

Abstract
Structure-based design was used to link zinc finger peptides and make poly-finger proteins that have dramatically enhanced affinity and specificity. Our studies focused on a fusion in which the three-finger Zif268 peptide was linked to a designed three-finger peptide (designated “NRE”) that specifically recognizes a nuclear hormone response element. Gel shift assays indicate that this six-finger peptide, 268//NRE, binds to a composite 18-bp DNA site with a dissociation constant in the femtomolar range. We find that the slightly longer linkers used in this fusion protein provide a dramatic improvement in DNA-binding affinity, working much better than the canonical “TGEKP” linkers that have been used in previous studies. Tissue culture transfection experiments also show that the 268//NRE peptide is an extremely effective repressor, giving 72-fold repression when targeted to a binding site close to the transcription start site. Using this strategy, and linking peptides selected via phage display, should allow the design of novel DNA-binding proteins—with extraordinary affinity and specificity—for use in biological research and gene therapy.
Zinc fingers belonging to the Cys2-His2 family constitute one of the most common DNA-binding motifs found in eukaryotes, and these zinc fingers have provided a very attractive framework for the design and selection of DNA-binding proteins with novel sequence specificities. Numerous studies have used phage display methods or design ideas to explore and systematically alter the specificity of zinc finger–DNA interactions (1–7). Structure-based design has been used to link Cys2-His2 zinc fingers with other DNA-binding domains to generate hybrid proteins that recognize extended sites (8, 9), with a GAL4 dimerization domain to develop homo- and heterodimers (10), and with a nuclease domain to generate restriction enzymes (11). [A zinc finger/homeodomain fusion is being tested for potential applications in gene therapy (12).] There also have been several attempts to increase affinity and specificity by adding additional fingers to a three-finger protein (13, 14) or by tandemly linking two three-finger proteins (15). However, these previous design strategies for poly-finger proteins—which all used canonical “TGEKP” linkers to connect the additional fingers—gave relatively modest increases in affinity. Equations describing the chelate effect or the “effective concentration” of the linked peptides (see Discussion) suggested that it should be possible to design poly-finger proteins that bind much more tightly and specifically.
Structural and biochemical analyses show that DNA often is slightly unwound when bound to zinc finger peptides (16–18). Modeling studies confirm that the canonical linker is a bit too short to allow favorable docking of Zif268 on ideal B-DNA (19); the DNA must be slightly unwound to interact with zinc fingers in the mode seen in the Zif268 complex. Essentially, it appears that the helical periodicity of the zinc fingers does not quite match the helical periodicity of B-DNA. Because the strain of unwinding may become a more serious problem when there are more fingers (the helical periodicities of the peptide and DNA may get progressively further out of phase), we decided to test longer, more flexible linkers in the design of poly-finger proteins. Here, we report the design and characterization of six-finger peptides that bind the appropriate 18-bp sites significantly more tightly (>6,000-fold) than three-finger peptides. We also show that these new poly-finger peptides can very effectively repress reporter gene expression in vivo when targeted to a site near the initiator element.
MATERIALS AND METHODS
Plasmid Construction.
Zinc finger expression plasmids used in transfection studies were constructed by PCR amplification of DNA segments encoding the desired fingers of the Zif268 peptide and/or the NRE peptide. These DNA segments were inserted into the HindIII and BamHI sites of pCS, which had been constructed by subcloning an oligonucleotide duplex (5′-AGCTACCATGGCCAAGGAAACCGCAGCTGCCAAATTCGAAAGACAGCATATGGATTCTAAGCTTCGCGGATCCT-3′ + 5′-CTAGAGGATCCGCGAAGCTTAGAATCCATATGCTGTCTTTCGAATTTGGCAGCTGCGGTTTCCTTGGCCATGGT-3′) into the HindIII and XbaI sites of pcDNA3 (Invitrogen). These expression plasmids were designed to produce zinc finger peptides with both an S-peptide tag (20, 21) and a nuclear localization signal from simian virus 40 (SV40) large T antigen (22) at their N terminus. Reporter plasmid were constructed by site-directed mutagenesis by using the QuikChange kit (Stratagene). Construction of the template plasmid (pGL3-TATA/Inr) for the mutagenesis was described previously (23). The DNA sequences of all constructs were confirmed by dideoxy sequencing.
Protein Production and Purification.
The DNA segments encoding the Zif268, NRE, and 268//NRE peptides were amplified by PCR and subcloned into pGEX-6P-3 (Pharmacia). The zinc finger proteins were expressed in Escherichia coli as fusions with glutathione S-transferase (GST) and were purified by using affinity chromatography according to the manufacturer’s protocol. (These constructs did not have an S-peptide tag or an SV40 nuclear localization signal.) GST was subsequently removed by digestion with PreScission Protease (Pharmacia). Protein concentrations were estimated by using SDS/PAGE with BSA as a standard (8). Concentrations of active zinc finger proteins were determined essentially as described (3). These two methods gave comparable results, indicating that almost all of the protein was active.
