

Abstract
We have developed a simple procedure to identify protein homologs in genomic databases. The program, called orf, is based on comparisons of predicted secondary structure. Protein structure is far better conserved than amino acid sequence, and structure-based methods have been effective in exploiting this fact to find homologs, even among proteins with scant sequence identity. orf is a secondary structure-based method that operates solely on predictions from sequence and requires no experimentally determined information about the structure. The approach is illustrated by an example: Thymidylate synthase, a highly conserved enzyme essential to thymidine biosynthesis in both prokaryotes and eukaryotes, is thought to be used by Archaea, but a corresponding gene has yet to be identified. Here, a candidate thymidylate synthase is identified as a previously unassigned open reading frame from the genome of Methanococcus jannaschii, viz., MJ0757. Using primary structure information alone, the optimally aligned sequence identity between MJ0757 and Escherichia coli thymidylate synthase is 7%, well below the threshold of sensitivity for detection by sequence-based methods.
At least 12 genomes now have been sequenced from diverse organisms, with many additions anticipated in coming weeks. How can this wealth of information best be used to address fundamental questions in biology? In particular, how can related protein domains be identified among organisms that diverged during the Cambrian explosion or earlier (1)? The mechanism of protein evolution gives rise to homologous sequences, with attendant redundancy. Computational biologists have exploited this fact in developing powerful recognition tools. Among these, sequence-based methods (2) to recognize homologs are well developed, but sensitivity falters as sequence similarity sinks into the “twilight zone,” a threshold near 30% sequence identity (3). Sensitivity can be extended by using information from multiple aligned sequence families (4, 5), local multiple alignment of blocks (6–9), and structure-based fold recognition such as threading (ref. 10 and references therein) and profiles (11).
Here we present a procedure for homolog recognition based on secondary structure prediction. The method is implemented in a computer program called orf, an acronym for Ostensible Recognition of Folds. Unlike many other fold recognition approaches, orf requires no three-dimensional template. In brief, orf operates solely on sequence information to predict the secondary structure of both an unknown protein and all entries in a database of interest and then uses this information in a query-against-all alignment to select likely candidates. The strategy is based on a simple idea: although sequence space is vast, the number of conceivable protein folds is small, of order 5,000 or fewer (12–15). Typically, such folds can be parsed into a linear sequence of repetitive secondary structure elements interconnected by intervening nonrepetitive regions (i.e., helices, β-strands, and everything else), whose orientation in three-dimensions establishes the fold. The order and size of these elements are expected to be similar in homologous proteins. The converse proposition—viz., elements of similar order and size imply similar proteins—is a likely conjecture (16–18), and we adopt it here.
To demonstrate the approach, we applied orf to a challenging problem, the identification of a thymidylate synthase (TS) from the archeon Methanococcus jannaschii. TS, an ancient and highly conserved enzyme (19), is essential in thymidine biosynthesis. Other enzymes in this pathway appear to have been identified in M. jannaschii [e.g., see The Institute for Genomic Research (TIGR) web site: http://www.tigr.org], and Archaea are believed to use a TS (20), but an authentic gene for the enzyme has yet to be documented in the literature.
By using orf, we have identified MJ0757 as a likely TS in M. jannaschii. The sequence identity between MJ0757 and TS in Escherichia coli is 11% when secondary structure is used to guide alignment (and only 7% from sequence alone). Once identified, a candidate of interest can be validated by methods that are independent of the orf search procedure. Accordingly, supporting evidence is presented, and a three-dimensional model is developed. Our hypothesis that MJ0757 is an authentic TS in M. jannaschii now awaits the attention of experimentalists, who alone can assess its validity.
METHODS
In orf, the secondary structure of a query protein is compared with that of all test proteins in a database of interest. An a-b-c classification of secondary structure is effected, where “a” is α-helix, “b” is β-strand, and “c” is coil (i.e., all else). Then, optimal pairwise secondary structure alignment of query and test proteins is performed, using dynamic programming (21). It remains an open question whether knowledge of the order and size of secondary structure elements is sufficient to identify a three-dimensional fold uniquely. Assertions to the contrary notwithstanding (22), helix capping studies, which reveal a link between secondary and supersecondary structure (23), are suggestive.
