

Abstract
Nociceptive stimulation, at an intensity that elicits pain-related behavior, attenuates recovery of locomotor and bladder functions, and increases tissue loss after a contusion injury. These data imply that nociceptive input (e.g., from tissue damage) can enhance the loss of function after injury, and that potential clinical treatments, such pretreatment with an analgesic, may protect the damaged system from further secondary injury. The current study examined this hypothesis and showed that a potential treatment (morphine) did not have a protective effect. In fact, morphine appeared to exacerbate the effects of nociceptive stimulation. Experiment 1 showed that after spinal cord injury 20 mg/kg of systemic morphine was necessary to induce strong antinociception and block behavioral reactivity to shock treatment, a dose that was much higher than that needed for sham controls. In Experiment 2, contused rats were given one of three doses of morphine (Vehicle, 10, 20 mg/kg) prior to exposure to uncontrollable electrical stimulation or restraint alone. Despite decreasing nociceptive reactivity, morphine did not attenuate the long-term consequences of shock. Rats treated with morphine and shock had higher mortality rates, and displayed allodynic responses to innocuous sensory stimuli three weeks later. Independent of shock, morphine per se undermined recovery of sensory function. Rats treated with morphine alone also had significantly larger lesions than those treated with saline. These results suggest that nociceptive stimulation affects recovery despite a blockade of pain-elicited behavior. The results are clinically important because they suggest that opiate treatment may adversely affect the recovery of function after injury.
Introduction
Activity in pain (nociceptive) fibers after a spinal injury can provide an uncontrollable source of over-excitation [1, 2, 3], and can lead to maladaptive alterations within the central nervous system. This central excitability is derived not only from the spinal injury itself, but also from accompanying peripheral tissue damage. The significance of uncontrollable nociceptive stimulation after injury is two-fold: first, it can severely impact recovery of function [4] and secondly, it can sensitize spinal neurons leading to the development of neuropathic pain [5, 6].
We have developed an animal model to empirically investigate the effects of uncontrollable nociceptive stimulation on recovery. Using electrical stimulation, we apply nociceptive input without inducing long-term peripheral tissue damage and its secondary consequences. Rats with a moderate contusion injury are exposed to uncontrollable stimulation, and their recovery of motor and sensory functions are monitored for 3–6 weeks [7]. Contused rats given uncontrollable stimulation showed poor recovery of locomotor function and reduced reactivity to thermal, tactile, and shock stimuli [4]. They also showed delayed recovery of bladder function, less weight gain, greater mortality, a higher incidence of spasticity, and increased tissue loss at the site of injury. Stimulation only had an adverse effect if 1) it occurred within a few days of injury, and 2) only if the stimulation was uncontrollable; subjects that received an equal amount of controllable shock (and exhibited greater movement) showed normal recovery. Grau et al. [4] concluded that uncontrollable nociceptive stimulation undermines recovery after a spinal injury.
Intermittent tailshock elicits a variety of behaviors in intact rats indicative of pain, including vocalization, struggling, escape behaviors, and conditioned freezing [8, 9]. A moderate contusion injury greatly reduces these behaviors but does not eliminate shock-elicited movement or vocalization. We hypothesized that the aversive quality of shock was central to its long-term effects on recovery. If this is true, then administration of an opiate analgesic should reduce the consequences of shock treatment. Opiate analgesics are currently recommended as one of 5 first-line medications for the treatment of chronic pain [10, 11] and are given soon after a spinal injury for attenuation of pain symptoms [12]. In the present study, a single injection of morphine sulfate (Vehicle, 5, 10, or 20 mg/kg) was given to contused rats 30 minutes prior to exposure to uncontrollable electrical stimulation. Experiment 1 showed that, for contused rats, 10 mg/kg produced a robust antinociception on the tail-flick test but 20 mg/kg was needed to block a supraspinally-mediated measure (vocalization) of shock-elicited pain. Despite the total blockade of pain-elicited behavior, morphine treatment did not attenuate the long-term consequences of shock on recovery (Experiment 2). Rather, acute morphine had a detrimental effect and exacerbated the adverse effects of uncontrollable shock.
General Methods
Subjects
Subjects were male Sprague-Dawley rats obtained from Harlan (Houston, TX). They were approximately 90–110 days old (350–400 g), and were individually housed in Plexiglas bins [45.7 (length) × 23.5 (width) × 20.3 (height) cm] with food and water continuously available. To facilitate access to the food and water, extra bedding was added to the bins after surgery and long mouse sipper tubes were used so that the rats could reach the water without rearing. Subjects were weighed on days that they were assessed for locomotor function, and were checked daily for signs of autophagia and spasticity. A subject was classified as having spasticity if the limb was in an extended, fixed position and was resistant to movement. Bladders were manually expressed in the morning (8–9.30 a.m.) and evening (6–7.30 p.m.) until subjects regained bladder control, which was operationally defined as three consecutive days with an empty bladder at the time of expression. The rats were maintained on a 12 hr light/dark cycle and tested during the last 6 hrs of the light cycle.
All of the experiments were reviewed and approved by the institutional animal care committee at Texas A&M and all NIH guidelines for the care and use of animal subjects were followed.
Surgery
Subjects received a contusion injury using the MASCIS device [13, 14]. Subjects were anesthetized with pentobarbital (50 mg/kg, i.p.). After a stable and comparable level of anesthesia was achieved, an area extending approximately 4.5 cm above and below the injury site was shaved and disinfected with iodine. A 7.0 cm incision was made over the spinal cord. Next, two incisions were made on either side of the vertebral column, extending about 3 cm rostral and caudal to the T12–T13 segment. The dorsal spinous processes at T12–T13 were removed (laminectomy), and the spinal tissue exposed. The dura remained intact. The vertebral column was fixed within the MASCIS device and a moderate injury (see Fig. 5) was produced by allowing the 10 g impactor (outfitted with a 2.5 mm tip) to drop 12.5 mm. Sham subjects underwent the same surgical procedures except the weight drop was not performed. After injury, the subject was removed from the device, placed on a heating pad, and the wound was closed with Michel clips. To help prevent infection, subjects were treated with 100,000 units/kg Pfizerpen (penicillin G potassium) immediately after surgery and again 2 days later. For the first 24 hrs after surgery, rats were placed in a recovery room maintained at 26.6°C. To compensate for fluid loss, subjects were given 2.5 ml of saline after surgery. Michel clips were removed 14 days after surgery.
FIG. 5.
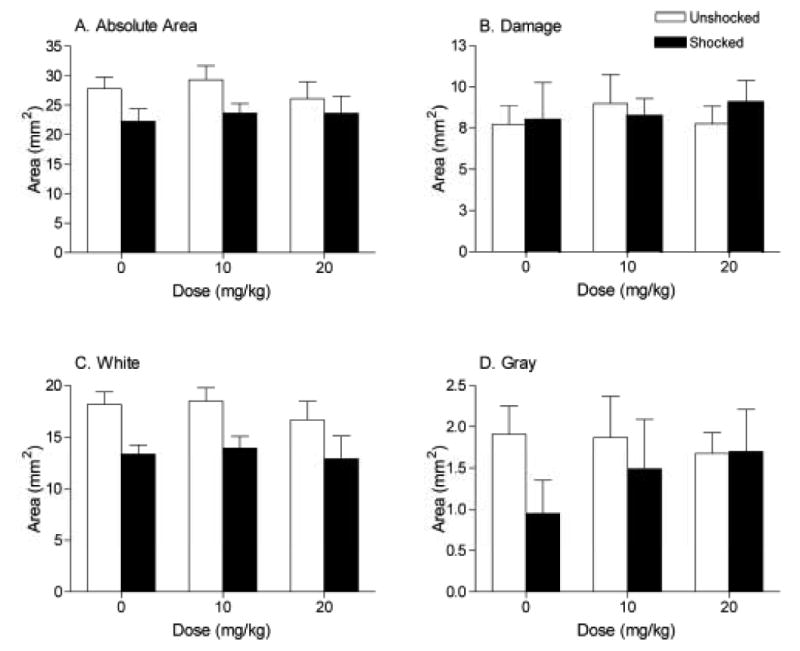
Shock significantly decreased the absolute area of the spinal cord remaining at the maximal injury site (A), an effect primarily due to loss of white matter (C). Shock did not affect the area of damage (B) remaining at the center of the lesion or the amount of gray matter remaining (D).
