

Abstract
Chronic wounds represent a substantial public health problem. The development of tools that would enable sophisticated scrutiny of clinical wound tissue material is highly desirable. This work presents evidence enabling rapid specific identification and laser capture of blood vessels from human tissue in a manner which lends itself to successful high-density (U133A) microarray analysis. Such screening of transcriptome followed by real-time PCR and immunohistochemical verification of candidate genes and their corresponding products were performed by using 3 mm biopsies. Of the 18,400 transcripts and variants screened, a focused set of 53 up-regulated and 24 down-regulated genes were noted in wound-derived blood vessels compared with blood vessels from intact human skin. The mean abundance of periostin in wound-site blood vessels was 96-fold higher. Periostin is known to be induced in response to vascular injury and its expression is associated with smooth muscle cell differentiation in vitro and promotes cell migration. Forty-fold higher expression of heparan sulfate 6-O-endosulfatase1 (Sulf1) was noted in wound-site vessels. Sulf1 has been recently recognized to be anti-angiogenic. During embryonic vasculogenesis, CD24 expression is down-regulated in human embryonic stem cells. Wound-site vessels had lower CD24 expression. The findings of this work provide a unique opportunity to appreciate the striking contrast in the transcriptome composition in blood vessels collected from the intact skin and from the wound-edge tissue. Sets of genes with known vascular functions but never connected to wound healing were identified to be differentially expressed in wound-derived blood vessels paving the way for innovative clinically relevant hypotheses.
Keywords: angiogenesis, microarray, wound healing
The public health impact of chronic wounds is staggering. An estimated 1.3–3 million US individuals are believed to have pressure ulcers; and as many as 10–15% of the 20 million individuals with diabetes are at risk of developing diabetic ulcers. Many more have had venous ulcers or wounds that result from arterial disease. Treating these wounds costs an estimated $5–10 billion each year (1). According to the National Institute of General Medical Sciences, a major sponsor of wound healing research in the United States, research to advance wound care is handicapped by the limitations of animal model systems in mimicking human wound healing (http://grants.nih.gov/grants/guide/rfa-files/RFA-GM-06-002.html). Indeed, a vast majority of current wound healing research is based on rodent and in vitro studies, which have limited resemblance with the chronic human wound (2). Few studies use the preclinical porcine model, which has been estimated to agree with human studies 78% of the time (2). The study of experimental acute biopsy wounds in humans is valuable in many ways but does not resemble the chronic wounds that represent the most significant problem (3). Although experimental models may be more traceable, direct investigation of the clinically presented chronic wound tissue could provide data that directly address public health. Thus, development of tools and approaches that would enable sophisticated scrutiny of clinical wound tissue material is highly desirable.
Biopsies collected from human cutaneous wounds presented in the clinic are highly heterogeneous in cellular composition. The nature of the tissue may vary from one collection to another complicating comparison of results derived from tissue homogenates. Thus, the utility of such tissue material is primarily limited to histological studies. Vasculopathy represents a major factor that complicates cutaneous wound healing (4, 5). The objective of this study was to develop an approach to specifically laser capture blood vessels from standard 3-mm human wound biopsies such that the captured blood vessel tissue element would lend itself to genomic screening and verification of candidate genes, using quantitative PCR.
Results
The novel approach standardized in this study enables the rapid detection of blood vessels in human tissues in a manner that lends itself to successful microarray analyses and validation. Rapid and reliable detection of blood vessels in human tissue samples was made possible by the use of Ulex Europaeus Agglutinin (UEA) I. UEA I staining, which was completed in 2 min, tightly agreed with the identification of blood vessels, using the classical von Willebrand factor (VWF) staining approach (Fig. 1a). Immunohistochemical staining, such as that of VWF, takes over 30 min and causes tissue contents such as nucleic acid to degrade (6). UEA I is a glycoprotein that binds to endothelial cells and endothelial progenitor cells (7). In human skin tissue, UEA1 was effective in specifically identifying blood vessels as a structure encircling a lumen. The PALM RoboMover hardware enables automated marking of such structures allowing a large number of vessels to be marked in seconds. The use of laser size as fine as 0.6–1.2 μM enable precise collection of vessel elements (Fig. 1b). Comparison of several fixation approaches identified the RNALater approach as being effective to preserve mRNA stability for our purposes (Fig. 2a). Dehydration of the tissue section also supported preservation of mRNA stability. After dehydration, it is desirable to rapidly perform laser microdissection pressure catapulting (LMPC). Holding the tissue for 1 h after dehydration caused significant loss of mRNA (Fig. 2b). Characterization of the blood vessel and non-blood vessel tissue elements included the quantitation of genes that are specific to each region. VWF, CD31, and smooth muscle actin represent integral and abundant components of the blood vessel. Measurement of the expression of these genes indicated high abundance of these genes in the vessel element compared with the nonvessel elements. Genes such keratin 14 and vimentin are expected in the human skin tissue but are not abundant in blood vessels. Indeed, these genes were found abundantly in the nonvessel tissue but not in the vessel elements (Fig. 3). These results demonstrate that the laser captured tissue elements were high in purity and that the procedure was effective in harvesting the blood vessel tissue with a high level of specificity.