In systematic validation tests to be published elsewhere (unpublished work), orf was applied initially to a set of diverse folds, using secondary structure assignments extracted from known three-dimensional structures (i.e., observed vs. observed matches). Comparisons among proteins with similar architecture resulted in high scores, with few false-positives, and comparisons between proteins with differing architecture exhibited low scores, with few false-negatives. In greater detail, the entire Protein Data Bank (24), including 22 different representative folds, was reduced to its corresponding a-b-c sequence by identifying secondary structure from coordinates and then rewriting the sequence in an a-b-c alphabet. Then, each representative fold was used, in turn, as a template to search the full set. False-positives are rare above a search score of +60 in a scale ranging from −100 to +100 (using the scoring matrix given below), and all are eliminated upon inclusion of a length filter that excludes candidates differing by more than ±20%. For example, the PDB includes a large number of two-helix fragments that align well against a subsequence in globins, but these false-positives are mismatched conspicuously in overall size. At this level of stringency, recovery of true positives ranges between 30 and 60% for the 22 representative folds, when the reference set of all like folds is taken to be those identified by vast (10). Almost all false-negatives can be attributed either to incomplete structures (e.g., only α-carbon coordinates or regions of missing density) or domain insertions that cause frame-shifts.
Next, tests were redone with predicted secondary structure assignments (i.e., predicted vs. predicted matches) by using the gor prediction method (25). However, the approach does not depend critically on the choice of gor, and we confirmed that several other currently available alternatives would have sufficed. Following the previous procedure for observed vs. observed matches, the PDB, including all 22 representative folds, was reduced to its corresponding a-b-c sequence, but in this instance secondary structure assignments were obtained from predictions based solely on the sequence; coordinates were ignored. Results were similar to those realized previously in observed vs. observed matches. In particular, false-positives are rare above a score of 60, and nearly all are eliminated by a size filter. Recovery of true-positives for the 22 representative folds ranges between 80 and 125% relative to observed vs. observed. Using this approach, many structural homologs with sequence identity as low as 5–10% were detected, regardless of the fold.
It is important to emphasize that the issue of prediction accuracy is not germane when assessing similarity between predicted structures. On first consideration, this crucial point may seem surprising. However, prediction accuracy is a measure of predicted vs. observed matches, whereas homolog identification by orf depends instead on the accuracy of predicted vs. predicted matches. At an extreme, an observed strand that is incorrectly but consistently predicted to be a helix in both a target of interest and its homologs would still be discriminatory.
Similar folds can embody dissimilar function. For this reason, secondary structure comparison is only used as a first-level screen. Structurally related candidates identified by orf are subjected to further, independent tests, such as identification of known functional “landmarks” (e.g., residues required for binding and/or catalysis). Although such additional validation can be time-consuming, it remains practicable because only a handful of likely candidates survive the initial orf screen. The putative TS from M. jannaschii identified here illustrates both steps in the approach.
Alignment and Scoring.
For an N-residue sequence, predicted secondary structure is represented as a string of length N in the three-letter alphabet: a, b, and c. Alignment of two strings, corresponding to query and test sequences, was performed using dynamic programming (21), with the score matrix:
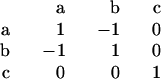 |
Gaps in a and b were penalized by 2 points; those in c by 1 point. Using these values, scores were sorted and alignments were evaluated. The percentage of sequence identity was calculated from the optimally aligned secondary structures.
Search for TS in the M. jannaschii Genome.
The database of M. jannaschii orfs was obtained from The Institute for Genomic Research (TIGR) web site: http://www.tigr.org. Searches were performed by using the TS sequence of both yeast and E. coli. MJ0757 scored high in each search, ranking fourth and fifth, respectively (81 for yeast and 63 for E. coli.) The number of likely candidates is a steep function of aligned sequence identity. For example, in yeast, the distribution of candidates by percentile includes one in the 90th percentile, four in the 80th (including MJ0757), none in the 70th, 39 in the 60th, 132 in the 55th, and so on. Further examination of the top candidates eliminated all but three, based on size. Of these, only MJ0757 was found to have a residue corresponding to the catalytic cysteine.
