

Abstract
Rhodopsin kinase (RK), a rod photoreceptor cytosolic enzyme, plays a key role in the normal deactivation and recovery of the photoreceptor after exposure to light. To date, three different mutations in the RK locus have been associated with Oguchi disease, an autosomal recessive form of stationary night blindness in man characterized in part by delayed photoreceptor recovery [Yamamoto, S., Sippel, K. C., Berson, E. L. & Dryja, T. P. (1997) Nat. Genet. 15, 175–178]. Two of the mutations involve exon 5, and the remaining mutation occurs in exon 7. Known exon 5 mutations include the deletion of the entire exon sequence [HRK(X5 del)] and a missense change leading to a Val380Asp substitution in the encoded product (HRKV380D). The mutation in exon 7 is a 4-bp deletion in codon 536 leading to premature termination of the encoded polypeptide [HRKS536(4-bp del)]. To provide biochemical evidence for pathogenicity of these mutations, wild-type human rhodopsin kinase (HRK) and mutant forms HRKV380D and HRKS536(4-bp del) were expressed in COS7 cells and their activities were compared. Wild-type HRK catalyzed light-dependent phosphorylation of rhodopsin efficiently. In contrast, both mutant proteins were markedly deficient in catalytic activity with HRKV380D showing virtually no detectible activity and HRKS536(4-bp del) only minimal light-dependent activity. These results provide biochemical evidence to support the pathogenicity of the RK mutations in man.
Keywords: phototransduction, retina, isoprenylation human, retinopathy
Phosphorylation of the light-activated form of the visual pigment rhodopsin plays a key role in the deactivation of the rod photoreceptor after exposure to light (2). Phototransduction initiated by light is quenched rapidly as a rod cytosolic enzyme, rhodopsin kinase (RK), recognizes and phosphorylates photoexcited rhodopsin. The phosphorylated photoactive rhodopsin is then bound by a regulatory cytosolic protein, arrestin, and is thereby uncoupled from the G protein transducin downstream in the phototransduction cascade. This simple rapid deactivation pathway, mediated in part by RK, readily restores sensitivity to the rod photoreceptor after exposure to light. Slower steps leading to the full recovery of the photoreceptor to its maximally sensitive state follow with the regeneration of all of the visual pigment from its activated state. Without functional RK catalyzing light-dependent phosphorylation of rhodopsin, a bright flash of light would saturate the photoreceptor with activated intermediates and render it insensitive for a prolonged period of time until all of the residual photoactivated visual pigment is cleared by the slower secondary recovery pathways. Recently, the physiologic importance of RK and arrestin in photoreceptor deactivation has been highlighted by the discovery of mutations in the loci encoding these proteins in patients with Oguchi disease, an autosomal recessive form of stationary night blindness (1, 3–5). The biochemical effects of these mutations have not been characterized previously.
RK is a specialized member of a family of G protein-dependent receptor kinases involved in the stimulus-dependent deactivation of a diverse group of signal transduction pathways in various cells (6). Like other G protein-dependent receptor kinases, RK is a serine/threonine kinase composed of a core catalytic domain flanked by an N- and C-terminal domain (7). The catalytic domain, which has invariant structural elements in common with other protein kinases, most likely contains the active site of RK (8). The N-terminal domain may play a role in binding of the RK to rhodopsin as suggested from immunochemical studies with antibodies against the N-terminal domain (9). The C-terminal domain contains the major autophosphorylation sites (Ser488 and Thr489 in bovine RK) thought to be involved in regulation of RK activity (10). Posttranslational processing of the C-terminal domain of bovine RK by farnesylation via Cys558 of the C-terminal prenylation consensus sequence, CaaX (11–13), has been implicated in the translocation of the enzyme from cytosol into the disk membranes upon light activation (14). Based on the above studies, it is likely that the N- and C-terminal domains of RK play roles in modulating substrate specificity, binding, and catalysis.