Fig. 1.

Rapid identification of blood vessel elements from human wound tissue. (a) Human wound sections stained with UEA I lectin (green) and anti-human VWF antibody (red) with DAPI nuclear stain (blue). (i) UEA I lectin and DAPI. (ii) VWF and DAPI. (iii) Colocalization of UEA I lectin with VWF. (iv) UEA lectin, VWF and DAPI. (Scale bars: 20 μm.) (b) Human wound section stained with UEA I lectin. (i) A vessel is identified and marked for capture and catapult (shown with a white arrow). (ii) Laser-assisted cutting and separation of the identified vessel. The vessel is ready to be catapulted. (iii) The tissue section after the cut vessel has been catapulted. (iv) Isolated vessel captured in chaotropic solution.
Fig. 2.

Optimization of vessel staining protocol: fixation and dehydration. (a) Human wound sections were subjected to standard fixation methods, i.e., RNALater, acetone, formalin (neutral buffered formalin), or ethanol (95% vol/vol ethanol) as shown. After fixation, sections were stained with UEA I lectin (green). The bar graph shows relative β-actin mRNA levels quantified by using real-time PCR. RNA was extracted from 400,000 μm2 of vessel elements captured after laser microdissection after specified fixation and UEA I lectin staining. *, P < 0.05 lower compared with the RNALater treated group. (b) UEA I stained dehydrated versus nondehydrated wound tissue sections. The bar graph shows relative β-actin mRNA levels quantified from tissue elements captured from RNALater fixed and dehydrated or nondehydrated. *, P < 0.05 lower compared with the dehydrated group. (c) Stability of β-actin transcript in tissue sections as a function of time after RNALater fixation, staining, and dehydration. Vessel area (400,000 μm2) was processed, and β-actin expression was quantified as described. *, P < 0.05 lower compared with 5-min group. (Scale bars: 200 μm.)
Fig. 3.

Characterization of captured vessel and nonvessel elements. (a) Images of vessel and nonvessel elements are marked by using the LMPC system. (b) Quantification of vessel-specific mRNA from 400,000 μm2 of vessel elements (VE) or nonvessel elements (non-VE) captured following laser microdissection from RNALater treated, UEA I stained and dehydrated human wound tissue. RNA was extracted from the captured tissue, amplified, and reverse transcribed into cDNA. Gene expression was quantified by using real-time PCR (normalized to β-actin). *, P < 0.05 compared with vessel element. SMA, smooth muscle actin; KRT14, keratin 14; VIM, vimentin.