Gel Shift Assay.
DNA-binding reactions contained the appropriate zinc finger peptide and binding site(s) in a solution of 20 mM Bis-Tris propane, pH 7.0/100 mM NaCl/5 mM MgCl2/20 μM ZnSO4/10% glycerol/0.1% Nonidet P-40/5 mM DTT/0.10 mg/ml BSA in a total volume of 10 μl. All binding experiments were performed at room temperature. The DNA sequences of the binding sites follow: N site, 5′-TCTGC AAGGGTTCA GGCGACACCAACCAA-3′; Z site, 5′-GTGTGTGTGTGATCT GCGTGGGCG GTAAG-3′; NZ site, 5′-TCTGC AAGGGTTCA GCGTGGGCG GTAAG-3′; N/Z site, 5′-TCTGC AAGGGTTCA G GCGTGGGCG GTAAG-3′; and N//Z site, 5′-TCTGC AAGGGTTCA GT GCGTGGGCG GTAAG-3′. (In each case, the 9-bp recognition sequences are underlined.) Labeled DNAs used in gel shift assays were prepared by Klenow extension or kinase reaction.
To determine dissociation constants, 3-fold serial dilutions of the Zif268 or NRE peptide were incubated with a labeled probe DNA (0.4–1.4 pM) at room temperature for 1 h, and then the reaction mixtures were subjected to gel electrophoresis. The radioactive signals were quantitated by PhosphorImager analysis; apparent dissociation constants were determined as described (3).
On-rates and off-rates also were determined by gel shift assay. To initiate the binding reaction when determining on-rate constants, a labeled probe DNA (final concentration, ≈0.4 pM) was added to the zinc finger peptide (final concentration, 5–10 pM) at room temperature, and aliquots were analyzed by gel electrophoresis at various time points (0–20 min). The fraction bound at time t was determined by PhosphorImager analysis of the gels. The data were then fit [kaleidagraph program (Synergy Software)] to the equation:
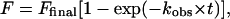 |
where F is the fraction bound at time t; Ffinal is the calculated fraction bound at the completion of the reaction; and kobs is the rate constant (24). The on-rate constant was calculated from the equation:
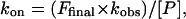 |
where [P] is the concentration of the zinc finger protein. Off-rate constants were determined essentially as described (9). Proteins (final concentration, 100 pM) were preincubated with a labeled probe DNA for 1 h, and then a large excess of unlabeled probe DNA (final concentration, 20 nM) was added. Aliquots were removed at various time points and analyzed by gel electrophoresis. The fraction of labeled site was normalized to the fraction found at the end of the 1-h preincubation period. The natural log of the normalized fraction bound was plotted against time, and the off-rate was determined from the slope. (All data points for fast on-rate and off-rate measurements were corrected for the electrophoresis dead time.)
Competition Binding Studies.
The 268//NRE peptide (final concentration, 5 pM) was first incubated for 1 h with various amounts of a cold competitor DNA (0, 0.05, 0.5, 5, and 50 nM), and then the labeled N/Z site (6–8 pM) was added. Samples were analyzed by gel electrophoresis after 2, 24, 48, 96, 190, and 600 h. Specificity ratios (Kdc/Kd) were calculated from the equation:
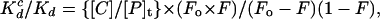 |
where Kdc is the dissociation constant for binding to the competitor DNA; Kd is the dissociation constant for binding to the intact chimeric site; [C] is the concentration of competitor DNA; [P]t is the total concentration of the protein; Fo is the fraction bound in the absence of the competitor DNA; and F is the fraction bound in the presence of the competitor DNA. This equation assumes that the concentration of free protein is significantly smaller than that of protein bound to DNA. [This criterion should readily be satisfied because the Kd of the 268//NRE peptide at the N/Z site is 3.8 fM (see Results), and we used 5 pM of the fusion peptide in these competition experiments.]
Competition experiments with salmon sperm DNA contained the 268//NRE or Zif268 peptide (200 pM), the labeled N/Z site, and a slight molar excess of unlabeled N/Z site. Various amounts of salmon sperm DNA were added, and samples were analyzed by gel electrophoresis after 2, 24, and 48 h of incubation. When calculating specificity ratios, we assumed that each base in the salmon sperm DNA represents the beginning of a potential (nonspecific) binding site.
Transient Cotransfection Assay.
The 293 cells were transfected by calcium phosphate precipitation with a glycerol shock as described (25). Transfection experiments typically used cells at 10–30% confluency in monolayer cultures (six-well plates), and the following plasmids were added: 0.2 μg of the empty expression plasmid (pCS) or of expression plasmids encoding zinc finger peptides; 0.2 μg of a reporter plasmid; 1 μg of activator plasmid (GAL4-VP16); 0.1 μg of β-galactosidase expression plasmid (pCMVβ; CLONTECH); and 2.5 μg of carrier plasmid (pUC19). The luciferase and β-galactosidase activities in the transfected cells were measured as described (9, 23). All the zinc finger peptides expressed in 293 cells were quantitated by using the S⋅Tag Rapid Assay kit (Novagen) (20, 21).