Recently, mutations in the RK locus have been discovered in patients with Oguchi disease, an autosomal recessive form of night blindness characterized by prolonged dark adaptation, abnormal sensitivity to light, and a golden-brown discoloration of the fundus upon light adaptation called the Mizuo phenomenon (1, 4, 5). Affected individuals with Oguchi disease in two pedigrees of European ancestry were found to be homozygous for a deletion encompassing exon 5 of the RK locus. In a third pedigree, the affected individuals were compound heterozygotes carrying a missense mutation in exon 5 of one allele leading to a Val380Asp substitution in the mutant protein (HRKV380D) and a 4-bp deletion in exon 7 of the other allele, designated Ser536(4-bp del), leading to the generation of a truncated protein [HRKS536(4-bp del)]. Although cosegregation of these mutations with the disease supports their pathogenicity, other definitive evidence as to how these mutations may alter the activity of RK has thus far not been available.
In this paper, we provide biochemical evidence in support of the pathogenicity of the above gene defects. We expressed the mutant proteins HRKV380D and HRKS536(4-bp del) and compared their activity to wild-type human rhodopsin kinase (HRK) demonstrating the mutant proteins to be markedly deficient in light-dependent rhodopsin phosphorylating activity.
MATERIALS AND METHODS
Construction of Expression Plasmids.
Most cDNA modifications and mutagenesis steps preceding the construction of the final eukaryotic expression plasmids were carried out in Bluescribe (Stratagene) vector. Sequence alterations were verified by dideoxy chain termination method using [α-33P] dideoxynucleotide triphosphates and the ThermoSequenase kit supplied by Amersham.
All expression plasmids were derived from the eukaryotic expression vector pCMV5 (15) modified to eliminate the EcoRI site. The expression plasmid pCMV-HRK (Fig. 1A) encoding wild-type HRK was constructed by ligating HindIII-XbaI digested vector to a HindIII-NheI cDNA cassette containing the wild-type HRK coding sequence flanked by 33 bases of 5′- and 85 bases of 3′- nontranslated sequence. The expression plasmid pCMV-HRKV380D was derived from pCMV-HRK by replacing the HindIII-EcoRI fragment with a comparable mutant fragment encoding the Val380Asp substitution. This mutant fragment was generated from the wild-type cDNA by in vitro mutagenesis using QuickChange kit (Stratagene) in conjunction with a pair of complementary mutagenic primers [sense primer: 5′-CTTTGCCCTGGGGGACACCCTGTATGAGATG-3′ (codons 375–385)]. The expression plasmid pCMV-HRKS536(4-bp del) was derived from pCMV-HRK by replacement of the EcoRI–Avr II fragment with a comparable fragment containing the Ser536(4-bp del) mutation amplified from the DNA of Oguchi patient 303–002 (1) (kindly provided by Dr. Dryja). The PCR amplification was carried out with Taq polymerase by using the primer pair: 5′-GTGGCCTTTGACAAAACAGA-3′ (codons 496–502) and 5′-TATGGGTCCTTTTCCTCCAC-3′ (3′-untranslated sequence, nucleotides 1707–1726) in the presence of 8% dimethyl sulfoxide essentially as described (16).
Figure 1.