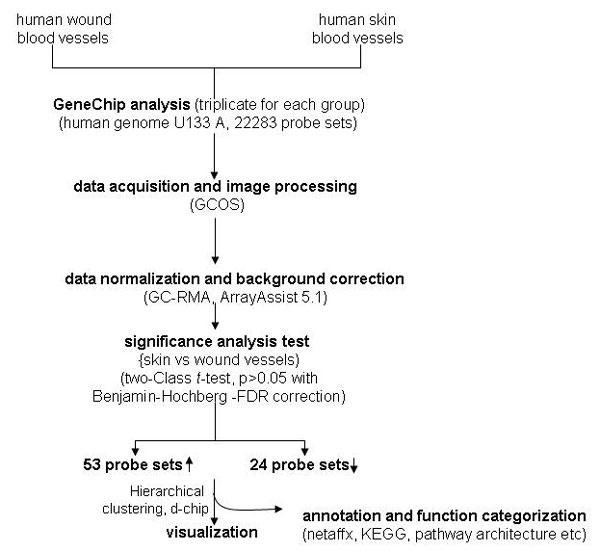
Genome-wide screening for transcripts in cutaneous blood vessels that are differentially expressed in wounds compared with blood vessels from the intact human skin resulted in the identification of a focused set of 53 up-regulated and 24 down-regulated genes (Fig. 4). These changes were statistically significant [see supporting information (SI) Tables 1 and 2]. These findings provide key insight into the definition of blood vessels at the wound site. Approximately half of all genes up-regulated in the wound-site blood vessels were affected at the level of 10-fold or more. For example, mean abundance of periostin in wound-site blood vessels was noted to be 96-fold higher than that noted in blood vessels from the intact skin. Periostin is known to be induced in response to vascular injury and its expression is associated with smooth muscle cell differentiation in vitro and promotes cell migration (8). Forty-fold higher expression of heparan sulfate 6-O-endosulfatase1 (Sulf1) was noted in blood vessels from wound-site compared with that from intact skin. Sulf1 has been recently recognized to be anti-angiogenic (9). The α-1 chain of collagen IV (also known as arresten) or COL4A1 was overexpressed in blood vessels of wound site by 37-fold compared with that in vessels from intact skin. COL4A1 is expressed in the endothelial cells of murine dermal wounds (10) and functions to resist angiogenesis (11). The inwardly rectifying potassium channel KCNJ8 (also known as kir6.1) is induced by abrupt loss of shear stress (12) as would be expected in blood vessels at a chronic wound site. KCNJ8 supports vasodilatation and blood flow by restricting the release of the vasoconstrictor endothelin-1 (13). KCNJ8 was up-regulated >25-fold in wound-site blood vessels compared with vessels of the intact skin. Endothelium of newly formed blood vessels abundantly expresses Thy-1, a major cell surface glycoprotein (14). Thy-1 gene expression in wound-site blood vessel was 24-fold higher than that in vessels from the intact human skin.
Fig. 4.

Hierarchical cluster images illustrating genes up- or down-regulated in vessels from human wound edge compared with that from intact skin. Gene expression data obtained by using GeneChip were subjected to t test analysis with false discovery rate correction (see SI Fig. 8). For comparative visualization, those genes that were expressed at significantly different levels in the wound-edge skin versus the intact skin were subjected to hierarchical clustering as shown. Line graphs at Right illustrate the average pattern of gene expression in the corresponding cluster. Red to green gradation in color represent higher to lower expression signal. A scale representing fold change is indicated at the bottom.
A >100-fold change was noted in a quarter of all genes down-regulated in the wound-site blood vessel compared with the abundance in blood vessels from intact skin. The gene for keratin 14, one of 10 known type I keratins, was found in low abundance in vessel elements as well as in cultured human microvascular endothelial cells (data not shown). This finding is consistent with the literature reporting keratin in endothelial cells (15). Abundance of this and another rare transcript secretoglobin (SCB2A2) was much lower in vessels from the wound site compared with that from intact human skin. Discussion of select genes in this category is limited to abundant genes with known vascular functions. Two soluble naturally occurring Wnt antagonists, frizzled-related proteins, FRP1 and FRP3, are expressed by vascular cells (16). Wnt growth factors function via Frizzled receptors to stimulate angiogenesis (17). The expression of secreted frizzled-related protein 1 (SFRP1) was strikingly down-regulated in wound-site blood vessels compared with that in vessels from intact skin. During embryonic vasculogenesis, CD24 expression is down-regulated in human embryonic stem cells (18). Wound-site vessels had lower CD24 expression compared with abundance of the transcript in vessels from intact skin.
The findings of this work provide an extraordinary opportunity to appreciate the striking contrast in the transcriptome composition in blood vessels collected from the intact skin and from the wound-edge tissue. Ten up-regulated and five down-regulated genes were verified by using quantitative PCR to test the validity of the microarray analysis performed (Fig. 5). The products of select differentially expressed genes were tested immunohistochemically (Fig. 6). Taken together, outcomes of quantitative PCR and immunohistochemical studies validate the findings of our microarray studies.
Fig. 5.