Model Building.
The program look (Molecular Applications Group, Palo Alto, CA) was used to build a three-dimensional model of MJ0757 from the x-ray-elucidated E. coli structure (ref. 26; PDB file 2TSC, chain A, 24). Structural alignment based on predicted secondary structure and multiple sequence alignment from clustal w (27) were used to guide model building. The extent to which hydrophobes are sequestered and polars are exposed was assessed from this initial model. The model then was refined by using 100 cycles of minimization in look followed by further minimization and simulated annealing by using x-plor (28). Coordinates for the final model are available on our web site (http://cherubino.med.jhmi.edu).
RESULTS
M. jannaschii is an obligate anaerobe isolated from a deep-sea vent (29). As the name implies, these archaebacteria are methanogenic, with a normal growth temperature of ≈85°C, and their proteins are thermostable. The M. jannaschii genome has been sequenced by Bult et al. (30). To our knowledge, previous sequence-based searches of all open reading frames failed to identify a TS.
Do Archaea Have a TS?
TS, a methyl-transferase, converts dUMP to TMP. TMP is essential in DNA synthesis, and therefore TS is believed to be present in all wild-type cells. However, a TS has yet to be documented in archaebacteria. In both eubacteria and eukaryotes, the methyl group is transferred from tetrahydrofolate to dUMP by TS, generating TMP and dihydrofolate. The folate cofactor is not present in most members of the Archaea domain, prompting the existence of a normal TS to be questioned (31). However, methanogens are known to use methanopterin, a modified folate (31). Studies using 13CO2 in a number of methanogens find that the pyrimidine biosynthesis pathway inferred from labeling patterns resembles corresponding pathways in eubacteria and eukaryotes (32). Recently, Nyce and White (33) reported the presence of a TS activity in cell lysates of the archaebacteria M. thermophila and Sulfolobus solfataricus. Finally, homologs of all other enzymes required for pyrimidine biosynthesis appear to have been identified in the M. jannaschii genome, with the sole exception of TS.
An ongoing effort to purify TS from Archaea is documented in the literature. TS activity in Methanobacterium thermoautotrophicum was identified previously by tritium exchange (34), and an N-terminal, 30-residue fragment of the protein was obtained. However, conversion of dUMP to TMP could not be demonstrated in purified fractions. Later, Vaupel et al. (35), analyzing a clone of N5,N10-methenyltetrahydromethanopterin cyclohydrolase, encoded by the gene mch in M. thermoautotrophicum, identified an upstream open reading frame with an N-terminal sequence identical to the one obtained by Krone et al. (34). This finding was poignant because, in both eubacteria and eukaryotes, TS is a tetrahydrofolate-dependent enzyme and genes for TS and dihydrofolate reductase are polycistronic. Vaupel et al. (35) tentatively identified this upstream reading frame as tysY, the gene for TS. The corresponding tysY gene in M. jannaschii has been assigned as MJ0511 (30), but the evidence is ambiguous. The mch gene appears to be transcribed monocistronically (35). Furthermore, MJ0511 does not score well as a TS, either by our search criteria or by multiple sequence alignment. We conjecture that MJ0511 is likely to be a homolog of deoxyuridylate-hydroxymethyltransferase, a related enzyme found in bacteriophage SPO1 (36). Although Archaea are not known to incorporate hydroxymethyluracil in their DNA, the MJ0511 protein may be implicated in tRNA methylation.
Together, the preceding considerations indicate that a TS in M. jannaschii will have scant sequence similarity to known homologs, if indeed the enzyme is present at all. Thus, the situation provides an inviting test case for orf, which, in turn, identified MJ0757 as a likely TS candidate.
Evidence that MJ0757 Is a TS.