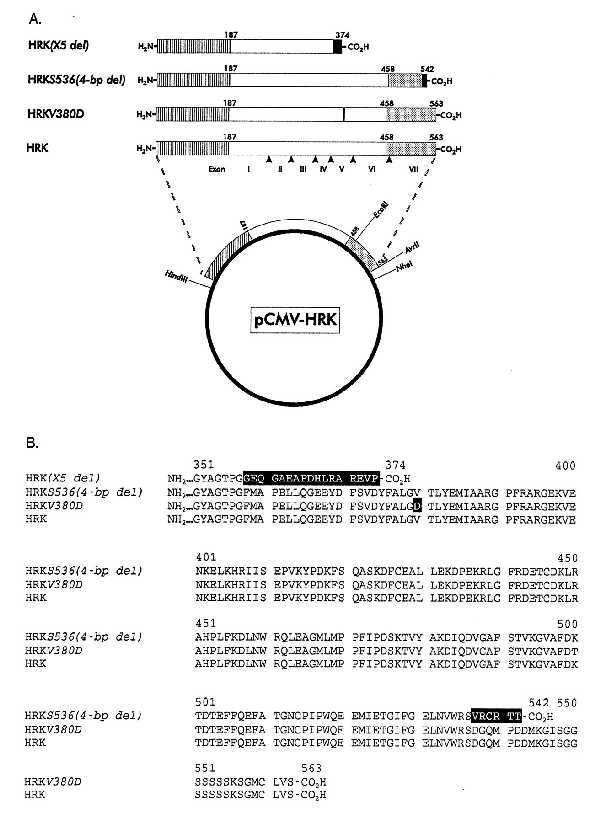
Architecture (A) and sequence (B) of wild-type HRK and the mutant proteins HRK(X5 del), HRKS536(4-bp del), and HRKV380D correlated with Oguchi disease. The pCMV5-based expression plasmids were constructed as described in Materials and Methods. Striped, clear, and shaded boxes represent the N-terminal, catalytic, and C-terminal domain of RK, respectively. Black boxes indicate the mutated sequences. Arrowheads mark the boundaries of the exons relative to the primary structure of the protein. Roman numerals indicate the exon number. Arabic numerals indicate the position of residues in the proteins. The relative positions of several unique restriction sites used for construction of expression plamids are indicated on the plasmid. Vector sequences are represented by a heavy line.
Heterologous Expression and Biochemical Analysis of Wild-Type and Mutant Proteins in COS7 Cells.
Transfection of the expression plasmids into subconfluent monolayers of COS7 cells grown in 175-cm2 flasks was carried out in the presence of diethylaminoethyl dextran by standard methods (15). Forty-eight hours after transfection the cells were disrupted in 0.5 ml of cold lysis buffer (10) containing 30 μM benzamidine and centrifuged at 140,000 × g (Beckman OptimaTM TLX) for 7 min at 4°C. To minimize proteolysis, the supernatants were assayed within 8 hr of the lysis for the presence of immunoreactive protein and light-dependent rhodopsin phosphorylating activity without further purification.
For immunoblot analysis, supernatants from COS7 cells were fractionated electrophoretically on 10% SDS-polyacrylamide minigels and transferred onto nitrocellulose membranes. Immunoblots were developed according to standard protocols using alkaline phosphatase-conjugated goat secondary antibody (Sigma) following incubation with either of two mouse mAbs against HRK (kindly provided by Dr. Palczewski, University of Washington), G8 against the C-terminal domain, or D11 against the N-terminal domain (17), each at 1:5,000 dilution. Anti-peptide RK antibodies GS16 against p216–237 and GS18 against p483–497 (9) at 1:10,000 dilution in 1% goat serum were used in some immunoblotting experiments as primary antibodies to further confirm the data obtained using mAbs.
Rhodopsin phosphorylating activities of the same COS7 cell supernatants were measured at 22°C using urea-washed bovine rod outer segments (20 μM) (18, 19) and [γ-32P]ATP (100 μM, 2,000 dpm/pmol) as substrates in 20 mM 1,3-Bis-[Tris(hydroxymethyl)methylamino]propane (pH 7.4) containing 5 mM MgCl2 (20). After incubation in light or darkness, the reactions were quenched by the addition of loading buffer (18, 21) and subjected to electrophoresis on 10% SDS-polyacrylamide gels followed by Coomassie staining, autoradiography, scintillation counting of excised rhodopsin bands, and phosphorescence imaging. Protein concentration was measured by the method of Bradford (22).
RESULTS AND DISCUSSION
Oguchi disease is an autosomal recessive form of stationary night blindness classically characterized by the phosphorescent metallic color of the light-adapted fundus (the Mizuo phenomenon) (4, 5) and marked impairment in the rate of dark adaptation. Recently, mutations in the photoreceptor recovery pathway involving the RK locus have been correlated with Oguchi disease (1). To determine the biochemical effect of these mutations, wild-type HRK and the mutant proteins HRKV380D and HRKS536(4-bp del) were expressed in COS7 cells and their activities were compared. The plasmid pCMV-HRK encodes wild-type HRK with an estimated molecular weight of 64 kDa (Fig. 1). Plasmids pCMV-HRKV380D and pCMV-HRKS536(4-bp del) encode the mutant proteins HRKV380D and HRKS536(4-bp del) with estimated molecular weights of 64 and 61 kDa, respectively.