Real-time PCR validation of GeneChip microarray expression analysis. Expression levels of selected genes identified by using GeneChip analysis were independently determined by using real-time PCR. For comparison, the real-time PCR data (normalized to β-actin, a housekeeping gene) were proportionately adjusted to fit to the scale with GeneChip expression values (normalized by using global scaling approach). *, P < 0.05 compared with corresponding wound vessels. Solid bars, wound-edge skin vessel elements; open bars, intact skin vessel elements. Shown are wound-edge skin vessels versus vessels in intact skin for up-regulated genes (a) and down-regulated genes (b). Sulf1, sulfatase 1; COL4A1, collagen 4A1; HLA-DRB4, major histocompatibility complex, class II, DR beta 4; THY1, Thy-1 T cell antigen; COL5A2, collagen 5A2; COL3A1, collagen 3A1; TIMP1, tissue inhibitor of metalloproteinases; CYR61, cysteine-rich, angiogenic inducer, 61; MMP1, matrix metalloproteinase-1; ANGPT2, angiopoietin 2; KRT14, keratin 14; SOX9, SRY (sex determining region Y)-box 9; KRT15, keratin 15; PROM1, CD133 antigen, or prominin 1.
Fig. 6.

Immunofluorescent staining of differentially expressed proteins (identified by using microarray analysis) in blood vessels from human wound edge and intact skin tissue. Human wound and skin tissue sections were stained with UEA I lectin (green) and coimmunostained with Cyr61 (red) (a), MMP1 (red) (b), ANGPT2 (red) (c), or Thy1 (red) (d) as shown. (Scale bars: 50 μm.) CYR61, cysteine-rich angiogenic inducer 61; MMP1, matrix metalloproteinase-1; ANGPT2, angiopoietin 2; THY1, Thy-1 T cell antigen.
Discussion
The ability to perform genome-wide screening of blood vessels captured directly from the disease affected tissue in patients represents a major advance in our approach to investigate vascular biology in any given disease setting. This work presents evidence demonstrating that that selective microdissection of blood vessels, high-density microarray analysis, quantitative PCR-based validation of microarray data, and immunohistochemistry can all be performed by using no more than one 3-mm punch biopsy from the affected tissue. Comparison of results from blood vessels at the edge of chronic wound tissue with that of vessels in intact human skin demonstrated a revealing contrast between the transcriptome of the two vessels. Results from such investigations are effective in developing experimental models and paradigms.
In the current study, unbiased genome-wide interrogation identified transcripts that were differentially expressed in the vessels of the wound site and provided insight into the vascular biology of chronic wounds in a clinical setting. Functionally, most candidate genes were linked to angiogenesis. Vascularization, or lack thereof, is a fate dictated by the net balance of angiogenic and angiostatic signals at the site of injury. Heretofore known as a facilitator of tumor angiogenesis (19), periostin emerged as the topmost gene up-regulated in wound-site blood vessels. Periostin up-regulates vascular endothelial growth factor (VEGF) receptor 2 expression (19). Indeed, vascular injury induces periostin (8). Up-regulation of periostin expression in rat carotid arteries after balloon injury and in cultured vascular smooth muscle cells after stimulation by growth factors is mediated through PI-3-kinase-dependent signaling pathway. Periostin protein secreted by vascular smooth muscle cells plays a significant role in regulating vascular smooth muscle cell migration in vitro.
Heparan sulfate proteoglycans act as coreceptors for numerous heparin-binding growth factors and cytokines and are involved in cell signaling. Heparan sulfate 6-O-endosulfatases, such as SULF1, selectively remove 6-O-sulfate groups from heparan sulfate. This activity modulates the effects of heparan sulfate by altering binding sites for signaling molecules. In cancer research, xenografts derived from SULF1-expressing stable clones of carcinoma cells show reduced vessel density, marked necrosis, and apoptosis, indicative of inhibition of angiogenesis. Furthermore, SULF1-expressing clonal lines showed reduced staining with the endothelial marker CD31 in a Matrigel plug assay, indicating that SULF1 expression inhibits angiogenesis. Of note, SULF1 expression in xenografts was associated with a reduced ability of vascular endothelial cell heparan sulfate to participate in a complex with FGF-2 and its receptor tyrosine kinase FGF receptor 1c. In vitro, short hairpin RNA-mediated down-regulation of SULF1 in human umbilical vein endothelial cells increased proliferation mediated by heparan sulfate-dependent FGF-2, hepatocyte growth factor, and vascular endothelial growth factor 165 (VEGF165) but not by heparan sulfate-independent VEGF121. Consistent with the role of heparan sulfate glycosaminoglycan sulfation in VEGF-mediated signaling, treatment of human umbilical vein endothelial cells with chlorate, which inhibits heparan sulfate glycosaminoglycan sulfation and therefore mimics SULF1 overexpression, led to an attenuated VEGF-mediated signaling. Thus, from studies in tumor biology, we know that SULF1 modulates the function of heparan sulfate binding VEGF165 in proliferation and angiogenesis (9). The current work provides a link between SULF1 and chronic wounds in a clinical setting. Thy-1 is an endothelial cell surface glycoprotein that marks adult but not embryonic angiogenesis. The up-regulation of Thy-1 by cytokines but not growth factors was noted as evidence supporting the importance of inflammation in the pathogenesis of adult angiogenesis (14). The current study noted that Thy-1 expression is markedly high in vessels from the wound site compared with that from the skin. Although itemized discussion of the functional significance of candidate genes is beyond the scope of this publication, it is clear that the approach adopted in the current study provides unprecedented power in understanding the vascular biology of human disease.
