. Insulin-like growth factor-I receptor signal transduction: at the interface between physiology and cell biology. Comp Biochem Physiol B Biochem Mol Biol. 1998;121:19–26. doi: 10.1016/s0305-0491(98)10106-2. [DOI] [PubMed] [Google Scholar]
- Byrne AT, Southgate J, Brison DR, Leese HJ. Regulation of apoptosis in the bovine blastocyst by insulin and the insulin-like growth factor (IGF) superfamily. Mol Reprod Dev. 2002;62:489–95. doi: 10.1002/mrd.10153. [DOI] [PubMed] [Google Scholar]
- Chen MC, Gong HY, Cheng CY, Wang JP, Hong JR, Wu JL. Cloning and characterization of zfBLP1, a Bcl-XL homologue from the zebrafish, Danio rerio. Biochim Biophys Acta. 2001;1519:127–33. doi: 10.1016/s0167-4781(01)00209-3. [DOI] [PubMed] [Google Scholar]
- Chong SW, Emelyanov A, Gong Z, Korzh V. Expression pattern of two zebrafish genes, cxcr4a and cxcr4b. Mech Dev. 2001;109:347–54. doi: 10.1016/s0925-4773(01)00520-2. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–59. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- Delaney CL, Cheng HL, Feldman EL. Insulin-like growth factor-I prevents caspase-mediated apoptosis in Schwann cells. J Neurobiol. 1999;41:540–8. doi: 10.1002/(sici)1097-4695(199912)41:4<540::aid-neu9>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, Meyer D, Esguerra CV, Leung T, Raz E. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 2002;111:647–59. doi: 10.1016/s0092-8674(02)01135-2. [DOI] [PubMed] [Google Scholar]
- Donovan PJ, de Miguel MP. Turning germ cells into stem cells. Curr Opin Genet Dev. 2003;13:463–71. doi: 10.1016/j.gde.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30:154–6. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- Dumstrei K, Mennecke R, Raz E. Signaling pathways controlling primordial germ cell migration in zebrafish. J Cell Sci. 2004;117:4787–95. doi: 10.1242/jcs.01362. [DOI] [PubMed] [Google Scholar]
- Her GM, Cheng CH, Hong JR, Sundaram GS, Wu JL. Imbalance in liver homeostasis leading to hyperplasia by overexpressing either one of the Bcl-2-related genes, zfBLP1 and zfMcl-1a. Dev Dyn. 2006;235:515–23. doi: 10.1002/dvdy.20624. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WH, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Knaut H, Pelegri F, Bohmann K, Schwarz H, Nüsslein-Volhard C. Zebrafish vasa RNA but not its protein is a component of the germ plasm and segregates asymmetrically before germline specification. J Cell Biol. 2000;149:875–888. doi: 10.1083/jcb.149.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaut H, Werz C, Geisler R, Nüsslein-Volhard C. A zebrafish homologue of the chemokine receptor Cxcr4 is a germ-cell guidance receptor. Nature. 2003;421:279–82. doi: 10.1038/nature01338. [DOI] [PubMed] [Google Scholar]
- LaFever L, Drummond-Barbosa D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science. 2005;309:1071–3. doi: 10.1126/science.1111410. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev. 2001;22:53–74. doi: 10.1210/edrv.22.1.0419. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (IGF-1) and type 1 IGF receptor (IGF-1R) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Loir M. Spermatogonia of rainbow trout: I. Morphological characterization, mitotic activity, and survival in primary cultures of testicular cells. Mol Reprod Dev. 1999;53:422–33. doi: 10.1002/(SICI)1098-2795(199908)53:4<422::AID-MRD8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Maures T, Chan SJ, Xu B, Sun H, Ding J, Duan C. Structural, biochemical, and expression analysis of two distinct insulin-like growth factor I receptors and their ligands in zebrafish. Endocrinology. 2002;143:1858–71. doi: 10.1210/endo.143.5.8768. [DOI] [PubMed] [Google Scholar]
- Molyneaux K, Wylie C. Primordial germ cell migration. Int J Dev Biol. 2004;48:537–44. doi: 10.1387/ijdb.041833km. [DOI] [PubMed] [Google Scholar]
- Molyneaux KA, Zinszner H, Kunwar PS, Schaible K, Stebler J, Sunshine MJ, O'Brien W, Raz E, Littman D, Wylie C, Lehmann R. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development. 2003;130:4279–86. doi: 10.1242/dev.00640. [DOI] [PubMed] [Google Scholar]
- Morita Y, Manganaro TF, Tao XJ, Martimbeau S, Donahoe PK, Tilly JL. Requirement for phosphatidylinositol-3'-kinase in cytokine-mediated germ cell survival during fetal oogenesis in the mouse. Endocrinology. 1999;140:941–9. doi: 10.1210/endo.140.2.6539. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818–35. doi: 10.1210/edrv.22.6.0452. [DOI] [PubMed] [Google Scholar]