Apparatus
Uncontrollable tail shock was applied while the subjects were restrained in opaque Plexiglas tubes [22 cm (length) × 6.8 cm (internal diameter)]. A sheet of Plexiglas formed a floor (5.5 cm wide, lying 5.3 cm from the top of the tube), on which the rats could lie. Constant current tailshock was generated with a 660-V transformer, in combination with a 2.03-MΩ series resistor, and applied through cutaneous shock electrodes constructed from a modified fuse clip. The electrode plates were lightly coated with electrode paste (Harvard apparatus) and taped 15 cm from the base of the rat's tail with Orthaletic tape. Unshocked rats were held in the restraining tubes for 30 min with the electrodes taped to their tails, but no shock was applied.
Locomotor behavior was assessed, using the Basso, Beattie and Bresnahan (BBB) scale [15], in an open enclosure (a blue children's wading pool, 99 cm diameter, 23 cm deep). Other measures of motor recovery, including beam and ladder walk performance, involved traversing black beams of varying widths (122.5 cm long and 17.5, 8.75, 4.375, or 1.875 cm wide) or a horizontal ladder (20 cm wide; 37 rungs at 2.5 cm spacing) to enter a black box.
Tactile reactivity was tested using Plexiglas tubes [7.0 cm (internal diameter) × 20 cm (length)] that had 6 (length) × 1.7 (width) cm notches removed from the sides of the base. These slots allowed the hindlegs to hang freely below the tube. Reactivity was assessed using von Frey stimuli formed from nylon monofilaments (Semmes-Weinstein Anesthesiometer; Stoelting Co., Chicago, IL) and applied to the plantar surface of the paw.
Thermal reactivity was tested using a 375-W movie light that was focused onto the rat's tail with a condenser lens positioned 8 cm below the light source. Shock thresholds were assessed using a manual shocker (BRS/LVE, Model SG-903) that allowed continuous variation of shock intensity between 0 and 2 mA (AC, constant current). Test shocks were applied 7 cm from the base of the tail through electrodes constructed from lightweight fuse clips. Test shock intensity was gradually incremented at a rate of 0.05 mA every 3 s. For testing shock and thermal reactivity, the subject's tail was positioned in a 0.5 cm deep groove that was cut into an aluminum block. Plastic sides (6 cm × 6.7 cm) were placed along the sides of the aluminum block to hold the rat's tail under the heat. A wire hook that was 10 cm long and covered with heat shrink tubing was taped to the last 2.5 cm of the tail. The hook was placed over an elastic band located 11 cm behind the aluminum block. The flexibility of the elastic band allowed for a tail-flick response while maintaining the rat's tail under the heat source. A photocell, located in the groove of the aluminum block, was used to automatically detect whether the rat moved its tail laterally 0.5 cm. To activate the photocell in the absence of radiant heat (on the shock test trials), a small 28-V light (General Instrumental, 1820) was positioned 3.5 cm above the photocell. The latency to vocalize was assessed using a microphone located at the front end of the tube. The vocalization threshold was set to 80 dB. A computer (Apple, Macintosh 8500) monitored the circuit controlled by the photocell and the output intensity from the microphone. After both movement and vocalization responses were detected, the shock or heat was terminated. If a subject failed to respond, the test trial was automatically terminated after 8 s of heat exposure or after shock intensity reached 1.2 mA.
Assessment of Motor/Sensory Recovery
Recovery of hindlimb stepping was assessed while subjects moved freely in an open field. Because rodents often remain motionless (freeze) when first introduced to a new apparatus, subjects were acclimated to the observation fields for 5 min per day for 3 days prior to surgery. Each subject was placed in the open field and observed for 4 min. During this period, locomotor behavior was scored using the procedure developed by Basso, Beattie, and Bresnahan [15]. Care was taken to ensure that the investigators' scoring behavior had high intra- and inter-observer reliability (all r's > 0.89) and that they were blind to the subject's experimental treatment.
Experiment 1
Forty-eight male Sprague-Dawley rats were subjects in this experiment. A 2 (sham versus contusion) × 4 (Vehicle, 5, 10, or 20 mg/kg of morphine) experimental design was used to examine sensory reactivity to nociceptive stimuli after treatment with systemic morphine. Tests were conducted 24 hours after surgery. First, the baseline motor function was assessed using the BBB scale. The rats were then injected with an assigned dose of morphine sulfate (Sigma-Aldrich, St. Louis) and, 30 minutes later, were placed in the restraining tubes and nociceptive reactivity was assessed. Nociceptive reactivity was assessed with gradually incremented shock and radiant heat, as described in prior studies [e.g., 9, 16, 17]. Briefly, subjects were placed in the restraining tubes, and the apparatus used to assess reactivity was attached to the tail. Next, subjects were allowed to acclimate to the apparatus for 15 min. Thermal and shock thresholds were then assessed at 2 min intervals, 2 times each, in an ABBA order. To confirm that subjects did not respond in the absence of the stimuli, blank trials were also performed. A ‘false alarm’ was recorded if subjects made a motor or vocalization response during the blank tests. The blank trials were performed 1 min before or after each sensory test (in counterbalanced fashion). No false alarms were recorded.
Finally, the rats were exposed to 1 min of intermittent tail shock (1.5 mA, 0.08 s duration, mean interstimulus interval of 2.0 s.) and their level of motor reactivity (Table 1) and the incidence of vocalization were recorded by an observer blind to the subject's surgery and drug treatment. The results were analyzed using factorial analyses of variance (ANOVAs). In cases where significant differences were obtained (main effect of a single variable), group means were compared using the Duncan's New Multiple Range Test (p < 0.05). Trend analyses were also used to identify dose-dependent changes in behavior.
Table 1.
Behavioral indices for scoring motor reactivity during uncontrollable electrical stimulation.
| Score | Description of behavior |
|---|---|
| 1 | Flicking tip of tail only |
| 2 | Reflexive tail and body movement |
| 3 | Hindlimb movement |
| 4 | Forelimb movement |
| 5 | Trunk movement |
Note that higher scores also include observation of each of the behaviors described for the preceding scores.
Experiment 2
This experiment examined the long-term effects of nociceptive stimulation, and morphine treatment, on functional recovery. Subjects were 58 contused, male Sprague-Dawley rats. Twenty-four hours after contusion surgery, the baseline motor function of the rats was assessed using the BBB scale. Then the rats were injected with morphine sulfate (10 or 20 mg/kg, Sigma-Aldrich, St. Louis) or saline (i.p.). Thirty minutes later, subjects were placed in the restraining tubes and nociceptive reactivity was assessed 3 times using the tail-flick test. These tests occurred at 2 minutes intervals, and the last 2 tests were used to compute the mean tail-flick latencies.
Shock Treatments
Next, the tail electrodes were secured with porous adhesive tape. Separate groups received nothing (0 s) or 30 min of uncontrollable, intermittent tailshock. The shocks were 1.5 mA, 0.08 s in duration, and occurred on a variable time schedule (range 0.2 to 3.8 s) with a mean interstimulus interval of 2.0 s. (Shock at an intensity of 1.5 mA [AC, constant current] is known to engage antinociceptive mechanisms within the spinal cord [18, 19], vigorous defensive behavior in intact rats, and pain in humans [see 8, 9].) An experimenter blind to the subject's drug treatment recorded whether the shocked rats vocalized during the first minute of stimulation. All subjects remained in the restraining tubes for an equivalent period (30 min).
Assessment of Motor/Sensory Recovery
The first behavioral assessment of locomotor recovery (BBB scale) was conducted 24 hrs after surgery, and prior to drug or shock treatment. Locomotor behavior was then scored once per day for 1 week (Days 2–7). Subjects were scored every other day from Day 9 to Day 15 and every 3rd day on Days 18 and 21. A video recording of each subject's performance in the open field was obtained on Days 1, 2, 4, 7, and 21.
Additional measures of motor recovery were obtained using ladder walk [20] and beam walk [21, 22, 23] tasks. Prior to testing, subjects were habituated to the experimental situation for 3 days (8 min per day). During this period of familiarization, they were trained to traverse a wide beam (48.3 cm) to enter a black box positioned at the end of the beam runway. The beginning of the runway is brightly lit, so subjects are motivated to move toward the box. They were left in the box for 2 min after they had traversed the beam. In testing, the subjects were required to cross a horizontal ladder in order to reach the black box. Using post hoc frame-by-frame video analyses, we then ascertained how many times the subjects did not successfully place their hindpaws (their paws slipped between the rungs). Based on the number of slips, the rats were given scores for the left and right leg separately; they received a score of 11 if they made no slips, 10 if they made one, reducing incrementally to 1 if they made more than 10 slips and 0 if they did not plantar place. Rats could receive a maximum score of 22 on this task (e.g., 11 points possible for each hind leg). The ladder task provides a measure of the extent to which experimental manipulations affect the fine motor abilities of the hindpaws.