This work provides evidence demonstrating that it is feasible to laser capture blood vessels from human tissues for microarray analysis and validation. This provides a powerful tool to interrogate blood vessels isolated from patients of different disease settings with the goal to understand the molecular aspects of vascular biology in actual clinical setting. The approach described herein is applicable to a broad range of clinical research and therefore represents a powerful tool to enable sophisticated translational research. It is now possible to compare, for example, blood vessels collected from the chronic wounds of healing and nonhealing patients with otherwise matched individual characteristics and clinical condition.
Materials and Methods
Human Subjects and Sample Collection.
Subjects participating in the study were nondiabetic males with or without chronic wounds. Protocols were approved by the Ohio State University's Institutional Review Board. Wound-edge skin (at the wound perimeter) or intact skin biopsies (3 mm) were obtained from individual subjects, immediately embedded in OCT compound (Tissue-Tek) and stored frozen in liquid N2 until further analysis.
LMPC of Tissue Vessels.
Tissue sectioning and fixation.
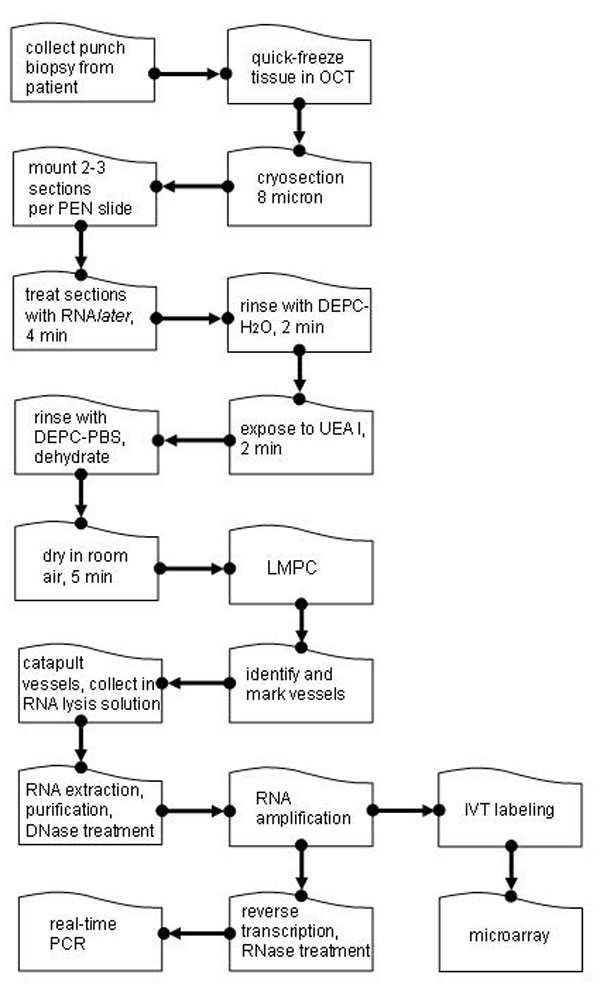
Frozen tissue blocks were cut into 8-μm sections. The sections (2–3) were mounted on each RNAZap-treated thermoplastic (polyethylene napthalate)-covered glass slide (PALM Technologies, Bernreid, Germany) and kept at −80°C until use. When needed, sections were thawed and fixed for 1 min in one of the following fixatives: 95% ethanol, 10% neutral-buffered formalin, or acetone (20). An additional section was treated with RNALater (Ambion, Austin, TX) for 4 min. After initial optimization studies for tissue fixation, RNALater was used for all other studies. See SI Fig. 7 for a flow chart outlining protocols used for tissue sectioning, staining, LMPC, and RNA processing.