- Nuttinck F, Charpigny G, Mermillod P, Loosfelt H, Meduri G, Freret S, Grimard B, Heyman Y. Expression of components of the insulin-like growth factor system and gonadotropin receptors in bovine cumulusoocyte complexes during oocyte maturation. Domest Anim Endocrinol. 2004;27:179–95. doi: 10.1016/j.domaniend.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Ohtsuki T, Otsuki M, Murakami Y, Maekawa T, Yamamoto T, Akasaka K, Takeuchi S, Takahashi S. Organ-specific and age-dependent expression of insulin-like growth factor-I (IGF-I) mRNA variants: IGF-IA and IB mRNAs in the mouse. Zoolog Sci. 2005;22:1011–21. doi: 10.2108/zsj.22.1011. [DOI] [PubMed] [Google Scholar]
- Onagbesan OM, Peddie MJ, Williams J. Regulation of cell proliferation and estrogen synthesis by ovine LH, IGF-I, and EGF in theca interstitial cells of the domestic hen cultured in defined media. Gen Comp Endocrinol. 1994;94:261–72. doi: 10.1006/gcen.1994.1082. [DOI] [PubMed] [Google Scholar]
- Otteson DC, Cirenza PF, Hitchcock PF. Persistent neurogenesis in the teleost retina: evidence for regulation by the growth-hormone/insulin-like growth factor-I axis. Mech Dev. 2002;117:137–49. doi: 10.1016/s0925-4773(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Park TS, Han JY. Derivation and characterization of pluripotent embryonic germ cells in chicken. Mol Reprod Dev. 2000;56:475–82. doi: 10.1002/1098-2795(200008)56:4<475::AID-MRD5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Perrot V, Moiseeva EB, Gozes Y, Chan SJ, Funkenstein B. Insulin-like growth factor receptors and their ligands in gonads of a hermaphroditic species, the gilthead seabream (Sparus aurata): expression and cellular localization. Biol Reprod. 2000;63:229–41. doi: 10.1095/biolreprod63.1.229. [DOI] [PubMed] [Google Scholar]
- Pesce M, Farrace MG, Piacentini M, Dolci S, De Felici M. Stem cell factor and leukemia inhibitory factor promote primordial germ cell survival by suppressing programmed cell death (apoptosis) Development. 1993;118:1089–94. doi: 10.1242/dev.118.4.1089. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozios KC, Ding J, Degger B, Upton Z, Duan C. IGFs stimulate zebrafish cell proliferation by activating MAP kinase and PI3-kinase-signaling pathways. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1230–9. doi: 10.1152/ajpregu.2001.280.4.R1230. [DOI] [PubMed] [Google Scholar]
- Schier AF. Chemokine signaling: rules of attraction. Curr Biol. 2003;13:R192–4. doi: 10.1016/s0960-9822(03)00122-2. [DOI] [PubMed] [Google Scholar]
- Schlueter PJ, Peng G, Westerfield M, Duan C. Insulin-like growth factor signaling regulates zebrafish embryonic growth and development by promoting cell survival and cell cycle progression. Cell Death and Differentiation. 2007 doi: 10.1038/sj.cdd.4402109. In press. [DOI] [PubMed] [Google Scholar]
- Schlueter PJ, Royer T, Farah MH, Laser B, Chan SJ, Steiner DF, Duan C. Gene duplication and functional divergence of the zebrafish insulin-like growth factor 1 receptors. FASEB J. 2006;20:1230–1232. doi: 10.1096/fj.05-3882fje. [DOI] [PubMed] [Google Scholar]
- Stebler J, Spieler D, Slanchev K, Molyneaux KA, Richter U, Cojocaru V, Tarabykin V, Wylie C, Kessel M, Raz E. Primordial germ cell migration in the chick and mouse embryo: the role of the chemokine SDF-1/CXCL12. Dev Biol. 2004;272:351–61. doi: 10.1016/j.ydbio.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Weidinger G, Wolke U, Köprunner M, Klinger M, Raz E. Identification of tissues and patterning events required for distinct steps in early migration of zebrafish primordial germ cells. Development. 1999;126:5295–5307. doi: 10.1242/dev.126.23.5295. [DOI] [PubMed] [Google Scholar]
- Weidinger G, Wolke U, Koprunner M, Thisse C, Thisse B, Raz E. Regulation of zebrafish primordial germ cell migration by attraction towards an intermediate target. Development. 2002;129:25–36. doi: 10.1242/dev.129.1.25. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book. University of Oregon Press; Eugene, OR: 1995. [Google Scholar]
- Wollmann HA. Growth hormone and growth factors during perinatal life. Horm Res. 2000;53(Suppl 1):50–4. doi: 10.1159/000053205. [DOI] [PubMed] [Google Scholar]
- Wood AW, Duan C, Bern HA. Insulin-like growth factor signaling in fish. Int Rev Cytol. 2005a;243:215–85. doi: 10.1016/S0074-7696(05)43004-1. [DOI] [PubMed] [Google Scholar]
- Wood AW, Schlueter PJ, Duan C. Targeted knockdown of insulin-like growth factor binding protein-2 disrupts cardiovascular development in zebrafish embryos. Mol Endocrinol. 2005b;19:1024–34. doi: 10.1210/me.2004-0392. [DOI] [PubMed] [Google Scholar]
- Yano K, Bauchat JR, Liimatta MB, Clemmons DR, Duan C. Down-regulation of protein kinase C inhibits insulin-like growth factor I-induced vascular smooth muscle cell proliferation, migration, and gene expression. Endocrinology. 1999;140:4622–32. doi: 10.1210/endo.140.10.7035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.