As seen from Fig. 2, COS7 cells transfected with the above expression plasmids expressed appropriate immunoreactive products. The immunoblot probed with the mAb D11, directed against the RK N-terminal domain, (Fig. 2A) reveals a band of 64 kDa in the lanes containing extracts from COS7 cells transfected with pCMV-HRK and pCMV-HRKV380D and a band of slightly lower Mr in the lane containing the extract from COS7 cells transfected with pCMV-HRKS536(4-bp del). Although the observed difference in electrophoretic mobility is slight, multiple immunoblotting experiments have confirmed the lower molecular weight of the immunoreactive band in COS7 cells transfected with pCMV-HRKS536(4-bp del) as compared with the wild-type protein. pCMV5-transfected cell extracts showed no immunoreactive bands as seen in Fig. 2A, confirming the specificity of the mAbs. The above data indicate the expression of polypeptide chains of expected size and immunoreactivity from the expression plasmids.
Figure 2.
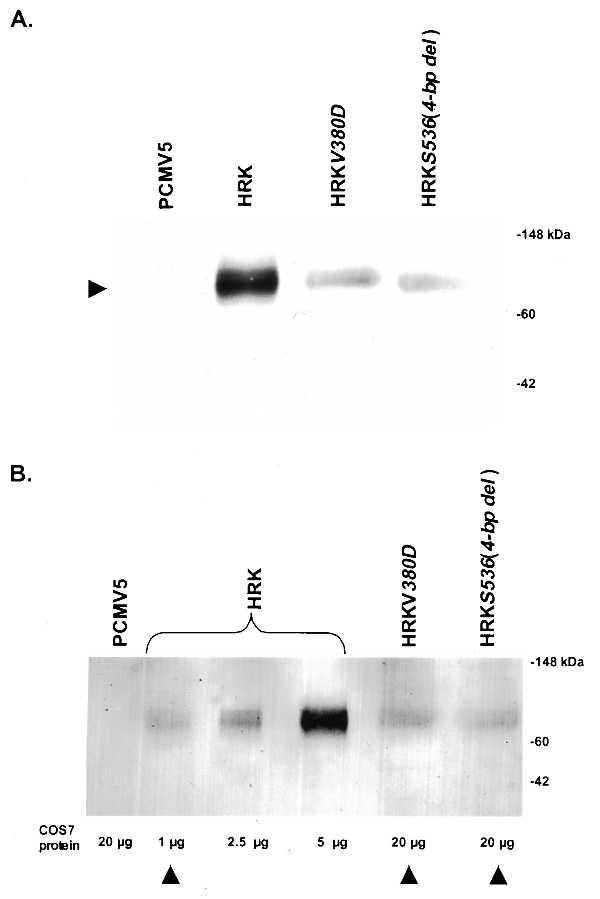
Expression (A) and relative content (B) of wild-type and mutant RKs in COS7 cells. Immunoblots of supernatants from COS7 cells transfected with pCMV5 plasmid or expression plasmids encoding wild-type HRK, HRKV380D, and HRKS536(4-bp del) were developed after reacting with mAbs G8 (A) and D11 (B) (17) against HRK. Seven micrograms of supernatant COS7 cells expressing HRK and 30 μg of the other COS7 supernatant protein were applied to each lane (A). The relative quantity of total protein loaded in each lane is indicated below each lane (B). The relative amounts of immunoreactive protein in the various COS cell supernatants is semiquantitatively determined by comparing the intensity of bands using a densitometer. The position of molecular weight standards are marked. Arrowheads (B) indicate the amount of each immunoreactive protein used for the corresponding activity assay (see Fig. 3).