TS structures from both Lactobacillus casei and E. coli have been solved by x-ray crystallography (26, 37). Although the two enzymes differ in size (318 residues in L. casei, 262 residues in E. coli), the two structures have nearly identical architecture. The E. coli TS structure is shown as a cartoon in Fig. 1, and its sequence and observed secondary structure are aligned against MJ0757 (260 residues in M. jannaschii) in Fig. 2. The secondary structure of MJ0757 predicted by both gor and phd (38) is included in Fig. 2.
Figure 1.

Cartoon of E. coli TS. A ribbon diagram of E. coli TS, with helices in red, strands in yellow, turns in blue, and coil in white (PDB file 2TSC, chain A, 26). The active site cleft, on the left, is shown with bound dUMP (thick wireframe) and anti-folate CB3717 (thin wireframe).
Figure 2.

Secondary structure alignment of E. coli TS and MJ0757. Sequence alignment (in single-letter code) generated by orf, based on predicted secondary structure. Observed secondary structure (blue), from x-ray coordinates used in Fig. 1, is shown above the E. coli sequence, with α-helices indicated by hatched boxes and β-strands by arrows. Secondary structure predicted by both gor (25) (blue) and phd (gray) (38) is shown below the MJ0757 sequence. The position of the catalytic cysteine is highlighted in yellow. Residues are numbered for convenience.
Evidence that the functionally important sites in TS are found in MJ0757 is described below and summarized in Table 1. In each case, the identification of such residues depends solely on optimal alignment of predicted secondary structure; no further restraint has been imposed. We adopted the convention that TS sequence numbers are keyed to the E. coli enzyme, with alternative L. casei numbering given in square brackets.
Table 1.
Equivalent residues in TS from L. casei, E. coli, and M. jannaschii identified by structural alignment
Cys146 [198] is the signature catalytic residue in TS. The thiol acts as a nucleophile that attacks C-6 of dUMP and activates the C-5 carbon for condensation with cofactor (19). After secondary structure alignment, Cys146 [198] coincides precisely with Cys152 in MJ0757. Among other residues important for dUMP binding and catalysis are two invariant arginines that correspond to an Arg and a Lys in MJ0757. Similarly, residues that bind the PABA ring of folate/methanopterin also are conserved. On the other hand, Tyr209 [261], another invariant residue, fails to align with a tyrosine in the MJ0757 sequence, although two tyrosines (viz., 201, 202) are situated nearby.
Residues that bind the pterin ring of the folate cofactor are not conserved in MJ0757. We suspect that this fact can be ascribed to differences between folate and methanopterin. In this regard, it is noteworthy that hydrogen bonds between the enzyme and the pterin ring are provided by backbone atoms, and, thus, they are not residue-specific. However, the two tryptophans that pack against the ring are conspicuously absent in the M. jannaschii candidate.
Multiple sequence alignment was performed, using TS sequences from Bacillus subtilis, Saccharomyces cerevisiae, and L. casei together with MJ0757. Results from the program clustal w (27) with default parameters are shown in Fig. 3. Again, residues essential to catalysis coincide, despite the fact that overall sequence identity is low. The best alignment to MJ0757 is obtained with yeast TS, where sequence identity is 16.8%.
Figure 3.

Multiple sequence alignment of TS with MJ0757. Optimal alignment of TS sequences B. subtilis, E. coli, S. cerevisiae, L. casei, and MJ0757 was generated with clustal w (27), using default parameters. The catalytic Cys and other residues from Table 1 are highlighted in yellow. The 17 absolutely conserved residues are annotated by an asterisk, and other strongly conserved residues are marked by a dot.
As a further independent check, the MJ0757 backbone was threaded onto E. coli TS (see Methods). The solvent accessibility of polar and hydrophobic residues was assessed in the model and appears to be plausible (Fig. 4), and the binding cavity was preserved upon addition of sidechains to the backbone model. It has been observed that proteins from thermophiles are enhanced in internal salt bridges (refs. 39 and 40 and reference therein) as well as larger side chains that facilitate tighter packing of the core and thereby promote stabilization at higher temperatures. This trend is observed in the TS model of MJ0757, with a higher proportion of β-branched residues in β-sheets than observed in the TS of either E. coli or L. casei. Also, two internal salt bridges are present although no attempt to restrain these residues was imposed on the model.