The beam walk test provides a comparative index of the postural stability of the subjects, as well as a gross measure of paw placement abilities. In this test, the subject's ability to traverse a series of progressively narrowing beams was observed. Rats are assigned points for the left and right limbs separately on each of the beams. For each leg, they received 1 point for successfully traversing a beam with plantar placement; 0.5 points for traversing more than half of a beam with plantar placing; 0 points for no hindpaw placement or traversing less than half of the beam. Rats could receive a maximum score of 8 on this task (e.g., 2 points possible on each of the 4 beams).
Sensory function was assessed on Day 1, prior to shock treatment, and after Day 21. Progressively stronger tactile stimuli (von Frey filaments) were applied sequentially at approximately 2 s intervals until subjects exhibited a paw withdrawal (motor response) and vocalization. If one or both responses were not observed, testing was terminated at a force of 300 g. Each subject was tested twice on each foot in a counterbalanced ABBA order. Test sequences were spaced 2 min apart. Stimulus intensity was reported using the formula provided by Semmes-Weinstein: Intensity = log10 (10,000 * g force). On an alternate day (test order was counter-balanced across groups), we assessed sensory reactivity using nociceptive stimuli (a gradually incremented shock and radiant heat) and the procedures described for Experiment 1.
Histology
At the end of behavioral testing, subjects were deeply anesthetized (100 mg/kg of pentobarbital, i.p.) and perfused (intracardially) with 4% paraformaldehyde. A 1 cm long segment of the spinal cord that included the lesion center was taken and embedded in paraffin prior to sectioning. The tissue was sectioned coronally in 10 μm thick sections and every 20th slice was preserved for staining. All sections were stained with cresyl violet for Nissl substance and luxol fast blue for myelin [24, 25].
The total cross-sectional area of the cord and spared tissue was assessed at the lesion center from camera lucida traces made by an experimenter who was blind to the subject's treatment condition. Sections ±600, 1200, and 1800 μm from the lesion center (rostral and caudal) were also traced and analyzed. Four indices of lesion magnitude were derived: damage area, area of residual gray matter, area of residual white matter, and width. For derivation of damage area, and the spared gray/white matter, the camera lucida drawings were scanned onto a Macintosh computer and imported into CANVAS 8 (Deneba systems Inc.). To determine the area of damage, an observer who was blind to the experimental treatments traced around the boundaries of cystic formations and areas of dense gliosis [26]. Nissl-stained areas that contained neurons and glia of approximately normal densities denoted residual gray matter. White matter was judged spared in myelin-stained areas lacking dense gliosis and swollen fibers. The total area of each cross-section was derived by summing the areas of damage, gray and white matter. Width was determined from the most lateral points along the transverse plane. These analyses yielded six parameters for each section: white matter area, gray matter area, spared tissue (white + gray), damaged tissue area, net area (white + gray + damage), and section width.
To control for variability in section area across subjects, two methods for computing the magnitude of the relative spinal lesion have traditionally been used. One method calculates the percent lesion from the ratio of lesioned tissue to net area [27]. The alternative procedure uses standards derived from undamaged cord sections [28]. These transformations can lead to some interpretational difficulties when an experimental manipulation affects section size (width or net area). Because preliminary analyses revealed that shock treatment affected section size it would be inappropriate to compare transformed data across shock treatments. However, within a shock condition neither the width nor the net area of the cords differed as a function of dose ([width, F(1, 21) = 1.60, < 1.0 respectively, p > 0.05][area, F(2, 21) < 1.0 in both cases, p > 0.05]). Given this we were able to control for individual variability within a shock condition by calculating a correction factor derived from standard undamaged cord sections. This correction factor is based on section widths and is multiplied by all area measurements to standardize area across analyses [see 4]. By standardizing area across sections we can also estimate the degree to which tissue is ‘missing’ (i.e., tissue loss from atrophy, necrosis, or apoptosis). An accurate assessment of the degree to which a treatment has impacted, or lesioned, the cord includes both the remaining ‘damaged’ tissue as well as resolved lesioned areas. When we sum the amount of ‘missing’ tissue and the measured ‘damaged’ area we can derive an index of the relative lesion in each section that is comparable across sections. This measure is highly correlated with various measures of behavioral performance including BBB locomotor scores, recovery of bladder function, and reactivity to shock [4].
Data Analyses
The results were analyzed using analysis of variance (ANOVA). In experiments with a continuous independent variable (e.g., exposure duration, recovery period, rostral-caudal histological sections), mixed-design ANOVAs were used. Trend analyses were also used to identify dose-dependent changes in behavior. In cases where significant between-subject differences were obtained (main effect of a single variable), group means were compared using the Duncan's New Multiple Range Test (p < 0.05).
Locomotor scores were transformed to help assure that the data were amendable to parametric analyses [29]. This transformation pools BBB scores 2–4, removing a discontinuity in the scale. The transformation also pools scores from a region of the scale (14–21) that is very seldom used under the present injury parameters. By pooling these scores, we obtain an ordered scale that is relatively continuous with units that have approximately equivalent interval durations. Meeting these criteria allows us to apply metric operations (computation of mean performance across legs), improves the justification for parametric statistical analyses, and increases statistical power.
Additional statistical power was also achieved by obtaining a measure of locomotor performance 24 hrs after injury, prior to shock treatment. This provides a behavioral index of the injury extent that is correlated with long-term recovery (r > 0.41, p < 0.05, [7]). By using Day 1 as a covariate in an analysis of covariance (ANCOVA), we substantially reduced unexplained variance and thereby increased statistical power.
Group differences on dichotomous variables (e.g., mortality) were evaluated using chi-square or Fisher exact probability tests. These tests allow for comparisons of simple (2 × 2) frequency tables with relatively small samples. Nonparametric Kruskal-Wallis and post hoc Mann-Whitney U tests were used to compare motor reactivity scores in Experiment 1.
Results
Experiment 1
As expected, the contusion injury produced a paraplegia that severely disrupted locomotor function relative to the sham controls (contused BBB score = 3.50 ± 0.45, sham BBB score = 18.48 ± 0.39). An ANOVA verified that there was a significant effect of injury (F(1, 40) = 6.26, p = 0.0001) and that there were no differences across groups prior to drug treatment.
The contusion injury did not alter morphine effectiveness on the spinally-mediated tail-flick test (Fig. 1a). There was a main effect of dose on the tail flick response to heat (F(3, 40) = 6.26, p = 0.0001), but no interaction between surgery and dose. All doses significantly attenuated the tail flick response in both sham and contused rats. It is acknowledged, however, that a ceiling effect may be masking interaction effects in these data: the thermal cut-off latency of 8 sec was reached by all groups treated with morphine. This cut-off was employed to avoid tissue damage, which could impact the recovery process.
FIG. 1.

Despite differences in locomotor abilities, there was no effect of surgery on motor reactivity to a noxious thermal stimulus (A). Independent of dose or surgery condition, morphine significantly increased motor reactivity thresholds when exposed to radiant heat. Morphine also increased vocal reactivity thresholds when rats were exposed to incremented shock stimuli (B). A quadratic relation was found between dose and vocal reactivity for both sham and contused rats. Contused rats required 20 mg/kg of morphine, however, to achieve levels of antinociception comparable to sham controls treated with 10 mg/kg of morphine. Similarly, there was a significant effect of dose on motor (C) and vocal (D) reactivity to uncontrollable electrical stimulation, with contused rats requiring higher doses of morphine to achieve levels of antinociception comparable to sham controls. ** indicates a significant difference (p ≤ 0.05) between groups connected with lines.
Given that the contusion injury also disrupts sensory function, we anticipated that less morphine would be needed to block brain mediated responses to nociceptive stimuli applied below the injury. However, the opposite trend was observed. Morphine treatment generally increased vocalization thresholds to electrical stimulation (F(3, 40) = 11.35, p = 0.0001, Fig. 1b). All 3 morphine doses significantly attenuated responses compared with vehicle controls. Subjects treated with 20 mg/kg of morphine also displayed significantly higher vocalization thresholds than those treated with 5 mg/kg (p < 0.05). Trend analyses revealed a significant quadratic relation between dose and surgery on shock thresholds required to elicit vocalization (F(1, 40) = 4.13, p < 0.05). Contused rats required significantly more morphine to achieve levels of antinociception comparable to sham controls when vocalization thresholds to incremented shock were assessed. Neither morphine nor surgery had a systematic effect on motor reactivity to a gradually incremented shock or vocalization to thermal stimulation (data not shown).