RESULTS
Structure-Based Design of Poly-Zinc Finger Peptides.
Our design strategy involved linking two three-finger peptides, using longer (noncanonical) linkers at the junction to avoid introducing any strain. To further reduce any risk of interference or collision between the fingers, we designed the linkers so they could accommodate composite binding sites with one or two additional base pairs inserted between the individual 9-bp binding sites. Studies reported in this paper used the three-finger Zif268 peptide (which recognizes the site 5′-GCG TGG GCG-3′) and a three-finger “NRE” peptide (a Zif268 variant previously selected via phage display) that binds tightly and specifically to part of a nuclear hormone response element (5′-AAG GGT TCA-3′) (7). The composite target site with one additional base pair at the center has the sequence 5′-AAG GGT TCA G GCG TGG GCG-3′ and is called the N/Z site. (N denotes the binding site for the NRE peptide and Z the binding site for Zif268.) The site with two additional base pairs at the center has the sequence 5′-AAG GGT TCA GT GCG TGG GCG-3′ and is called the N//Z site.
Structure-based design, with the Zif268 complex (16, 19) as a model, was used to determine the appropriate length of linkers for making poly-finger proteins that could recognize each binding site. At the N/Z site, it appeared that having 8 residues between the Leu at the α-helical end of the first peptide and the Tyr residue at the first β-sheet of the next peptide would allow sufficient flexibility. [A canonical “TGEKP” linker has 4 residues (i.e., Gly-Glu-Lys-Pro) in this region.] At the N//Z site, it seemed reasonable to use 11 residues between the Leu and the Tyr (Figs. 1 and 2A). Each linker (Fig. 2A) contained sequences that naturally flank the N terminus and C terminus of the three-finger Zif268 peptide. To allow additional flexibility, a glycine was included in the shorter linker (which still is 4 residues longer than a canonical linker), and a Gly-Gly-Gly-Ser sequence was included in the longer linker (which is 7 residues longer than a canonical linker). Using a notation analogous to that for the binding sites, we denote the fusion protein with the shorter linker as 268/NRE and the fusion protein with the longer linker as 268//NRE.
Figure 1.

Structure-based design of a six-finger peptide, 268//NRE. The cocrystal structure of the Zif268–DNA complex (16, 19) and the template B-DNA (used at the junction) were aligned by superimposing phosphates. In this model, two three-finger peptides bind to corresponding 9-bp sites (bases shown in white) separated by a 2-bp gap (bases shown in gray). Note that the orientation of one three-finger peptide almost exactly matches that of the other three-finger peptide because one helical turn of this underwound DNA contains 11 bp.
Figure 2.

Schematic representations of zinc finger peptides and of reporter constructs used in transfection studies. (A) Zinc finger peptides. Each finger is represented by a circle. The amino acid sequence of a linker in the Zif268 peptide (which has a canonical “TGEKP” linker) is shown, and longer linkers used to connect the three-finger peptides are indicated below. In each case, the box on the left denotes the helical region and includes the second of the conserved His residues of the finger; the zigzag line denotes the first β-sheet of the next finger, which includes the first of the conserved Cys residues. (B) Promoters of luciferase reporter genes. The nucleotide positions of the TATA box, the start codon, and zinc finger binding sites are numbered with respect to the transcription start site (+1).
Gel Shift Assays to Determine Dissociation Constants and Half-Lives of Protein–DNA Complexes.
The Zif268, NRE, and 268//NRE zinc finger peptides were expressed and purified from E. coli and used in several sets of gel shift experiments. A preliminary set of experiments was designed simply to determine whether two three-finger proteins could bind at adjacent 9-bp sites. (Any interference in binding of the unlinked peptides could reduce the affinity of a poly-finger protein for the composite sites.) Our first experiments used a DNA fragment—referred to as the NZ site—with the NRE- and Zif268-binding sites directly juxtaposed (5′-AAG GGT TCA GCG TGG GCG-3′). Various amounts of the NRE peptide were incubated with labeled NZ site in the presence or absence of Zif268 (Fig. 3). We find that the three-finger NRE peptide actually binds slightly more tightly to the NZ site with prebound Zif268 than to the free site. The apparent dissociation constant (Kd) of the NRE peptide is 180 pM when it binds alone but 60 pM when Zif268 is prebound to the neighboring site. Similar results were obtained at the N/Z site. These experiments prove that there is no collision between peptides bound at adjacent sites and suggest that there may even be some modest cooperative effect. It appears that previous limits in the affinity of poly-finger proteins (13–15) must have been due to problems with linker design.
Figure 3.

Gel shift assay. Various amounts (0, 0.01, 0.1, and 1 nM) of the NRE peptide were incubated for 1 h with free binding sites (lanes 1–4) or binding sites preincubated with 0.1 nM of the Zif268 peptide for 0.5 h (lanes 5–8). The positions of the free DNA and the protein–DNA complexes are indicated.