Figure 4.

Three-dimensional model of MJ0757. Using the program look 2.0, the MJ0757 sequence was threaded onto the E. coli structure. Side chain placement was refined in look 2.0 and further refined in x-plor (28). The model is visualized in rasmol (53) and color-coded by residue property: hydrophobic (V, I, L, M, F, W, and C), polar (S, T, N, and Q), special backbone (A, P, and G), positively charged (K and R), and negatively charged (D and E). The E. coli x-ray structure (Upper) and MJ0757 modeled structure (Lower) are shown separately for comparison.
Finally, in analogy to two-dimensional gel electrophoresis (where molecular weight vs. pI are used as identifiers), the pI of MJ0757 is calculated to be 5.4, near that of TS from E. coli (pIcalc = 5.5) and L. casei (pIcalc = 5.3). In general, pI values ranging from 4.7 to 5.2 have been reported for TS (41, 42). The combined filters of molecular mass and pI can bracket a protein of interest within a surprisingly narrow range. In the M. jannaschii genome, there are only 15 open reading frames with a calculated pI between 5.0 and 5.7 that lie in the size range 29.5–31.0 × 103 Da.
Issues of speed are of concern when searching whole genomes. In its current implementation on a workstation of medium speed (SGI R4400 Indigo), orf can search the M. jannaschii genome in 7–8 min and GenBank, which contains a quarter-million open reading frames, in 8 h.
DISCUSSION
We have developed a search tool, called orf, that uses predicted secondary structure to detect protein homologs with scant sequence identity. To illustrate the method, a candidate TS (MJ0757) was identified in an ancient organism, M. jannaschii. Once identified, the validity of a candidate can be assessed independently, without further involvement of the search tool. Indeed, when such an assessment is made, many TS residues known to be essential in catalysis and binding are found to be conserved in the identified candidate. The optimally aligned sequence identity between MJ0757 and any TS of known structure is well below the threshold required for detection by traditional sequence-based methods.
Two related structure-based approaches to fold recognition have met with considerable success: profiles and threading. Profile-based methods (see, e.g., refs. 11 and 43) are based on a property matrix (i.e., a profile) that is computed for a query sequence and each entry in a library of known structures. Typical properties include secondary structure, solvent accessibility, and residue contact energies. Dynamic programming then is used to identify the optimal match between the profile value of the query sequence and corresponding values for entries from the library. A related approach based on hidden Markov models uses aligned multiple sequence families to develop a statistical profile (44).
In threading, a query sequence is built (i.e., threaded) onto a template of known structure (10, 45). Typically, a library of such templates is tried, and each is evaluated and ranked by using a pseudo-energy potential. Templates with sufficiently favorable scores represent preferred matches. Recent analysis by several groups has raised questions about the validity of threading potentials (46–49). Nevertheless, profiles and threading emerged as the most successful predictive methods in the recent Critical Assessment of Structure Prediction (CASP2) meeting (50). The performance of both methods improves with increased structural similarity between the query sequence and the template (50).
Does Secondary Structure Imply Tertiary Structure?
orf is based on the premise that the secondary structure of a protein determines its tertiary structure, at least in large part. This beguiling premise has cycled through the folding literature for years and has been applied successfully in recent studies (16–18, 43, 51), despite cautionary argument (22). Additional support for this premise comes from our analysis of helix capping (23).
The application of structure to genomics can make a substantial difference in the search strategy. As an example, the evaluation of point mutations is expected to be inherently context-dependent. Replacement of Asp by Glu is a conservative mutation in a β-strand but not in an α-helix (see, e.g., ref. 52). Important to note, such differences are reflected in the GOR (or any equivalent) procedure and therefore in orf, but they are neglected in the substitution matrices of typical alignment procedures.