Motor reactivity to intermittent electrical stimulation was assessed using the scale outlined in Table 1. The reliability of this was confirmed by independent observers who were blind to the subjects' drug treatment. A nonparametric Kruskal-Wallis test revealed a significant main effect of condition (dose × surgery) on motor reactivity (KW = 34.44, p < 0.0001, Fig. 1c). Post hoc Mann-Whitney U tests revealed that, compared with vehicle controls, all doses of morphine significantly lowered motor reactivity in the sham groups (p < 0.05). In the contused groups, only the 10 and 20 mg/kg doses significantly lowered reactivity compared to vehicle controls (p < 0.05). As can be seen in Figure 1c, for both surgery groups motor reactivity progressively decreased with increasing doses of morphine, but the contused rats showed a slower decline than sham controls. An observer also recorded whether subjects exhibited an audible vocalization during the period of intermittent stimulation. Again, there was perfect agreement across observers who were unaware of the subjects' drug treatment. As can be seen in Figure 1d, contused rats required 20 mg/kg of morphine to reduce supraspinally-mediated vocalization to intermittent electrical stimulation to a level comparable to sham controls treated with 10 mg/kg of morphine. There was a significant effect of dose on vocal reactivity (χ2 = 30.97, p < 0.0001, df = 3; Fig. 1d). Fisher's Exact tests corrected for multiple comparisons (Bonferonni correction, p = 0.008) revealed significant differences between groups of sham rats treated with saline and those treated with 10 or 20 mg/kg of morphine (Fisher's Exact, p = 0.001). For contused groups, subjects treated with saline differed significantly from the 20 mg/kg group only (Fisher's Exact, p = 0.001).
Experiment 2
Baseline Assessments of Motor and Sensory Function
To verify the effectiveness of drug treatment, both a spinal (tail withdrawal from a noxious thermal stimulus) and supraspinal (vocalization to shock) measure of nociceptive reactivity was recorded. Again, morphine inhibited tail withdrawal from a thermal stimulus (F(2, 42) = 12.46, p = 0.001) prior to exposure to uncontrollable nociceptive stimulation. Both 10 and 20 mg/kg of morphine reduced reactivity when compared to saline controls (p < 0.05; Fig. 2a). During nociceptive stimulation rats treated with 20 mg/kg of morphine were significantly less likely to vocalize to uncontrollable shock, when compared to those treated with 10 mg/kg of morphine or saline (χ2 = 8.06, p < 0.05, df = 2; Fig. 2b).
FIG. 2.
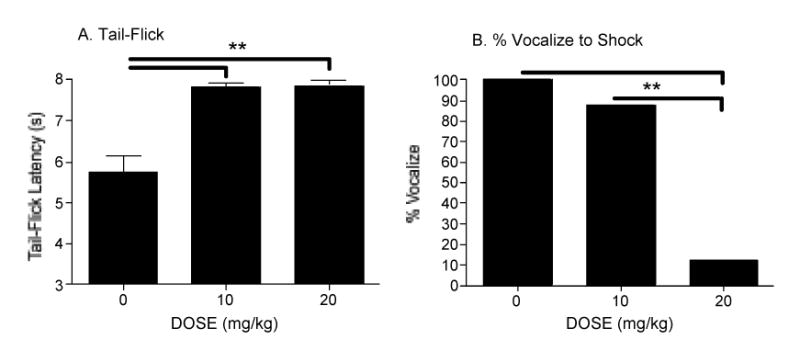
Nociceptive reactivity was assessed on Day 1 after administration of morphine. Both doses of morphine significantly attenuated reactivity on the tail-flick test to radiant heat (A), increasing the latency to move the tail. During uncontrollable, intermittent shock treatment, rats given an injection of 20 mg/kg of morphine were also significantly less likely to vocalize (B), which is also indicative of a reduction in the pain experience.
Recovery of Motor Function
Baseline BBB scores, collected before treatment on day 1, did not differ across groups (F(1, 42) < 1.0, p > 0.05). Using Day 1 scores as a covariate, there was a significant effect of shock on the recovery of locomotor function (F(11, 451) = 9.00, p < 0.001, Fig. 3). Shock attenuated the overall recovery of locomotor function (p < 0.05). There was no overall effect of morphine on locomotor recovery in the shocked or unshocked groups. However, as can be seen in Figure 3 (see inset), morphine did affect locomotor performance 24–48 hours after treatment (Day 2–3). Comparing unshocked rats only, both doses of morphine caused a short-term dip in recovery, relative to saline controls, on Days 2 and 3 (F = 5.19 and 7.00, for 10 and 20 mg/kg, respectively, p < 0.05).
FIG. 3.
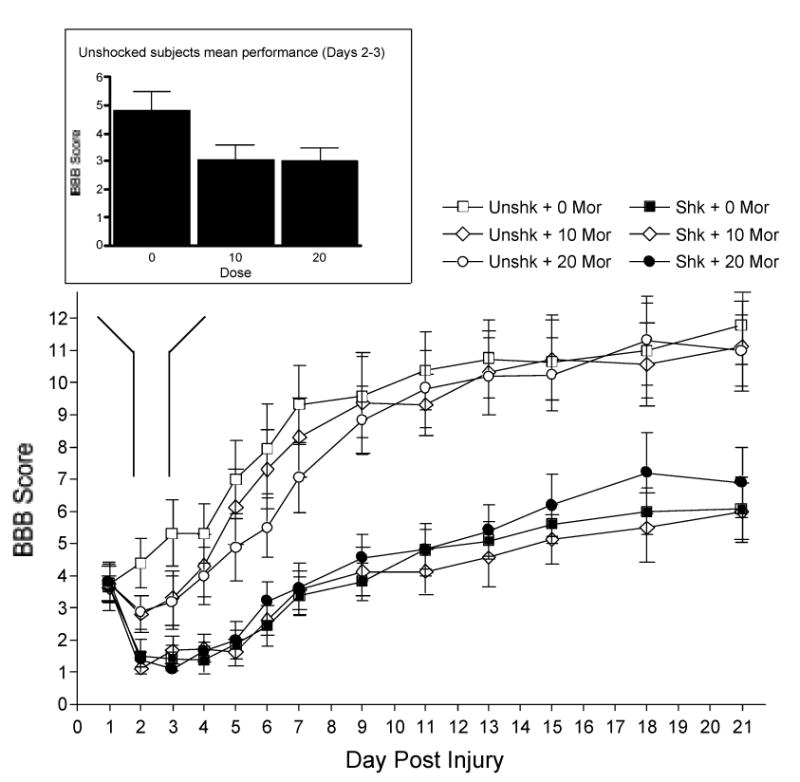
The mean locomotor performance of the rats over days. Shocked rats obtained significantly lower BBB scores throughout the recovery period. Morphine treatment alone (unshocked rats only) also caused a short-term dip in motor recovery on days 2 and 3 (see inset). ** indicates a significant difference (p ≤ 0.05) between groups connected with lines.
There was a significant main effect of shock on the beam and ladder tasks conducted 3 weeks after morphine and/or shock treatment (F(1, 41) = 52.63, p < 0.001, F(1, 40) = 21.96, p < 0.001, for beam and ladder respectively). Shock decreased performance on both of these tasks (p < 0.05). Shocked animals were unable to traverse the narrower beams (Fig. 4a) and displayed more footfalls (Fig. 4b). Morphine treatment did not significantly affect performance on the beam and ladder tasks.
FIG. 4.

Performances on the beam (A) and ladder (B) walk tasks were assessed approximately 25 days after injury. Shock significantly reduced performances on both tasks. Morphine (20 mg/kg) also lowered tactile withdrawal thresholds 21 days after treatment (C), indicating that these rats had developed allodynia. Conversely, shock and morphine, independently, increased vocalization thresholds to shock suggesting that both treatments had a detrimental effect on sensory function (D). Finally, morphine treated shocked rats exhibited increased mortality (E). Both shock and morphine treatment also reduced weight gain after injury (F). ** indicates a significant difference (p ≤ 0.05) between groups connected with lines. Note that main effects are not depicted on the graphs.