A second set of binding studies confirms the efficacy of our new linker design. Equilibrium titrations show that the 268//NRE peptide has significantly higher affinity for the composite sites than for the individual 9-bp sites (Table 1). The fusion protein binds to the isolated 9-bp sites with Kd values similar to those of the NRE peptide (180 pM) and the Zif268 peptide (14 pM) for their binding sites. In contrast, the 268//NRE fusion protein binds composite sites so tightly that dissociation constants are too small to readily be determined by protein titration. (At least 0.4 pM of labeled probe DNA was needed in these gel shift experiments, making it difficult to accurately determine Kd values of <1 pM.) Given these technical difficulties, we decided to measure the on-rate and off-rate for binding of the 268//NRE peptide and to use these rates to estimate the equilibrium binding constant (Table 1). Parallel studies with the three-finger peptides provided useful controls. On-rates for the 268//NRE, NRE, and Zif268 peptides were fast and were close to the diffusion-controlled limit (108–109 M−1·s−1) (see ref. 26). The off-rates showed amazing differences: The three-finger peptides have half-lives of ≤39 s, whereas the 268//NRE peptide has a half-life of 370 h at the NZ site. Control studies show that the 268//NRE peptide forms a much less stable complex with a single 9-bp site (thus the half-life = 150 s at the N site). Both the NRE fingers and the Zif268 fingers must bind their respective 9-bp subsites to form the extraordinarily stable complex observed with the 268//NRE peptide at the NZ site.
Table 1.
Dissociation constants and rate data
| Protein | Binding site | Kd, pM | kon, M−1·s−1 | koff, s−1 | t1/2, s |
|---|---|---|---|---|---|
| 268//NRE | N | 190 ± 50 | 2.5 ± 0.4 × 107 | 4.7 ± 2.9 × 10−3 | 150 |
| 268//NRE | Z | 10* | |||
| 268//NRE | NZ | <1.0† | 2.5 ± 0.2 × 108 | 5.2 ± 0.9 × 10−7 | 1.3 × 106 |
| 268//NRE | N/Z | <1.0† | 2.5 ± 0.2 × 108 | 9.2 ± 0.7 × 10−7 | 7.5 × 105 |
| 268//NRE | N//Z | <1.0† | 2.6 ± 0.6 × 108 | 7.7 ± 1.3 × 10−7 | 9.0 × 105 |
| NRE | N/Z | 180 ± 43 | >7.3 × 107 | >5.9 × 10−2 | <12 |
| Zif268 | NZ | 12 ± 3 | |||
| Zif268 | N/Z | 14 ± 4 | >7.0 × 108 | 1.4 ± 0.4 × 10−2 | 39 |
| Zif268 | N//Z | 14 ± 1 |
All the constants were determined in at least two separate experiments, and the SEM is indicated.
An exact Kd value could not be determined because this complex gave a smeared band on the gels.
As explained in the text, these Kdvalues could not be measured directly. Estimating Kdfrom the ratio koff/kon gives values of 2.1 fM at the NZ site, 3.7 fM at the N/Z site, and 3.0 fM at the N//Z site.
In all cases where parallel measurements could be performed, Kd values calculated from the ratio of kinetic constants (koff/kon) were in good agreement with those determined from equilibrium studies (Table 1). This gave us confidence in using the kinetic data to determine Kd values in cases where direct titration was impracticable. Calculations show that the 268//NRE peptide has femtomolar affinity for the composite binding sites, with a Kd of 2.1 × 10−15 M (2.1 fM) at the NZ site, 3.7 fM at the N/Z site, and 3.0 fM at the N//Z site. (The consistency of these three Kd values also is encouraging because we would expect that the longer, flexible linker should readily accommodate any of these spacings.) Although we defer a more detailed analysis to Discussion, our data show that the new linker design is quite effective: the 268//NRE fusion peptide binds far more tightly (5,000- to 95,000-fold) to the composite site than to the individual 9-bp sites, and it binds far more tightly (6,000- to 90,000-fold) than either of the original three-finger peptides.
Competition Binding Studies to Measure Affinity and Specificity.