Blood vessel staining.
After rinsing slides in diethyl pyrocarbonate (DEPC)-treated water for 2 min, sections were exposed to 5 mg/ml fluorescein-labeled UEA I (Vector Labs, Burlingame, CA) diluted 1:50 in DEPC water for 2 min. The sections were rinsed in DEPC phosphate buffer (PBS) for 2 min, sequentially dehydrated (70% ethanol for 30 s, 95% ethanol for 30 s, and 100% ethanol for 1 min), and then placed in histological grade Xylene (Fisher Scientific, Waltham, MA) for 2 min. The slides were air-dried for 5 min and immediately observed under a fluorescent microscope for LMPC processing. To examine the effects of dehydration on RNA degradation and visualization, some slides were rinsed in DEPC PBS after staining and then allowed to air dry for 5 min, bypassing the sequential ethanol and Xylene washes.
LMPC.
LMPC was performed by using the laser microdissection system from PALM Technologies (Bernreid, Germany) containing a PALM MicroBeam and RoboStage for high-throughput sample collection and a PALM RoboMover (PALM Robo software, Version 2.2) (6). Typical settings used for laser cutting were UV-Energy of 75–85 and UV-Focus of 52. Blood vessels and nonvessel area were typically cut and captured under a 40× ocular lens, using a fluorescent lamp. Cut elements were catapulted into 25 μl of RNA extraction buffer (PicoPure RNA Isolation Kit; Arcturus, Sunnyvale, CA). Approximately 400,000 μm2 of tissue area was captured into each cap. Upon completion of microdissection, the captured material was spun down into a 0.5-ml tube, combined with an additional 25 μl of extraction buffer, and incubated at 42°C for 30 min. The extract was then held at −80°C until RNA isolation.
RNA Isolation.
Total RNA from LMPC samples was isolated by using the PicoPure RNA Isolation Kit (Arcturus). RNA quantity was measured by using the NanoDrop system (NanoDrop Technologies, Wilmington, DE). Pooled RNA was reduced to 10 μl, using Speed-Vac and then amplified one round, using either the RiboAmp RNA Amplification Kit (Arcturus) for real-time PCR samples or the RiboAmp OA RNA Amplification Kit (Arcturus) for GeneChip analyses.
Reverse Transcription and Quantitative Real-time PCR.
Amplified RNA was reverse transcribed into cDNA, using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Reactions were done by using random hexamer priming. One microliter each of random hexamers (50 ng/μl) and dNTPs (10 mM) was added to ≈10 μl of RNA solution, and the resulting solution was incubated for 5 min at 65°C and then placed on ice for at least 1 min. Ten microliters of 2× reaction mixture containing Tris·HCl (pH 7.4), 25 mM magnesium chloride, 0.1 M DTT, 40 units RNase Out, and 200 units of SuperScript III enzyme were then added. The resulting solution was incubated at 25°C for 10 min, followed by 50 min at 50°C, and finally at 85°C for 5 min. RNA was degraded by incubating for 20 min at 37°C with RNase H. The synthesized cDNA was used directly for real-time PCR analysis (MX3000P system; Stratagene, La Jolla, CA). The PCR included 5 μl of diluted cDNA solution, 7.3 μl of nuclease-free water, 0.1 μl of each primer solution (50 μM), and 12.5 μl of SYBR green real-time PCR mixture (Applied Biosystems, Foster City, CA) The solution was initially incubated at 50°C for 2 min, followed by 10 min incubation at 95°C to activate the polymerase. cDNA standards were used to determine relative quantities and to compare dissociation temperatures to partially ensure correct product formation. β-Actin gene expression was measured to correct for differences in extraction efficiency between samples. The experimental groups consisted of replicates for statistical purposes. Primer sequences are listed in SI Table 3.
GeneChip Probe Array Analyses.
Target labeling for GeneChip analysis, using nanogram amounts of RNA samples.
Amplified RNA was processed as follows: after the second round of cDNA synthesis and purification in the RiboAmp OA protocol, 20 μl of eluted cDNA was added to the GeneChip IVT Labeling Kit (Affymetrix, Santa Clara, CA) in vitro transcription reaction to generate biotinylated cRNA. This protocol has been optimized to reproducibly amplify RNA from nanogram quantities of RNA and is based on the principle of performing two cycles of cDNA synthesis and in vitro transcription for target amplification (21).
Hybridization.