Recovery of Sensory Functions
Independent of shock treatment, morphine-treated rats exhibited a tactile allodynia (enhanced limb withdrawal) 3 weeks after drug treatment (Fig. 4c). There was a significant main effect of morphine treatment on the motor response to von Frey stimulation (F(2, 42) = 3.65, p < 0.05). Rats treated with 20 mg/kg of morphine displayed significantly lower mechanical thresholds than those treated with 10 mg/kg morphine and saline respectively (p < 0.05). Rats treated with 10 mg/kg and saline did not differ on this measure.
Limb withdrawal from mechanical stimulation can be elicited after spinal transection, suggesting that the change in tactile reactivity reported above could involve an intraspinal modification related to the sensitization of spinal neurons [30, 31, 32]. Vocalization to shock provides a supraspinal measure of sensory function and it revealed a different pattern of results; both prior shock and morphine treatment increased vocalization thresholds, suggesting both treatments increased the loss of sensory fibers. There was a significant Morphine X Shock interaction when latencies to vocalize were compared (F(2, 42) = 4.25, p < 0.05). As can be seen in Fig. 4d, shock treatment significantly increased vocalization thresholds in the vehicle treated (0 mg/kg) group (F = 9.79, p < 0.005), with morphine decreasing thresholds in these groups. Inspection of the data from the unshocked rats revealed that morphine treatment increased vocalization thresholds (F = 4.94, p < 0.05). Other measures of sensory recovery did not reveal any significant difference (all Fs < 3.02, p > 0.05).
Other Behavioral Indices of Recovery
As shown in Fig. 4e, shocked rats treated with 20 mg/kg of morphine were significantly more likely to die than those in any other condition (χ2 = 18.75, p < 0.005, df = 5). Eight of the 16 subjects allocated to this condition died during the recovery period. Two rats treated with 10 mg/kg of morphine and shock died. Rats died an average of 4.6 days after morphine treatment, long after the drug was pharmacologically active. Opiate overdose from factors such as respiratory depression generally occurs during the period of drug action within 30–120 minutes after injection. None of the morphine treated rats died during this period. Drug-related mortality did not emerge until a few hours after treatment, well after the behavioral signs of opiate administration had waned. Five of the 10 rats died within a day of treatment, while the remaining 5 died 6–11 days after treatment. There was no mortality in the saline-treated or unshocked conditions. (To achieve a balanced design, additional subjects were folded into the experimental groups when a subject died. To assess whether this affected the results obtained for locomotor recovery, we examined whether the pattern of results changed when we excluded the added subjects. An ANCOVA verified that there was no change in the overall pattern of results.)
Both shock (F(11, 451) = 3.70, p < 0.001) and morphine (F(22, 451) = 1.69, p < 0.05) treatment significantly reduced weight gain after injury (Fig. 4f). There was also a significant main effect of shock on bladder function (F(1, 42) = 5.25, p < 0.05). Unshocked rats recovered bladder function between 12 and 16 days post-surgery (mean = 12.5 ± 2.45), whereas shocked rats showed recovery 15 to 20 days after surgery (mean = 17.5 ± 1.5). Morphine did not affect the recovery of bladder function. None of the rats developed spasticity, and the incidence of autophagia was not affected by shock or morphine.
Histological Analyses
Lesion Center
At the lesion center, there was a significant main effect of shock on the absolute area (F(1, 42) = 4.82, p < 0.05, Fig. 5a) of the sections (Fig. 6). The main effect of shock on the width of sections also approached significance (F(1, 42) = 3.88, p = 0.06). Further, as found previously, shock significantly affected the total tissue remaining at the lesion center (F(1, 42) = 10.74, p < 0.005), an effect attributable to a loss of white matter (F(1, 42) = 11.36, p = 0.001; Fig. 5c). These data confirm the results of our previous studies [4].
FIG. 6.

Shock significantly decreased the amount of total tissue remaining at the lesion center (d–f). Morphine also, independently of shock, reduced the amount of total tissue remaining at the lesion center (a–c). In unshocked subjects, both 10 and 20 mg/kg doses of morphine produced a larger relative lesion and significantly decreased the amount of white tissue remaining for measurement.
To further examine the effects of morphine, we analyzed the data for the shocked and unshocked groups independently. By removing shock as a variable in these analyses, we were able to determine whether morphine has histological effects that do not depend on shock treatment. Because within a shocked/unshocked condition neither width nor area varied as a function of drug treatment, a correction factor could be applied to control for individual variability (see methods). Analyses, controlling for individual variability, revealed a number of effects of morphine treatment in the unshocked controls. Comparison of the unshocked groups (Fig. 7) revealed a significant effect of morphine on the amount of white matter remaining at the lesion center. A linear contrast confirmed morphine treatment led to greater white matter loss (F = 5.67, p < 0.05; Fig. 7c. Also see Fig. 6) and a larger relative lesion (F = 5.05, p < 0.05; Fig. 7a). Significantly more tissue was denoted as ‘missing’ in the morphine treated, unshocked rats (F = 4.32, p = 0.05; Fig. 7b). Analyses of the shocked groups did not reveal any morphine-related, additional, damage at the lesion center.
FIG. 7.
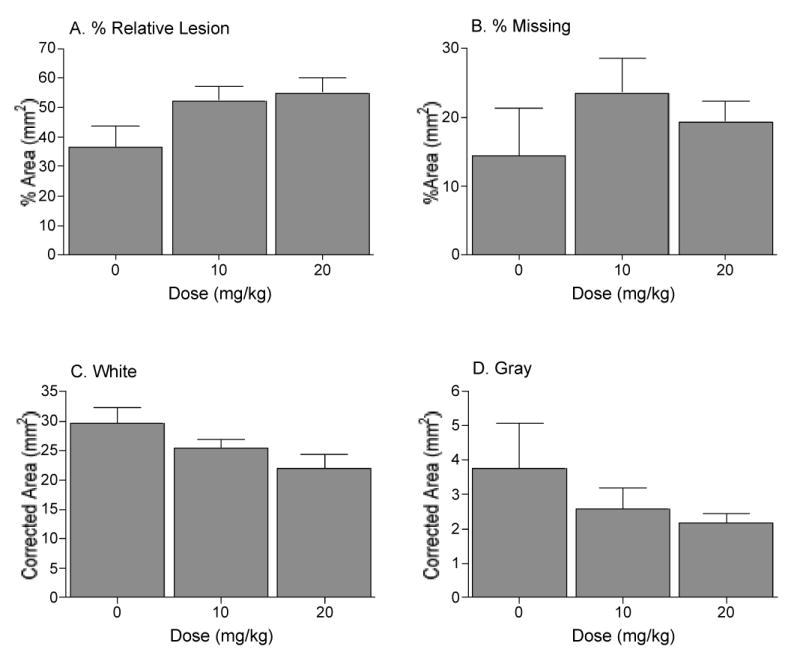
In unshocked subjects, morphine treatment significantly increased the size of the relative lesion (damaged + missing area) (A) at the maximal point of injury. Significantly more tissue was ‘missing’ in morphine treated groups (B), an effect again attributable to a loss of white matter (C). Morphine did not affect the amount of gray matter remaining (D) at the center of the lesion.
Rostral-Caudal Comparisons
A repeated measures ANOVA across the rostral-caudal extent of the lesion revealed a significant effect of shock on the absolute area (F(1, 38) = 6.33, p < 0.05; Fig. 8a) and the width (F(1, 38) = 4.50, p < 0.05) of the spinal cord sections. Shock increased the volume of damage (F(1, 38) = 11.75, p < 0.005; Fig. 8b), with significantly less white (F(1, 38) = 7.16, p < 0.05; Fig. 8c) and gray matter (F(1, 38) = 11.31, p < 0.005; Fig. 8d) remaining across the rostral-caudal extent of the lesion.
FIG. 8.