A mature science of structure-based genomics would use the predicted three-dimensional structure of the query sequence and a library of known or predicted three-dimensional structures for all relevant genomes. However, predicted secondary structure is a more realistic goal at present, and, as exemplified by TS, it may be sufficient for use in comparative genomics.
Acknowledgments
We are grateful to Jeremy Berg, who suggested TS as a useful test case. We thank him, Rajgopal Srinivasan, Teresa Przytycka, Venkatesh Murthy, and an anonymous referee for many helpful suggestions. This work was supported by grants from the National Institutes of Health.
ABBREVIATIONS
- TS
thymidylate synthase
- PDB
Protein Data Bank
References
- 1.Doolittle R F. Annu Rev Biochem. 1995;64:287–314. doi: 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- 2.Gribskov M, Devereux J. Sequence Analysis Primer. New York: Freeman; 1992. [Google Scholar]
- 3.Doolittle R F. Of Urfs and Orfs. Mill Valley, CA: Univ. Sci. Books; 1986. [Google Scholar]
- 4.Altschul S F, Boguski M S, Gish W, Wootton J C. Nat Genet. 1994;6:119–129. doi: 10.1038/ng0294-119. [DOI] [PubMed] [Google Scholar]
- 5.Lipman D J, Pearson W R. Science. 1985;227:1435–1441. doi: 10.1126/science.2983426. [DOI] [PubMed] [Google Scholar]
- 6.Smith H O, Annau T M, Chandrasegaran S. Proc Natl Acad Sci USA. 1990;87:826–830. doi: 10.1073/pnas.87.2.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatusov R L, Altschul S F, Koonin E V. Proc Natl Acad Sci USA. 1994;91:12091–12095. doi: 10.1073/pnas.91.25.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neuwald A F, Liu J S, Lipman D J, Lawrence C E. Nucleic Acids Res. 1997;25:1665–1677. doi: 10.1093/nar/25.9.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henikoff S, Henikoff J G. Protein Sci. 1997;6:698–705. doi: 10.1002/pro.5560060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibrat J-F, Madej T, Bryant S H. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 11.Luthy R, Bowie J U, Eisenberg D. Nature (London) 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 12.Murzin A G, Brenner S E, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 13.Orengo C A, Jones D T, Thornton J M. Nature (London) 1994;373:631–634. doi: 10.1038/372631a0. [DOI] [PubMed] [Google Scholar]
- 14.Holm L, Sander C. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z X. Proteins. 1996;26:186–191. doi: 10.1002/(SICI)1097-0134(199610)26:2<186::AID-PROT8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Russell R B, Copley R R, Barton G J. J Mol Biol. 1996;259:349–365. doi: 10.1006/jmbi.1996.0325. [DOI] [PubMed] [Google Scholar]
- 17.Rost B, Schneider R, Sander C. J Mol Biol. 1997;270:471–480. doi: 10.1006/jmbi.1997.1101. [DOI] [PubMed] [Google Scholar]
- 18.Di Francesco V, Garnier J, Munson P J. J Mol Biol. 1997;267:446–463. doi: 10.1006/jmbi.1996.0874. [DOI] [PubMed] [Google Scholar]
- 19.Carreras C W, Santi D V. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 20.White R H. J Bacteriol. 1997;179:3374–3377. doi: 10.1128/jb.179.10.3374-3377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Needleman S B, Wunsch C D. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 22.Havel T F, Crippen G M, Kuntz I D. Biopolymers. 1979;18:73–81. [Google Scholar]
- 23.Aurora R, Rose G D. Prot Sci. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernstein F C, Koetzle T G, Williams G, Meyer E, Jr, Brice M D, Rogers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 25.Garnier J, Robson B. In: The GOR Method. Fasman G, editor. New York: Plenum; 1989. pp. 417–465. [Google Scholar]