Based on semiquantitative and qualitative comparisons of band intensities in Fig. 2B, the levels of immunoreactive protein in COS7 cells transfected with mutant expression plasmids encoding HRKV380D and HRKS536(4-bp del) are ≈10% of that of cells transfected with the wild-type HRK expression plasmid. Using two different anti-peptide antibodies GS16 and GS18, directed against known RK epitopes (9), similar results were obtained in general, confirming the observation that the immunoreactive protein content in COS7 cells expressing mutant proteins is lower as compared with cells expressing wild-type RK (data not shown). Although the difference in the levels of immunoreactive protein in different COS7 cell cultures can be a function of transfection efficiencies, the consistently lower immunoreactive protein content in COS7 cells expressing mutant proteins may implicate increased susceptibility of the mutant proteins to proteolysis as an additional mechanism for the inactivation of the gene product in vivo.
To determine whether mutations in the RK locus found in patients with Oguchi disease lead to catalytic deficiency, light-dependent rhodopsin phosphorylating activities of the wild-type RK and its mutant forms were compared. As indicated by arrowheads in Fig. 2B, we intentionally used approximately equal amounts of the two mutant proteins in the corresponding activity assays as compared with the wild-type protein to ensure that any observed deficit in the rhodopsin phosphorylating activity that may be anticipated with mutant proteins would be unequivocally attributable to a deficiency in their specific activity rather than in their content in COS7 cell supernatants. As seen from the autoradiogram in Fig. 3, the supernatants from COS7 cells expressing wild-type HRK catalyzed efficient incorporation of 32P into bovine rhodopsin in the light. In contrast, the supernatants from the cells expressing the mutant protein HRKV380D failed to show any detectable rhodopsin phosphorylating activity. The supernatants from COS7 cells transfected with pCMV-HRKS536(4-bp del) also were markedly deficient in light-dependent activity exhibiting <3% of the wild-type activity. These data clearly suggest a marked deficit in the activity of mutant proteins HRKV380D and HRKS536(4-bp del) as compared with the wild-type HRK.
Figure 3.
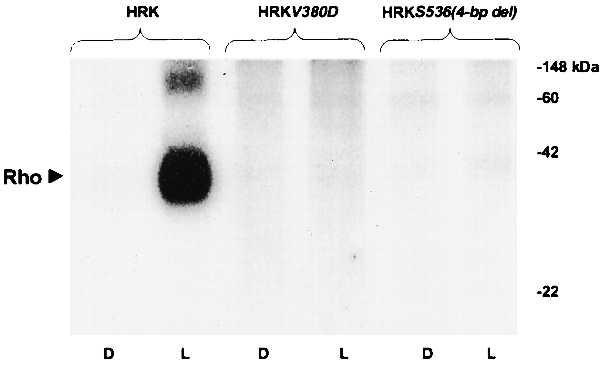
Rhodopsin phosphorylation by wild-type RK and the mutant forms HRKV380D and HRKS536(4-bp del). Autoradiogram of electrophoretically fractionated urea-washed rod outer segments incubated with supernatants from COS7 cells transfected with expression plasmids encoding wild-type HRK, HRKV380D, and HRKS536(4-bp del) in the presence of [γ-32P]ATP for 20 min in the dark (D) or the light (L). The enzyme assays were run at saturating substrate concentrations as detailed in Materials and Methods. The levels of RK immunoreactive protein in each assay are indicated by the arrowheads in Fig. 2B. The reactions were quenched by addition of SDS loading buffer at room temperature and an aliquot from each, containing 0.7 nmol of rhodopsin, was fractionated on 10% SDS-polyacrylamide gels before autoradiography. The positions of rhodopsin and molecular weight standards are marked.
The above study provides compelling evidence for the inactivation of RK by the Oguchi mutations involving exon 5 of the RK gene. Our data directly demonstrate that the single point mutation in the region of the RK catalytic domain encoded by exon 5 leading to the Val380Asp substitution is sufficient to render the gene product HRKV380D nonfunctional in vitro. Based on these data, we can also extrapolate the inactivity of any gene product that may be derived from a defective RK locus altered by a gross deletion encompassing the entire exon 5 as reported in three separate pedigrees with Oguchi disease (1, 21, 23). Recently, the inactivity of the polypeptide derived from this locus has been confirmed directly by biochemical studies similar to the ones reported here (23).