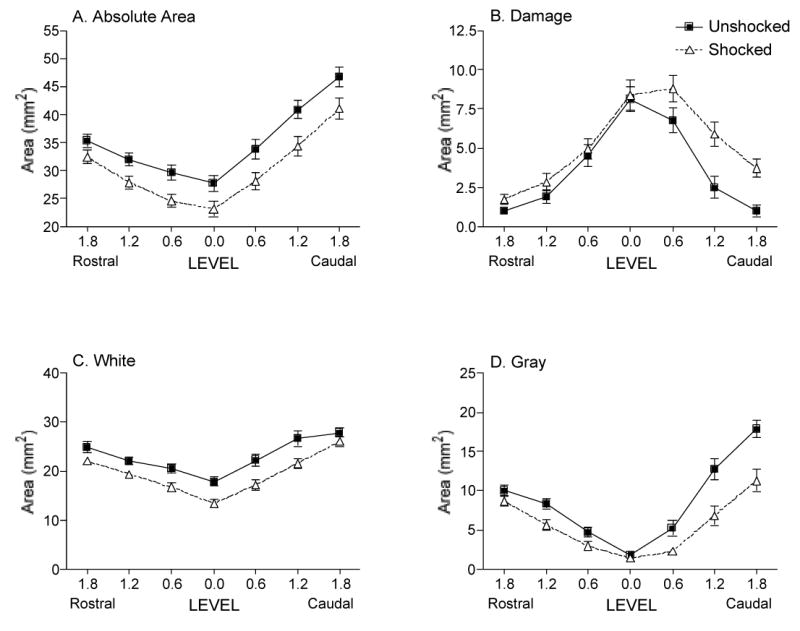
Shock significantly decreased the volume of the spinal cord across the rostral-caudal extent of the lesion (A). It led to a significant increase in the volume of damaged tissue remaining (B), with a consequent decrease in both white (C) and gray (D) matter remaining across the lesioned area.
Analyzing the rostral-caudal extent of the lesion in shocked and unshocked subjects separately (corrected for individual variability) yielded similar trends to those found for the analyses at the lesion center. The only exception was a significant linear contrast for the volume of damage in the shocked subjects (F = 5.12, p < 0.05). Morphine dose-dependently increased damage caudal to the injury in shocked subjects (Fig. 9).
FIG. 9.
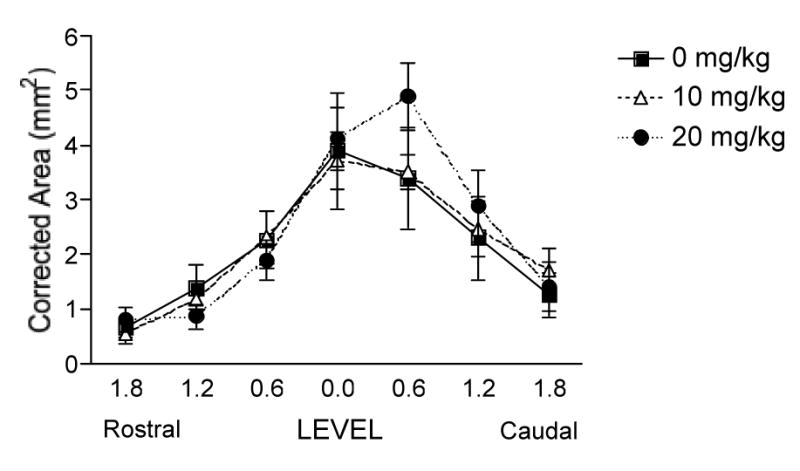
Morphine combined with shock (shocked subjects only) dose-dependently increased the total amount of damaged tissue remaining across the rostral-caudal extent of the lesion.
Discussion
The present study was designed to test whether suppression of nociceptive responses with morphine would protect the injured spinal cord against the adverse effects of uncontrollable electrical stimulation. It did not. In fact, morphine appeared to undermine sensory recovery and led to greater histological damage following the contusion. In addition, Experiment 1 showed that contused rats required more morphine to achieve levels of antinociception comparable to sham controls. Only the 20 mg/kg dose abolished segmental and supraspinal reactivity to intermittent electrical stimulation in contused rats, whereas 10 mg/kg morphine produced antinociception in the sham controls. This finding is counter-intuitive because, if anything, the contusion injury itself should decrease supraspinal reactivity, an effect we expected would combine in an additive fashion with morphine treatment. Yet, the opposite was observed. The significance of this finding is two-fold. First, from a clinical perspective it suggests that injured spinal patients would need significantly larger doses of morphine to attenuate pain, doses that could adversely affect recovery. Secondly, it provides further insight into the mechanisms underlying the effects of morphine. Inflammation, which is inherent to a contusion injury, and the development of central sensitization may change the efficacy and molecular actions of morphine.
As found by Grau et al. [4], uncontrollable shock undermined recovery across a range of measures. Shocked rats displayed decreased locomotor recovery, less weight gain, and delayed recovery of bladder function. Even three weeks after exposure to uncontrollable stimulation, contused rats displayed attenuated performances on beam and ladder tests, underscoring the critical impact of the afferent barrage on axial musculature and fine motor control. Shock also affected the amount of spinal tissue remaining after the injury, producing a significant decrease in the amount of white matter at the lesion center and increasing the amount of damage across the rostral-caudal extent of the lesion. Shock significantly reduced the amount of gray matter remaining rostral and caudal to the center of the lesion.
Rather than attenuating the detrimental effects of shock, morphine exacerbated some of these effects. Rats treated with morphine and shock were significantly more likely to die than saline-treated rats. While none of the saline-treated rats died during the 3-week recovery period, nearly half of the rats that received the highest dose of morphine and shock died. Of the rats that died, 50% died in the first 24 hours while the remaining 50% died approximately one week after treatment. The cause of these deaths is unclear but, given the timing of mortality, it seems unlikely to be due to the direct effects (e.g., respiratory distress) of morphine per se. Subjects tended to die long after (mean = 4.6 days) the drug was pharmacologically active. Shocked rats treated with morphine also displayed tactile allodynia and hyperalgesic responses to shock. The combined effects of electrical stimulation and morphine appear to have sensitized neurons that contribute to spinally mediated reflexes. Increased reactivity to painful, and previously innocuous, stimuli is a significant clinical outcome that could contribute to neuropathic pain symptoms in spinally injured patients.
Our results suggest that the adverse effect uncontrollable stimulation has on recovery develops independent of its ability to engage nociceptive behavior. Morphine dramatically reduced pain-elicited behavior, as measured using both a spinal reflex (tail-withdrawal from radiant heat) and a brain-mediated response (vocalization) to shock. Yet, morphine did not reduce the long-term adverse effects of shock treatment on recovery. Moreover, the adverse effects shock has on recovery are not due to shock-elicited movements; morphine eliminated behavioral reactivity to shock, but this did not restore recovery. Clearly, uncontrollable electrical stimulation can affect recovery independent of its capacity to engage pain-elicited behavior.
Afferent input derived from electrical stimulation may be mediated, in part, by the morphine-insensitive Aβ-fibers. Indeed, the tactile allodynia observed suggests that Aβ-fiber-related neural networks have been significantly altered. Tactile allodynia, an index of neuropathic pain and central sensitization, depends on large diameter Aβ-fibers, and is not susceptible to modulation by spinal opioids [33, 34]. This does not explain, however, the deficits induced by morphine alone, independent of shock exposure. For example, subjects previously treated with morphine had significantly more lesioned tissue than those treated with saline. Clearly, another process is being engaged in the injured system.
Inflammatory processes characteristic of the acute stage of injury are likely to alter the actions of morphine. Pro-inflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), are significantly upregulated 1, 3, and 6 hrs after a contusion injury, returning to background levels 1–2 days after injury [35, 36]. Morphine appears to further increase pro-inflammatory cytokine levels by activating μ- receptors on spinal glial cells [37, 38, 39, 40]. An increased concentration of pro-inflammatory cytokines may be significant at two levels. First pro-inflammatory cytokines appear to block the analgesic effects of opioids [41, 42], which may explain the decreased efficacy of morphine in Experiment 1. Second, pro-inflammatory cytokines have been linked to cytotoxic effects in the nervous system [43, 44, 45]. Yang et al. [36] suggest that there is a critical balance between beneficial and toxic effects that depends on cytokine concentrations.
The increased lesion size observed in morphine treated subjects may also result from NMDA-mediated excitoxicity in the compromised neural environment. Chronic opioid treatment reduces glial glutamate transporter expression [46], which may lead to sustained increases in synaptic excitatory amino acids. Increased synaptic excitatory amino acids, such as glutamate, would potentiate the NMDA receptor. Further, it has been suggested that large doses of systemic morphine may potentiate excitotoxic cell death through a kappa-opiate mediated cascade [47, 48, 49, 50, 51] potentiating NMDA receptor function and enhancing neurotransmitter release in the spinal cord [46, 52, 53]. Interactions between opioid exposure and sensitization of primary afferent nociceptive neurons (i.e., by tissue injury or inflammation) have been previously reported [for review see 54, 55]. Generally, the development of hyperalgesia resulting from injury undermines the antinociceptive effectiveness of opioids [56, 57, 58]. The current study suggests that the administration of opiates may also promote the development of allodynia in the injured system. Binding of morphine to the μ-receptor on the post-synaptic membrane is thought to initiate G-protein mediated protein kinase C (PKC) translocation and activation, promoting the removal of the NMDA receptor Mg2+ plug [59]. With this blockade removed even a small amount of excitatory amino acid release from the presynaptic cell allows Ca2+ influx. Potentiation of the NMDA receptor could promote the development of hyperalgesia/allodynia in the neural system, and could lead to excitotoxic cell death [46]. From a clinical perspective, these results suggest that analgesia induced with morphine may enhance vulnerability to the development of neuropathic pain and to excitotoxic cell death in a compromised (injured) system.