- 26.Montfort W R, Perry K M, Fauman E B, Finer-Moore J S, Maley G F, Hardy L, Maley F, Stroud R M. Biochemistry. 1990;29:6964–6977. doi: 10.1021/bi00482a004. [DOI] [PubMed] [Google Scholar]
- 27.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brünger A T. X-PLOR, Version 3.8. A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ.; 1996. [Google Scholar]
- 29.Jones W J, Leigh J A, Mayer F, Woese C R, Wolfe R S. Arch Microbiol. 1983;136:254–261. [Google Scholar]
- 30.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 31.White R H. J Bacteriol. 1993;175:3661–3663. doi: 10.1128/jb.175.11.3661-3663.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choquet C G, Richards J C, Patel G B, Sprott G D. Arch Microbiol. 1994;161:471–480. [Google Scholar]
- 33.Nyce G W, White R H. J Bacteriol. 1996;178:914–916. doi: 10.1128/jb.178.3.914-916.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krone U E, McFarlan S C, Hogenkamp H P. Eur J Biochem. 1994;220:789–794. doi: 10.1111/j.1432-1033.1994.tb18680.x. [DOI] [PubMed] [Google Scholar]
- 35.Vaupel M, Dietz H, Linder D, Thauer R K. Eur J Biochem. 1996;236:294–300. doi: 10.1111/j.1432-1033.1996.00294.x. [DOI] [PubMed] [Google Scholar]
- 36.Wilhelm K, Rüger W. Virology. 1992;189:640–646. doi: 10.1016/0042-6822(92)90587-f. [DOI] [PubMed] [Google Scholar]
- 37.Finer-Moore J, Fauman E B, Foster P G, Perry K M, Santi D V, Stroud R M. J Mol Biol. 1993;232:1101–1116. doi: 10.1006/jmbi.1993.1463. [DOI] [PubMed] [Google Scholar]
- 38.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 39.Vogt G, Argos P. Fold Des. 1997;2:S40–S46. doi: 10.1016/s1359-0278(97)00062-x. [DOI] [PubMed] [Google Scholar]
- 40.Jaenicke R, Schurig H, Beaucamp N, Ostendorp R. Adv Protein Chem. 1996;48:181–269. doi: 10.1016/s0065-3233(08)60363-0. [DOI] [PubMed] [Google Scholar]
- 41.Dunlap R B, Harding N G, Huennekens F M. Ann N Y Acad Sci. 1971;186:153–165. doi: 10.1111/j.1749-6632.1971.tb46966.x. [DOI] [PubMed] [Google Scholar]
- 42.Haertle T, Wohlrab F, Guschlbauer W. Eur J Biochem. 1979;102:223–230. doi: 10.1111/j.1432-1033.1979.tb06283.x. [DOI] [PubMed] [Google Scholar]
- 43.Rice D W, Eisenberg D. J Mol Biol. 1997;267:1026–1038. doi: 10.1006/jmbi.1997.0924. [DOI] [PubMed] [Google Scholar]
- 44.Eddy S R. Curr Opin Struct Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- 45.Torda A E. Curr Opin Struct Biol. 1997;7:200–205. doi: 10.1016/s0959-440x(97)80026-7. [DOI] [PubMed] [Google Scholar]
- 46.Crippen G M. Proteins Struct Funct Genet. 1996;26:167–171. doi: 10.1002/(SICI)1097-0134(199610)26:2<167::AID-PROT6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 47.Thomas P D, Dill K A. J Mol Biol. 1996;257:457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- 48.Godzik A. Protein Eng. 1995;8:409–416. doi: 10.1093/protein/8.5.409. [DOI] [PubMed] [Google Scholar]
- 49.Godzik A. Protein Sci. 1996;5:1325–1338. doi: 10.1002/pro.5560050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchler-Bauer A, Bryant S H. Trends Biochem Sci. 1997;22:236–240. doi: 10.1016/s0968-0004(97)01078-5. [DOI] [PubMed] [Google Scholar]
- 51.Fischer D, Tsai C-J, Nussinov R, Wolfson H. Protein Eng. 1995;8:981–997. doi: 10.1093/protein/8.10.981. [DOI] [PubMed] [Google Scholar]
- 52.Chou P Y, Fasman G D. Adv Enzymol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 53.Sayle R A, Milner-White E J. Trends Biochem Sci. 1995;220:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]