The exact basis for the observed severe functional deficit of HRKS536(4-bp del) remains somewhat speculative. The 4-bp deletion at codon 536 of the HRK gene associated with Oguchi disease leads to a gene product which is truncated at the C-terminus by only 21 residues and lacks a prenylation consensus sequence as compared with the wild-type protein. The absence of a typical C-terminal prenylation consensus sequence (13) and consequent loss of prenylation may in part explain the severe deficit observed in the activity of the encoded protein. Normally, isoprenylation plays a key role in the interaction of the wild-type RK with the light-activated form of rhodopsin. However, nonprenylated bovine RK engineered with a Cys558Ser substitution retains at least 20% of its light-dependent activity (14), a far greater proportion of the wild-type activity as compared with the mutant studied here having <3% of the activity of the wild-type RK. Loss of other functionally important structural elements in the C-terminal domain of RK in addition to the prenylation site may therefore need to be implicated as the cause for the observed marked deficiency in the activity of HRKS536(4-bp del).
According to the above data, the defects seen in the RK locus of Oguchi patients are in effect all null mutations and should therefore lead to very similar clinical phenotypes. The comparison of two Oguchi patients, 303–003 and 303–002, with distinct known genotypes (1, 24) has revealed virtually indistinguishable clinical characteristics with comparable dark adaptation and photoreceptor recovery kinetics in both patients. Patient 303–003, with a large deletion encompassing exon 5 of RK in the homozygous state would not be expected to have any RK activity in his photoreceptors. Patient 303–002, a compound heterozygote with a missense Val380Asp mutation in one allele and Ser536(4-bp del) in the other, would be expected to have essentially no functional RK activity in the photoreceptors as well based on our in vitro experiments. The equivalence in the reported phenotypes of these two patients is consistent with our results showing virtual inactivation of the RK by the mutations seen in Oguchi patients.
The study reported here provides biochemical evidence in support of the pathogenicity of the RK mutations found in patients with Oguchi disease. In addition to genotypic variation, substantial phenotypic heterogeneity has been reported among Oguchi patients of unknown genotype. Absence of rod adaptation (25), persistently elevated rod threshold (26), and cone system abnormalities (25) are some of the reported phenotypic variations in this disorder. To date, no other pathogenic mutations in the RK locus have been reported although a few mutations in this locus have been associated with retinitis pigmentosa (27). The basis of the phenotypic heterogeneity in Oguchi disease remains to be explored in detail at the molecular and biochemical level. It is also tempting to speculate that similar forms of stationary night blindness such as fundus albipunctatus (28), characterized by prolongation of both photoreceptor recovery and dark adaptation phase, may also be caused by mutations in the deactivation pathway.
Acknowledgments
We thank Dr. A. Albert in whose lab numerous initial experiments were performed. We also are grateful to Dr. K. Palczewski for his helpful advice, critical comments on the manuscript, and the generous gift of monoclonal and polyclonal antipeptide antibodies for RK. We also thank Dr. T. Dryja for providing the DNA from an Oguchi patient. This work was supported in part by an internal grant (BRSG 150-8323) and Grant GIA 97021, which was funded by Fight For Sight, Inc., NY Research Division of Prevent Blindness America in memory of Silas Adelsheim and wife Peggy.