Similarly disturbing are the results indicating that, independent of shock treatment, morphine inhibited some measures of motor and sensory recovery and further undermined the physiological integrity of the cord. Morphine treatment produced a short-term dip in motor recovery (Days 2–3). Kakinohana et al. [60] found that morphine (30 μg) potentiates transient motor dysfunction following a noninjurious interval of spinal ischemia. In the current study, morphine also impaired supraspinal responses to gradually incremented shock stimuli 3 weeks later. This finding suggests that morphine treatment further undermined communication between the brain and spinal cord. As noted previously, unshocked rats treated with morphine had a significantly larger relative lesion at the maximal point of impact, a result attributable to ‘missing’ white matter. Therefore, at a physiological level, the connection between the brain and spinal cord was further disrupted by morphine. Finally, morphine-treated rats gained less weight, which is suggestive of a reduction in general health.
These results are alarming given the widespread use of morphine for the clinical treatment of pain after a spinal cord injury [61, 62]. If one injection of morphine can affect recovery, the potential consequences of long-term treatment could be substantial. Current recommendations suggest that clinicians should monitor the effects of morphine on pain and function [10], modifying treatment if there is reduction of pain without improvement in function. The problem with this is that the assessment period may last 4–6 weeks, and during this period morphine could profoundly affect the plasticity of the spinal system. Also, while humans should not be prescribed a dose of 20 mg/kg of morphine, self-administration and abuse of morphine is not uncommon. While a high dose of morphine alone did not affect mortality, spinal injuries are often accompanied by tissue damage and inflammation (both sources of uncontrollable nociceptive stimulation that affect the plasticity of the neural system, [63]), which may interact with opiate treatment. Given these observations, it is critical that we understand the potential outcomes of combining nociceptive stimulation and morphine in this trauma model. To our knowledge, there have been no clinical trials of morphine and its interaction with recovery of function after a spinal injury. In fact, there have been no empirical studies on the effects of any analgesics on the recovery of function after a spinal injury.
In conclusion, the results of the present study suggest that the negative consequences of electrical stimulation on recovery [4] arise independent of the aversive psychological impact of these stimuli. Systemic morphine administration significantly reduced nociceptive reactivity, but it did not block the devastating effects of uncontrollable stimulation. Morphine also appeared to have an adverse effect on long-term recovery. This was especially alarming given that the injury appeared to reduce morphine effectiveness, which could lead to injured patients self-administering (or requesting) higher doses to alleviate pain. Perhaps as a result of shared cellular mechanisms mediating the effects of uncontrollable stimulation and morphine [46], morphine further undermines functional and physiological recovery in a contusion model.
Acknowledgments
This study was supported by NS41548 and DA020596. We would like to thank Cynthia Lin, Marissa Maultsby, Christine Petrich, and Denise Puga for their comments on an earlier draft of this manuscript. A portion of the data from this study has been previously presented in abstract form.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Christensen MD, Hulsebosch CE. Chronic central pain after spinal cord injury. J Neurotrauma. 1997;14:517–37. doi: 10.1089/neu.1997.14.517. [DOI] [PubMed] [Google Scholar]
- 2.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Zhang FX, Huang F, Lu YJ, Li GD, Bao L, Xiao HS, Zhang X. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. Eur J Neurosci. 2004;19:871–83. doi: 10.1111/j.0953-816x.2004.03121.x. [DOI] [PubMed] [Google Scholar]
- 4.Grau JW, Washburn SN, Hook MA, Ferguson AR, Crown ED, Garcia G, Bolding KA, Miranda RC. Uncontrollable stimulation undermines recovery after spinal cord injury. J Neurotrauma. 2004;21:1795–817. doi: 10.1089/neu.2004.21.1795. [DOI] [PubMed] [Google Scholar]
- 5.Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci. 1986;6:1433–42. doi: 10.1523/JNEUROSCI.06-05-01433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sluka KA, Rees H, Chen PS, Tsuruoka M, Willis WD. Capsaicin-induced sensitization of primate spinothalamic tract cells is prevented by a protein kinase C inhibitor. Brain Res. 1997;772:82–6. doi: 10.1016/s0006-8993(97)00876-7. [DOI] [PubMed] [Google Scholar]
- 7.Hook MA, Ferguson AR, Garcia G, Washburn SN, Koehly LM, Grau JW. Monitoring recovery after injury: procedures for deriving the optimal test window. J Neurotrauma. 2004;21:109–18. doi: 10.1089/089771504772695995. [DOI] [PubMed] [Google Scholar]
- 8.Illich PA, King TA, Grau JW. Impact of shock on pain reactivity: I. Whether hypo- or hyperalgesia is observed depends on how pain reactivity is tested. J Exp Psychol Anim Behav Processes. 1995;21:331–47. doi: 10.1037//0097-7403.21.4.331. [DOI] [PubMed] [Google Scholar]
- 9.King TE, Joynes RL, Meagher MW, Grau JW. Impact of shock on pain reactivity: II. Evidence for enhanced pain. J Exp Psychol Anim Behav Processes. 1996;22:265–78. doi: 10.1037//0097-7403.22.3.265. [DOI] [PubMed] [Google Scholar]
- 10.Dworkin RH, Backonja M, Rowbotham MC, Allen RR, Argoff CR, Bennett GJ, Bushnell MC, Farrar JT, Galer BS, Haythornthwaite JA, Hewitt DJ, Loeser JD, Max MB, Saltarelli M, Schmader KE, Stein C, Thompson D, Turk DC, Wallace MS, Watkins LR, Weinstein SM. Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol. 2003;60:1524–34. doi: 10.1001/archneur.60.11.1524. [DOI] [PubMed] [Google Scholar]
- 11.McCarberg B. Contemporary management of chronic pain disorders. J Fam Pract. 2004;53:S11–22. [PubMed] [Google Scholar]
- 12.NWRSCIS, Current and future management of SCI. In NWRSCIS SCI Forum 2000 [On-line]. Available: http://depts.washington.edu/rehab/sci/current_management.html
- 13.Constantini S, Young W. The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg. 1994;80:97–111. doi: 10.3171/jns.1994.80.1.0097. [DOI] [PubMed] [Google Scholar]
- 14.Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–6. doi: 10.1089/neu.1992.9.123. discussion 126–8. [DOI] [PubMed] [Google Scholar]
- 15.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 16.Crown ED, King TE, Meagher MW, Grau JW. Shock-induced hyperalgesia: III. Role of the bed nucleus of the stria terminalis and amygdaloid nuclei. Behav Neurosci. 2000;114:561–73. [PubMed] [Google Scholar]
- 17.McLemore S, Crown ED, Meagher MW, Grau JW. Shock-induced hyperalgesia: II. Role of the dorsolateral periaqueductal gray. Behav Neurosci. 1999;113:539–49. doi: 10.1037//0735-7044.113.3.539. [DOI] [PubMed] [Google Scholar]
- 18.Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord: IV. Induction and retention of the behavioral deficit observed after noncontingent shock. Behav Neurosci. 2002;116:1032–51. doi: 10.1037//0735-7044.116.6.1032. [DOI] [PubMed] [Google Scholar]
- 19.Meagher MW, Chen PS, Salinas JA, Grau JW. Activation of the opioid and nonopioid hypoalgesic systems at the level of the brainstem and spinal cord: does a coulometric relation predict the emergence or form of environmentally induced hypoalgesia? Behav Neurosci. 1993;107:493–505. doi: 10.1037//0735-7044.107.3.493. [DOI] [PubMed] [Google Scholar]
- 20.Soblosky JS, Colgin LL, Chorney-Lane D, Davidson JF, Carey ME. Ladder beam and camera video recording system for evaluating forelimb and hindlimb deficits after sensorimotor cortex injury in rats. J Neurosci Methods. 1997;78:75–83. doi: 10.1016/s0165-0270(97)00131-3. [DOI] [PubMed] [Google Scholar]
- 21.Hicks SP, D'Amato CJ. Motor-sensory cortex-corticospinal system and developing locomotion and placing in rats. Am J Anat. 1975;143:1–42. doi: 10.1002/aja.1001430102. [DOI] [PubMed] [Google Scholar]
- 22.Von Euler M, Akesson E, Samuelsson EB, Seiger A, Sundstrom E. Motor performance score: a new algorithm for accurate behavioral testing of spinal cord injury in rats. Exp Neurol. 1996;137:242–54. doi: 10.1006/exnr.1996.0023. [DOI] [PubMed] [Google Scholar]