Samples were hybridized to Affymetrix Human Genome arrays (HG-U133A) for the screening of >22,000 genes and ESTs. The arrays were washed, stained with streptavidin-phycoerythrin and scanned with the GeneArray scanner (Affymetrix) in our own facilities as described in refs. 21–25.
Data analyses.
Raw data were collected and analyzed by using Stratagene ArrayAssist Expression software, Version 5.1 (Stratagene). Additional processing of data were performed by using dChip software, Version 1.3 (Harvard University, Cambridge, MA) (24). Data acquisition and image processing was performed by using Gene Chip Operating Software (Affymetrix). Data normalization and background corrections were performed by using GC-RMA. Differentially expressed genes were identified by using a two-class t test where significance level was set at P > 0.05 with Benjamin–Hochberg false discovery rate correction (24). Genes that were >1.5 fold up- or down-regulated compared with skin sample were selected. A detailed analysis scheme has been illustrated in Figure S2. For data visualization, statistically significant (t test) genes were subjected to hierarchical clustering, using dChip software, Version 1.3. Gene annotation was performed by using Pathway architecture software (Stratagene). Selected differentially expressed candidates were verified by using quantitative real-time PCR.
Immunohistochemistry.
Tissue specimens were sectioned (10 μm) and mounted on positively charged Superfrost slides (Fisher Scientific). Immunohistochemical staining was performed as described in ref. 22, using the following primary antibodies: rabbit polyclonal anti-human VWF (DakoCytomation, Denmark), goat polyclonal anti-human Ang-2 and rabbit polyclonal anti-human Cyr61 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA), mouse monoclonal anti-human Thy1, rabbit polyclonal anti-human Collagen IV and rabbit polyclonal anti-human MMP1 antibodies from Abcam (Cambridge, MA). The primary antibody was detected by using a fluorescencently tagged secondary antibody.
Statistics.
In bar graphs, results are presented as mean ± SD. In these cases, difference between means was tested by using Student's t test. Microarray data processing is described above under GeneChip probe array analyses.
Supplementary Material
Acknowledgments
We thank our clinical research staff Samantha Bellamy and Lynn Lambert for patient enrollment and sample collection. This work was supported by National Institutes of Health Grants RO1 HL073087, GM 077185, and GM 069589 (to C.K.S.); a National Science Foundation award under agreement no. 0112050; General Clinical Research Center Grant M01-RR00034 from the National Center of Research Resources of the National Institutes of Health at The Ohio State University. Partnership of National Healing Corporation in enabling a Chronic Wound Tissue Bank, which was used for this work, is gratefully acknowledged.
Abbreviations
- LMPC
laser microdissection pressure catapulting
- UEA
Ulex Europaeus Agglutinin
- VEGF
vascular endothelial growth factor
- VWF
von Willebrand factor.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706793104/DC1.
References
- 1.Kuehn BM. Jama. 2007;297:938–939. doi: 10.1001/jama.297.9.938. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan TP, Eaglstein WH, Davis SC, Mertz P. Wound Repair Regen. 2001;9:66–76. doi: 10.1046/j.1524-475x.2001.00066.x. [DOI] [PubMed] [Google Scholar]
- 3.Kiecolt-Glaser JK, Marucha PT, Malarkey WB, Mercado AM, Glaser R. Lancet. 1995;346:1194–1196. doi: 10.1016/s0140-6736(95)92899-5. [DOI] [PubMed] [Google Scholar]