We also used competition experiments to further study the affinity and specificity of the six-finger 268//NRE peptide. One set of experiments directly tested how well the 9-bp N and Z sites could compete with the composite N/Z site for binding to the fusion peptide. In these experiments, various amounts of cold N or Z site were mixed with a limiting amount of the 268//NRE peptide. After 1 h of incubation, we added a slightly molar excess (relative to the total amount of fusion protein) of labeled N/Z site. Under these conditions, about 70% of the labeled DNA is shifted in the absence of competitor DNA (Fig. 4A, lane 2). Samples taken at various time points were analyzed by gel electrophoresis. Because the 268//NRE peptide concentration in this experiment (5 pM) is a few orders of magnitude higher than the peptide’s dissociation constant for the N/Z site, almost all the peptide binds to the N/Z site when no competitor DNA is added. Any decrease in the amount of shifted N/Z site in the presence of competitor DNA reflects binding of the 268//NRE peptide to the competing site. Equilibration in these experiments requires hundreds of hours, and the stability of the purified protein actually becomes a significant concern. (The composite site is added last, and equilibration takes a long time because the fusion protein may encounter cold Z sites hundreds or thousands of times before it first encounters a labeled N/Z site!) After preequilibration with high concentrations of cold N or Z site, we find that the fraction of N/Z label shifted increases steadily with increasing incubation times of up to about 600 h. After 600 h of incubation, a significant fraction of the labeled N/Z site is shifted even in the presence of a 10,000-fold molar excess of cold N or Z site (Fig. 4A, lanes 10 and 14, respectively). Specificity ratios (calculated as described in Materials and Methods) indicate that the 268//NRE peptide prefers the composite site over the N site by a factor of at least 3,800 ± 1,600 and that the fusion peptide prefers the composite site over the Z site by a factor of at least 320 ± 44. These experiments directly confirm the remarkable specificity of the six-finger peptide, but these values are only lower bounds on the specificity ratios. The protein sample loses some activity during the long incubation time required by these experiments (the activity of the free protein has a half-life of about 2 days under these conditions), and denatured protein will never have a chance to shift the labeled N/Z site.
Figure 4.

Competition binding studies. (A) The 268//NRE peptide (5 pM) was preincubated with various amounts (0.05, 0.5, 5, and 50 nM) of cold competitor DNAs (lanes 3–14) for 1 h, and then a slight molar excess (over the peptide concentration) of the labeled N/Z site (6–8 pM) was added to the reaction mixture. Aliquots were analyzed by gel electrophoresis at various time points, and this gel shows the result after 600 h of incubation at room temperature. (B) The 268//NRE (lanes 2–6) or Zif268 peptide (lanes 7–11) was mixed with the labeled N/Z site, a slight molar excess (over the peptide concentration) of unlabeled N/Z site was added (so that ≈70% of the labeled site would be shifted in the absence of salmon sperm DNA), and various amounts of salmon sperm DNA (0, 0.1, 1, 10, and 100 μg/ml) were included. Samples were analyzed by gel electrophoresis after 24 h of incubation.
Competition experiments with salmon sperm DNA were used to estimate the ratio of specific/nonspecific binding constants for the 268//NRE peptide (Fig. 4B). These experiments showed that the 268//NRE peptide discriminates very effectively against nonspecific DNA and indicate a specificity ratio (Kdns/Kd) of 8.8 ± 1.5 × 106. Parallel experiments with the three-finger Zif268 peptide give a specificity ratio of 1.2 ± 0.1 × 105. [Previous studies, using calf thymus DNA as a competitor and slightly different conditions, had given a specificity ratio of 0.31 × 105 for the Zif268 peptide (7).] Taken together, our data on the affinity and specificity of the six-finger 268//NRE fusion peptide suggested that it might serve as a very effective repressor and certainly indicated that it would be an excellent candidate for further analysis in vivo.
Transcriptional Repression in Vivo with Our Six-Finger Peptides.
We used transient cotransfection studies in the 293 human cell line to see whether our new poly-finger peptides could effectively repress transcription from reporter genes. In a previous study, we had shown that the Zif268 peptide could efficiently repress both basal and VP16-activated transcription when the Zif268 peptide bound to a site near the TATA box or the initiator element (23). In this current study, we used a luciferase reporter and similar promoter constructs in which appropriate binding sites (Z, N, N/Z, and N//Z) were incorporated at comparable positions near the initiator element (Fig. 2B).
We find that the 268//NRE peptide gives 72-fold repression of VP16-activated transcription at a promoter containing the N/Z site and 47-fold repression at a promoter containing the N//Z site (Fig. 5). The 268/NRE peptide gives 68-fold repression at the N/Z site. Clearly, these fusion peptides are very effective repressors at sites with the appropriate spacings. Parallel experiments with the three-finger peptides show repression but indicate that they are considerably less effective than the fusion peptides. Thus, the NRE peptide gives 1.9-fold repression with an N site in the promoter; 1.8-fold repression with an N/Z site; 2.7-fold repression with an N//Z site; and no repression with an isolated Z site. The Zif268 peptide gives 13-fold repression from the Z promoter; 8.9-fold repression from the N/Z promoter; 15-fold repression from the N//Z promoter; and no repression with an isolated N site. Further experiments prove that covalent coupling is needed to achieve the much higher repression levels obtained with the fusion proteins at the N/Z site. Thus, coexpressing the Zif268 and NRE peptides as separate polypeptide chains (by including both expression plasmids in the cotransfection assays) gives only 8.5-fold repression at the N/Z site, a level comparable (within experimental error) to the 8.9-fold repression obtained at this site with the isolated Zif268 peptide. This is far less than the 68-fold and 72-fold repression that the 268/NRE and 268//NRE fusion proteins give at the N/Z site, and it is clear that these “synergistic” effects require covalent linkage.