- 23.Von Euler M, Seiger A, Sundstrom E. Clip compression injury in the spinal cord: a correlative study of neurological and morphological alterations. Exp Neurol. 1997;145:502–10. doi: 10.1006/exnr.1997.6481. [DOI] [PubMed] [Google Scholar]
- 24.Beattie MS. Anatomic and behavioral outcome after spinal cord injury produced by a displacement controlled impact device. J Neurotrauma. 1992;9:157–9. doi: 10.1089/neu.1992.9.157. discussion 159–60. [DOI] [PubMed] [Google Scholar]
- 25.Behrmann DL, Bresnahan JC, Beattie MS, Shah BR. Spinal cord injury produced by consistent mechanical displacement of the cord in rats: behavioral and histologic analysis. J Neurotrauma. 1992;9:197–217. doi: 10.1089/neu.1992.9.197. [DOI] [PubMed] [Google Scholar]
- 26.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996;139:244–56. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 27.Bresnahan JC, Beattie MS, Todd FD, 3rd, Noyes DH. A behavioral and anatomical analysis of spinal cord injury produced by a feedback-controlled impaction device. Exp Neurol. 1987;95:548–70. doi: 10.1016/0014-4886(87)90299-8. [DOI] [PubMed] [Google Scholar]
- 28.Olby NJ, Blakemore WF. A new method of quantifying the extent of tissue loss following spinal cord injury in the rat. Exp Neurol. 1996;138:82–92. doi: 10.1006/exnr.1996.0049. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson AR, Hook MA, Garcia G, Bresnahan JC, Beattie MS, Grau JW. A simple post hoc transformation that improves the metric properties of the BBB scale for rats with moderate to severe spinal cord injury. J Neurotrauma. 2004;21:1601–13. doi: 10.1089/neu.2004.21.1601. [DOI] [PubMed] [Google Scholar]
- 30.Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 31.Baranauskas G, Nistri A. Sensitization of pain pathways in the spinal cord: Cellular mechanisms. Prog Neurobiol. 1998;54:349–365. doi: 10.1016/s0301-0082(97)00067-1. [DOI] [PubMed] [Google Scholar]
- 32.Riedel W, Neeck G. Nociception, pain, and antinociception: Current concepts. Z Rheumatol. 2001;60:404–415. doi: 10.1007/s003930170003. [DOI] [PubMed] [Google Scholar]
- 33.Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–23. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- 34.Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–33. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- 35.Yang L, Blumbergs PC, Jones NR, Manavis J, Sarvestani GT, Ghabriel MN. Early expression and cellular localization of proinflammatory cytokines interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in human traumatic spinal cord injury. Spine. 2004;29:966–71. doi: 10.1097/00007632-200405010-00004. [DOI] [PubMed] [Google Scholar]
- 36.Yang L, Jones NR, Blumbergs PC, Van Den Heuvel C, Moore EJ, Manavis J, Sarvestani GT, Ghabriel MN. Severity-dependent expression of pro-inflammatory cytokines in traumatic spinal cord injury in the rat. J Clin Neurosci. 2005;12:276–84. doi: 10.1016/j.jocn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 37.Maderspach K, Solomonia R. Glial and neuronal opioid receptors: apparent positive cooperativity observed in intact cultured cells. Brain Res. 1988;441:41–7. doi: 10.1016/0006-8993(88)91381-9. [DOI] [PubMed] [Google Scholar]
- 38.Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–6. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 39.Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–65. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–34. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- 41.Gul H, Yildiz O, Dogrul A, Yesilyurt O, Isimer A. The interaction between IL-1beta and morphine: possible mechanism of the deficiency of morphine-induced analgesia in diabetic mice. Pain. 2000;89:39–45. doi: 10.1016/S0304-3959(00)00343-2. [DOI] [PubMed] [Google Scholar]
- 42.Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A. 2002;99:10276–81. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–8. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 44.Gruol DL, Nelson TE. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol Neurobiol. 1997;15:307–39. doi: 10.1007/BF02740665. [DOI] [PubMed] [Google Scholar]
- 45.Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–25. doi: 10.1016/s0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- 46.Mao J, Sung B, Ji RR, Lim G. Neuronal apoptosis associated with morphine tolerance: evidence for an opioid-induced neurotoxic mechanism. J Neurosci. 2002;22:7650–61. doi: 10.1523/JNEUROSCI.22-17-07650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faden AI. Opioid and nonopioid mechanisms may contribute to dynorphin's pathophysiological actions in spinal cord injury. Ann Neurol. 1990;27:67–74. doi: 10.1002/ana.410270111. [DOI] [PubMed] [Google Scholar]
- 48.Chen ZR, Irvine RJ, Somogyi AA, Bochner F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991;48:2165–71. [Google Scholar]
- 49.Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271:1630–7. [PubMed] [Google Scholar]
- 50.Mignat C, Wille U, Ziegler A. Affinity profiles of morphine, codeine, dihydrocodeine and their glucuronides at opioid receptor subtypes. Life Sci. 1995;56:793–9. doi: 10.1016/0024-3205(95)00010-4. [DOI] [PubMed] [Google Scholar]
- 51.Mao J. NMDA and opioid receptors: their interactions in antinociception, tolerance and neuroplasticity. Brain Res Brain Res Rev. 1999;30:289–304. doi: 10.1016/s0165-0173(99)00020-x. [DOI] [PubMed] [Google Scholar]
- Skilling SR, Sun X, Kurtz HJ, Larson AA. Selective potentiation of NMDA-induced activity and release of excitatory amino acids by dynorphin: possible roles in paralysis and neurotoxicity. Brain Res. 1992;575:272–8. doi: 10.1016/0006-8993(92)90090-v. [DOI] [PubMed] [Google Scholar]
- 53.Koetzner L, Hua XY, Lai J, Porreca F, Yaksh T. Nonopioid actions of intrathecal dynorphin evoke spinal excitatory amino acid and prostaglandin E2 release mediated by cyclooxygenase-1 and -2. J Neurosci. 2004;24:1451–8. doi: 10.1523/JNEUROSCI.1517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dickenson AH. Plasticity: implications for opioid and other pharmacological interventions in specific pain states. Behav Brain Sci. 1997;20:392–403. doi: 10.1017/s0140525x97241488. discussion 435–513. [DOI] [PubMed] [Google Scholar]
- 55.Mao J, Mayer DJ. Spinal cord neuroplasticity following repeated opioid exposure and its relation to pathological pain. Ann NY Acad Sci. 2001;933:175–84. doi: 10.1111/j.1749-6632.2001.tb05823.x. [DOI] [PubMed] [Google Scholar]
- 56.Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995;62:259–74. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 57.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. Inhibition by spinal morphine of the tail-flick response is attenuated in rats with nerve ligation injury. Neurosci Lett. 1995;199:83–6. doi: 10.1016/0304-3940(95)12026-z. [DOI] [PubMed] [Google Scholar]
- 58.Wegert S, Ossipov MH, Nichols ML, Bian D, Vanderah TW, Malan TP, Jr, Porreca F. Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]
- 59.Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–3. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 60.Kakinohana M, Marsala M, Carter C, Davison JK, Yaksh TL. Neuraxial morphine may trigger transient motor dysfunction after a noninjurious interval of spinal cord ischemia: a clinical and experimental study. Anesthesiology. 2003;98:862–70. doi: 10.1097/00000542-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 61.Warms CA, Turner JA, Marshall HM, Cardenas DD. Treatments for chronic pain associated with spinal cord injuries: many are tried, few are helpful. Clin J Pain. 2002;18:154–63. doi: 10.1097/00002508-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Widerstrom-Noga EG, Turk DC. Types and effectiveness of treatments used by people with chronic pain associated with spinal cord injuries: influence of pain and psychosocial characteristics. Spinal Cord. 2003;41:600–9. doi: 10.1038/sj.sc.3101511. [DOI] [PubMed] [Google Scholar]
- 63.Hook MA, Huie JR, Grau JW. Program No. 783.12. 2005. Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience; Instrumental learning in spinalized rats: the induction of central sensitization undermines behavioral plasticity in the spinal cord. CD-ROM. [Google Scholar]