ABBREVIATIONS
- RK
rhodopsin kinase
- HRK
human rhodopsin kinase
References
- 1.Yamamoto S, Sippel K C, Berson E L, Dryja T P. Nat Genet. 1997;15:175–178. doi: 10.1038/ng0297-175. [DOI] [PubMed] [Google Scholar]
- 2.Palczewski K. Eur J Biochem. 1997;248:261–269. doi: 10.1111/j.1432-1033.1997.00261.x. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. Nat Genet. 1995;10:360–362. doi: 10.1038/ng0795-360. [DOI] [PubMed] [Google Scholar]
- 4.Berson E L. In: Principles and Practice of Ophthalmology. Albert D M, Jakobiec F A, editors. Philadelphia: Saunders; 1994. pp. 1214–1237. [Google Scholar]
- 5.Carr R E. In: Principles and Practice of Clinical Electrophysiology of Vision. Heckenlively J R, Arden G B, editors. St. Louis: Mosby; 1991. pp. 713–720. [Google Scholar]
- 6.Premont R T, Inglese J, Lefkowitz R J. FASEB J. 1995;9:175–181. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- 7.Inglese J, Freedman N J, Koch W J, Lefkowitz R J. J Biol Chem. 1993;268:23735–23738. [PubMed] [Google Scholar]
- 8.Hanks S A, Quinn A M. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- 9.Palczewski K, Buczyłko J, Lebioda L, Crabb J W, Polans A S. J Biol Chem. 1993;268:6004–6013. [PubMed] [Google Scholar]
- 10.Palczewski K, Ohguro H, Premont R T, Inglese J. J Biol Chem. 1995;270:15294–15298. doi: 10.1074/jbc.270.25.15294. [DOI] [PubMed] [Google Scholar]
- 11.Lorenz W, Inglese J, Palczewski K, Onorato J J, Caron M G, Lefkowitz R J. Proc Natl Acad Sci USA. 1991;88:8715–8719. doi: 10.1073/pnas.88.19.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inglese J, Glickman J F, Lorenz W, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:1422–1425. [PubMed] [Google Scholar]
- 13.Clarke S. Annu Rev Biochem. 1992;61:355–386. doi: 10.1146/annurev.bi.61.070192.002035. [DOI] [PubMed] [Google Scholar]
- 14.Inglese J, Koch W J, Caron M G, Lefkowitz R J. Nature (London) 1992;359:147–150. doi: 10.1038/359147a0. [DOI] [PubMed] [Google Scholar]
- 15.Andersson S, Davis D L, Dahlbäck H, Jörnvall H, Russell D W. J Biol Chem. 1989;264:8222–8229. [PubMed] [Google Scholar]
- 16.Khani S C, Abitbol M, Yamamoto S, Maravic-Magovcevic I, Dryja T P. Genomics. 1996;35:571–576. doi: 10.1006/geno.1996.0399. [DOI] [PubMed] [Google Scholar]
- 17.Zhao, X., Huang, J., Khani, S. C. & Palczewski, K. (1998) J. Biol. Chem. (in press). [DOI] [PubMed]
- 18.Papermaster D S, Dreyer W J. Biochemistry. 1974;13:2438–2444. doi: 10.1021/bi00708a031. [DOI] [PubMed] [Google Scholar]
- 19.Shichi H, Somers R L. J Biol Chem. 1978;253:7040–7046. [PubMed] [Google Scholar]
- 20.Palczewski K. Methods Neurosci. 1993;15:217–225. [Google Scholar]
- 21.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Bradford M. Anal Biochem. 1976;72:248–252. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Cideciyan A V, Zhao X, Nelson L, Khani S C, Jacobson S G, Palczewski K. Proc Natl Acad Sci USA. 1998;95:328–333. doi: 10.1073/pnas.95.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carr R E, Gouras P. Arch Ophthalmol. 1965;73:646–656. doi: 10.1001/archopht.1965.00970030648010. [DOI] [PubMed] [Google Scholar]
- 25.Dosesschate J T, Alpern M, Lee G B, Heyner F. Docum Ophthalmol. 1966;20:406–419. doi: 10.1007/BF00165431. [DOI] [PubMed] [Google Scholar]
- 26.Sharp D M, Arden G B, Kemp C M, Hogg C R, Bird A C. Clin Vision Sci. 1990;5:217–230. [Google Scholar]
- 27.Yamamoto S, Khani S C, Berson E L, Dryja T P. Exp Eye Res. 1997;65:249–253. doi: 10.1006/exer.1997.9998. [DOI] [PubMed] [Google Scholar]
- 28.Carr R E, Ripps H, Siegel I M. Doc Ophthalmol Proc Ser. 1974;4:193–204. [Google Scholar]