Figure 5.

Transcriptional repression in vivo by zinc finger peptides. Human 293 cells were transfected as described (25) by using the calcium phosphate precipitation method. Luciferase and β-galactosidase activities were measured 48 h later. The luciferase activities were divided by corresponding β-galactosidase activities to yield the relative luciferase activities. Repression levels (fold repression) were obtained by dividing (i) the relative luciferase activities from the cells transfected with the empty expression plasmid by (ii) those from the cells transfected with zinc finger expression plasmids. Note different scales used in graphs for the different reporters. [The 68/NR, 68/NRE, 68//NR, and 68//NRE peptides are variants of our six-finger fusion proteins that are missing one or two of the terminal fingers. Thus, the 68/NR peptide contains fingers 2 and 3 of the Zif268 peptide fused (via the shorter of our two linkers) to fingers 1 and 2 of the NRE peptide (see Fig. 2A for comparison with the six-finger constructs).] The data represent an average of three independent experiments, and the SEM is shown.
We note in passing that the additional fingers in the fusion peptides may have some modest repressive effects even in cases where only three of the fingers can bind specifically. Thus, the six-finger peptides (268/NRE and 268//NRE) give 21- to 23-fold repression from the Z promoter. A similar (22-fold) repression level is obtained with the 268/NRE peptide at the N//Z site. (Modeling suggests that the linker is too short to allow specific binding of all six fingers at this site.) These repression levels are consistently somewhat higher than the level observed with the isolated Zif268 peptide at the Z site (13-fold repression). It seems possible—when the 268//NRE peptide binds to the Z site—that (i) the NRE fingers are free and yet sterically interfere with assembly of the transcription complex or that (ii) the NRE fingers make weak, nonspecific contacts with the DNA and thus slightly enhance the stability of the complex. [Note: Further studies indicate that all peptides are expressed at comparable levels. The zinc finger peptides expressed in 293 cells had an S-peptide tag, and we quantitated the amount of peptide by using a ribonuclease assay after activating with S-protein (20, 21). A conservative estimate indicates that the expression levels of the peptides in cells are significantly higher (at least 100-fold) than the dissociation constants of the three-finger peptides.] Although they are not central to the main conclusions of this paper, we also constructed plasmids that would encode four- and five-finger variants of the 268/NRE and 268//NRE peptides. These were tested in tissue culture transfection studies, and they typically gave repression levels intermediate between those obtained with the three-finger peptides and those obtained with the six-finger peptides (Fig. 5).
DISCUSSION
Studies of Cys2-His2 zinc finger proteins have shown a wide variation in the number of fingers per protein [the Xenopus Xfin protein (27) has 37!], and there are interesting questions about how many fingers actually contact the DNA and how this affects the affinity and specificity of recognition. Zif268 has three fingers, and each finger makes a critical set of base contacts (16, 19). The human glioblastoma protein has five fingers, but only two of these (fingers 4 and 5) make extensive base contacts (28). TFIIIA has nine fingers, but a peptide containing fingers 1–3 binds DNA almost as tightly as the intact protein, and fingers 4–7 seem to be especially important for RNA binding (29, 30). Many additional examples could be cited, and further research certainly is needed, but it appears that fingers in these natural poly-finger proteins can play a diversity of other roles. Not every finger will—like the fingers of Zif268—make a set of sequence-specific base contacts.
Analogous questions arise in design. Can we increase the affinity and specificity of designer proteins by simply adding more fingers? Can poly-finger proteins be designed in a way that allows each finger to make a full set of base contacts? Simple arguments from physical chemistry suggest that dramatic increases in DNA-binding affinity might be obtained by using the “chelate effect” and covalently linking two three-finger peptides. Such fusion proteins should recognize extended (18-bp) binding sites and thus may bind to a unique site (or a small number of sites) in the human genome (15). As observed with the two subdomains of the Oct-1 POU domain (31), covalent linkage of two three-finger peptides should facilitate binding via a chelate effect that maintains a high local concentration of the two DNA-binding modules. (Whenever one set of fingers is near the DNA, the linker will tether the other set of fingers to a nearby region of space and hold it—at a high local concentration—near the DNA.) Using an equation analogous to that used in studying Oct-1 (31), a value for the effective concentration in this system can be calculated as:
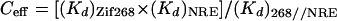 |
where (Kd)Zif268 is the dissociation constant of the three-finger Zif268 peptide (12 pM); (Kd)NRE is the dissociation constant of the three-finger NRE peptide in the presence of bound Zif268 peptide (60 pM); and (Kd)268//NRE is the dissociation constant of the fusion protein at the NZ site (2.1 fM). Substituting these values shows that the “effective concentration” of either peptide in an intermediate complex (when one set of fingers is bound to the DNA but the other set is still free) is 0.36 μM. Because this effective concentration is significantly larger than the dissociation constant of either three-finger module, it appears that any intermediate complex (with only three of the six fingers bound) will be thermodynamically unstable and will only be present as a transient intermediate, as during binding or release of the 268//NRE peptide at a composite binding site. The flexible linker clearly makes a significant contribution to binding. However, this estimate for the effective concentration (0.34 μM) also raises some interesting questions. One might expect—based on the length of the linker and on analogy with the Oct-1 POU domain—that a short, flexible linker would give an effective concentration in the millimolar range and thus give an even higher binding constant. Further experiments will be necessary to explore these issues, but it is possible (i) that there still is some strain in the linker region in our new designs (although this seems unlikely because binding of the 268//NRE peptide to the NZ, N/Z, and N//Z sites is so similar); (ii) that there is some adventitious alternative interaction competing with DNA binding (such as favorable contacts between the two three-finger peptides); or (iii) that our measurements have significantly underestimated the lifetime of the complex with the fusion protein at the hybrid site and that the fusion protein actually binds more tightly than we have estimated. (For example, it is conceivable that the fusion protein almost never really “dissociates” but moves from a labeled to an unlabeled DNA site by temporarily forming a bridging complex with three fingers bound to each DNA fragment. Determining whether the apparent half-life depends on the concentration of cold DNA should help clarify this issue.)