- 4.Zutt M, Haas E, Kruger U, Distler M, Neumann C. Dermatology. 2007;214:319–324. doi: 10.1159/000100882. [DOI] [PubMed] [Google Scholar]
- 5.Smith PC. Int J Low Extrem Wounds. 2006;5:160–168. doi: 10.1177/1534734606292429. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn DE, Roy S, Radtke J, Khanna S, Sen CK. Am J Physiol. 2007;292:H1245–H1253. doi: 10.1152/ajpheart.01069.2006. [DOI] [PubMed] [Google Scholar]
- 7.Zhao X, Huang L, Yin Y, Fang Y, Zhou Y. Transpl Int. 2007;20:712–721. doi: 10.1111/j.1432-2277.2007.00497.x. [DOI] [PubMed] [Google Scholar]
- 8.Lindner V, Wang Q, Conley BA, Friesel RE, Vary CP. Arterioscler Thromb Vasc Biol. 2005;25:77–83. doi: 10.1161/01.ATV.0000149141.81230.c6. [DOI] [PubMed] [Google Scholar]
- 9.Narita K, Staub J, Chien J, Meyer K, Bauer M, Friedl A, Ramakrishnan S, Shridhar V. Cancer Res. 2006;66:6025–6032. doi: 10.1158/0008-5472.CAN-05-3582. [DOI] [PubMed] [Google Scholar]
- 10.Darby IA, Bisucci T, Raghoenath S, Olsson J, Muscat GE, Koopman P. Lab Invest. 2001;81:937–943. doi: 10.1038/labinvest.3780304. [DOI] [PubMed] [Google Scholar]
- 11.Sudhakar A, Nyberg P, Keshamouni VG, Mannam AP, Li J, Sugimoto H, Cosgrove D, Kalluri R. J Clin Invest. 2005;115:2801–2810. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Chatterjee S, Levitan I, Wei Z, Fisher AB. Microcirculation. 2006;13:633–644. doi: 10.1080/10739680600930255. [DOI] [PubMed] [Google Scholar]
- 13.Malester B, Tong X, Ghiu I, Kontogeorgis A, Gutstein DE, Xu J, Hendricks-Munoz KD, Coetzee WA. FASEB J. 2007;21:2162–2172. doi: 10.1096/fj.06-7821com. [DOI] [PubMed] [Google Scholar]
- 14.Lee WS, Jain MK, Arkonac BM, Zhang D, Shaw SY, Kashiki S, Maemura K, Lee SL, Hollenberg NK, Lee ME, Haber E. Circ Res. 1998;82:845–851. doi: 10.1161/01.res.82.8.845. [DOI] [PubMed] [Google Scholar]
- 15.Traweek ST, Liu J, Battifora H. Am J Pathol. 1993;142:1111–1118. [PMC free article] [PubMed] [Google Scholar]
- 16.Goodwin AM, D'Amore PA. Angiogenesis. 2002;5:1–9. doi: 10.1023/a:1021563510866. [DOI] [PubMed] [Google Scholar]
- 17.Masckauchan TN, Kitajewski J. Physiology (Bethesda) 2006;21:181–188. doi: 10.1152/physiol.00058.2005. [DOI] [PubMed] [Google Scholar]
- 18.Gerecht-Nir S, Dazard JE, Golan-Mashiach M, Osenberg S, Botvinnik A, Amariglio N, Domany E, Rechavi G, Givol D, Itskovitz-Eldor J. Dev Dyn. 2005;232:487–497. doi: 10.1002/dvdy.20247. [DOI] [PubMed] [Google Scholar]
- 19.Shao R, Bao S, Bai X, Blanchette C, Anderson RM, Dang T, Gishizky ML, Marks JR, Wang XF. Mol Cell Biol. 2004;24:3992–4003. doi: 10.1128/MCB.24.9.3992-4003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldsworthy SM, Stockton PS, Trempus CS, Foley JF, Maronpot RR. Mol Carcinog. 1999;25:86–91. [PubMed] [Google Scholar]
- 21.Roy S, Khanna S, Shah H, Rink C, Phillips C, Preuss H, Subbaraju GV, Trimurtulu G, Krishnaraju AV, Bagchi M, et al. DNA Cell Biology. 2005;24:244–255. doi: 10.1089/dna.2005.24.244. [DOI] [PubMed] [Google Scholar]
- 22.Roy S, Khanna S, Kuhn DE, Rink C, Williams WT, Zweier JL, Sen CK. Physiol Genomics. 2006;25:364–374. doi: 10.1152/physiolgenomics.00013.2006. [DOI] [PubMed] [Google Scholar]
- 23.Roy S, Khanna S, Wallace WA, Lappalainen J, Rink C, Cardounel AJ, Zweier JL, Sen CK. J Biol Chem. 2003;278:47129–47135. doi: 10.1074/jbc.M308703200. [DOI] [PubMed] [Google Scholar]
- 24.Verducci JS, Melfi VF, Lin S, Wang Z, Roy S, Sen CK. Physiol Genomics. 2006;25:355–363. doi: 10.1152/physiolgenomics.00314.2004. [DOI] [PubMed] [Google Scholar]
- 25.Roy S, Khanna S, Yeh PE, Rink C, Malarkey WB, Kiecolt-Glaser J, Laskowski B, Glaser R, Sen CK. Gene Expr. 2005;12:273–287. doi: 10.3727/000000005783992025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}