Our new six-finger peptides bind far more tightly than previously reported poly-finger proteins that used a conventional “TGEKP” linker to connect two three-finger modules or to add additional fingers to a three-finger protein. Poly-finger proteins with canonical linkers had been tested by Rebar (13), by Shi (14), and by Liu et al. (15). Each study compared binding of the new poly-finger protein (at the appropriate extended site) with binding of the original three-finger peptide. Using canonical linkers, a four-finger peptide bound 6.3 times more tightly than the corresponding three-finger peptide (13), a five-finger construct showed no improvement in Kd over the original three-finger peptide (14), and six-finger peptides bound 58- to 74-fold more tightly than the corresponding three-finger peptides (15). In contrast, our peptides bind 6,000- to 90,000-fold more tightly than the original three-finger peptides. It seems clear that the longer linkers used in our 268/NRE and 268//NRE constructs must relieve some strain that accumulates when a larger set of fingers all are connected with canonical linkers. (Presumably this involves a slight mismatch in the helical periodicity of the DNA and the preferred helical periodicity of the zinc fingers, causing them to fall slightly out of register when four or more fingers are connected via canonical linkers.)
It is interesting to note that structure-based design fails to reveal any obvious problem with connecting additional modules via a canonical “TGEKP” linker. We had used explicit computer modeling in design of our four-finger peptide (13), and Liu et al. (15) also used explicit modeling in design of their six-finger constructs. This experience seems to highlight some subtle difficulties with structure-based design. Juxtaposing two substructures [as we have done in the design of the zinc finger/homeodomain fusion (8), the zinc finger/TBP fusion (9), zinc finger/GAL4 dimerization domain fusions (10), and these zinc finger/zinc finger fusions] can suggest a reasonable overall arrangement for the chimeric protein and the hybrid DNA-binding site. However, there is an inherent limit to the accuracy of these structural predictions: One cannot predict the exact three-dimensional structure of the fused DNA site or of the covalently connected protein. In linking the proteins, it seems risky to make overly precise (but possibly inaccurate!) predictions, which may introduce strain. Including some additional flexibility in the linker regions may help account for the uncertainty (on the 1–2 Å level?) inherent in the design process. We certainly believe that having longer linkers, which can accommodate for uncertainty in the precise spacing of the three-finger modules, accounts for the success of our 268/NRE and 268//NRE constructs. Having some leeway at this junction may also make our design more versatile for use with a wide variety of related fingers and sites.
Covalently linked poly-finger proteins that bind with such remarkable affinities allow us to pose many interesting questions about gene regulation, evolution of DNA-binding proteins, and gene therapy. Our data clearly show that such fusion proteins can function effectively as repressors, and this might be further enhanced by the addition of repression domains (15). In thinking about the in vivo studies, we also note that expressing our zinc finger peptides at lower levels might enhance the relative advantage of the six-finger peptides over the corresponding three-finger peptides. Further experiments also will be needed to determine precisely what advantages (or disadvantages) may be inherent in having DNA-binding proteins that bind with such remarkably long half-lives. One wonders: Do any of the natural zinc finger proteins make such an extensive set of contacts and bind with such extraordinary affinities? Can covalently linked proteins—such as these new poly-finger proteins—achieve a level of specificity comparable to that achieved via “combinatorial control” in typical biological regulatory circuits? Is a graded regulatory response harder to achieve when there is a single protein that binds with such unusual avidity? Our fusion proteins will allow us to address many fundamental questions about gene regulation and will provide powerful new reagents for potential applications in gene therapy, including targeted repression of viral genomes.
Acknowledgments
We thank H. A. Greisman and S. T. Smale for kindly providing plasmids used in these studies, A. Dunn for help preparing the manuscript, and P. A. Sharp for generously allowing us to use tissue culture facilities in the Center for Cancer Research at the Massachusetts Institute of Technology. Modeling studies used equipment purchased with support from the Pew Charitable Trusts, and this research was supported by the Howard Hughes Medical Institute.
References
- 1.Desjarlais J R, Berg J M. Proteins Struct Funct Genet. 1992;12:101–104. doi: 10.1002/prot.340120202. [DOI] [PubMed] [Google Scholar]
- 2.Desjarlais J R, Berg J M. Proc Natl Acad Sci USA. 1993;90:2256–2260. doi: 10.1073/pnas.90.6.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rebar E J, Pabo C O. Science. 1994;263:671–673. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 4.Jamieson A C, Kim S-H, Wells J A. Biochemistry. 1994;33:5689–5695. doi: 10.1021/bi00185a004. [DOI] [PubMed] [Google Scholar]
- 5.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163–11167. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H, Yang W-P, Barbas C F., III Proc Natl Acad Sci USA. 1995;92:344–348. doi: 10.1073/pnas.92.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greisman H A, Pabo C O. Science. 1997;275:657–661. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- 8.Pomerantz J L, Sharp P A, Pabo C O. Science. 1995;267:93–96. doi: 10.1126/science.7809612. [DOI] [PubMed] [Google Scholar]
- 9.Kim J-S, Kim J, Cepek K L, Sharp P A, Pabo C O. Proc Natl Acad Sci USA. 1997;94:3616–3620. doi: 10.1073/pnas.94.8.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pomerantz J L, Wolfe S A, Pabo C O. Biochemistry. 1998;4:965–970. doi: 10.1021/bi972464o. [DOI] [PubMed] [Google Scholar]
- 11.Kim Y-G, Cha J, Chandrasegaran S. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivera V M, Clackson T, Natesan S, Pollock R, Amara J F, Keenan T, Magari S R, Phillips T, Courage N L, Cerasoli F, Jr, et al. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 13.Rebar E J. Ph.D. thesis. Cambridge: Massachusetts Institute of Technology; 1997. [Google Scholar]
- 14.Shi Y. Ph.D. thesis. Baltimore, MD: Johns Hopkins University; 1995. [Google Scholar]
- 15.Liu Q, Segal D J, Ghiara J B, Barbas C F., III Proc Natl Acad Sci USA. 1997;94:5525–5530. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavletich N P, Pabo C O. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Berg J M. Biochemistry. 1996;35:3845–3848. doi: 10.1021/bi952384p. [DOI] [PubMed] [Google Scholar]
- 18.Nekludova L, Pabo C O. Proc Natl Acad Sci USA. 1994;91:6948–6952. doi: 10.1073/pnas.91.15.6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elrod-Erickson M, Rould M A, Nekludova L, Pabo C O. Structure. 1996;4:1171–1180. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 20.Kim J-S, Raines R T. Protein Sci. 1993;2:348–356. doi: 10.1002/pro.5560020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J-S, Raines R T. Anal Biochem. 1995;219:165–166. doi: 10.1006/abio.1994.1251. [DOI] [PubMed] [Google Scholar]
- 22.Kalderon D, Roberts B L, Richardson W D, Smith A E. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 23.Kim J-S, Pabo C O. J Biol Chem. 1997;272:29795–29800. doi: 10.1074/jbc.272.47.29795. [DOI] [PubMed] [Google Scholar]
- 24.Hoopes B C, LeBlanc J F, Hawley D K. J Biol Chem. 1992;267:11539–11547. [PubMed] [Google Scholar]
- 25.Cepek K L, Chasman D I, Sharp P A. Genes Dev. 1996;10:2079–2088. doi: 10.1101/gad.10.16.2079. [DOI] [PubMed] [Google Scholar]
- 26.von Hippel P H, Berg O G. J Biol Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 27.Ruiz i Altaba A, Perry-O’Keefe H, Melton D A. EMBO J. 1987;6:3065–3070. doi: 10.1002/j.1460-2075.1987.tb02613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavletich N P, Pabo C O. Science. 1993;261:1701–1707. doi: 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
- 29.Clemens K R, Zhang P, Liao X, McBryant S J, Wright P E, Gottesfeld J M. J Mol Biol. 1994;244:23–35. doi: 10.1006/jmbi.1994.1701. [DOI] [PubMed] [Google Scholar]
- 30.Clemens K R, Wolf V, McBryant S J, Zhang P, Liao X, Wright P E, Gottesfeld J M. Science. 1993;260:530–533. doi: 10.1126/science.8475383. [DOI] [PubMed] [Google Scholar]
- 31.Klemm J D, Pabo C O. Genes Dev. 1996;10:27–36. doi: 10.1101/gad.10.1.27. [DOI] [PubMed] [Google Scholar]