

Abstract
Site-specific proteolysis is an important regulatory mechanism in basic cellular and viral processes. Using the protease of the HIV as a model, a genetic system has been developed for the isolation and characterization of site-specific proteases. The system utilizes the well defined bacteriophage λ regulatory circuit where the viral repressor, cI, is specifically cleaved to initiate the lysogenic-to-lytic switch. The model system is rapid, highly specific, and demonstrates the ability to isolate and characterize enzymes of limited expression or activity. In addition, the system has a significant potential for the selection of clinically relevant mutant enzymes and in the development of anti-protease therapeutics.
Site-specific proteolytic processing plays a significant role in the regulation of cellular processes as diverse as signal transduction (1–4), RNA transcription (5–9), apoptosis (10–14), and development (15–19). In addition, specific processing of viral polypeptides is a critical stage in the replication and maturation of infectious particles (20). Although many general proteases have been identified, few of the enzymes responsible for site-specific processing events have been isolated. As there are inherent difficulties in the purification and characterization of these proteins by classical methodologies, a genetic screen has been developed by using a model enzyme encoded by HIV.
The system is based on the highly characterized bacteriophage λ lytic–lysogenic cycle (21). Expression of the phage-encoded repressor (cI) results in repression of the bacteriophage’s lytic functions. Induction from the lysogenic state is initiated by specific recA-mediated cleavage of the repressor, reducing the steady-state levels. The subsequent expression of cro results in the complete repression of cI expression, the induction of the phage’s replicative functions, and the ultimate lysis of the host cell. This paradigm has been adapted as illustrated in Fig. 1. A recombinant cI repressor containing a specific proteolytic processing site is expressed in Escherichia coli and infected with a λ-cDNA expression library. The expression of a random cDNA has no effect on the function of the recombinant cI repressor (Fig. 1 Left), which prohibits the expression of the phage lytic replication functions. If, however, a phage encodes the appropriate specific protease (Fig. 1 Right), the target repressor is cleaved and the phage enters the lytic replication cycle, resulting in selective isolation of the appropriate phage. Adaptation of the λ system provides several significant advantages: (i) the structure and regulation of the λ cI repressor is well characterized; (ii) the system adapts the dual λ cI-cro regulatory circuit that is sensitively controlled by specific proteolysis; and (iii) as λ has been a valuable research tool, an extensive array of reagents is available.
Figure 1.
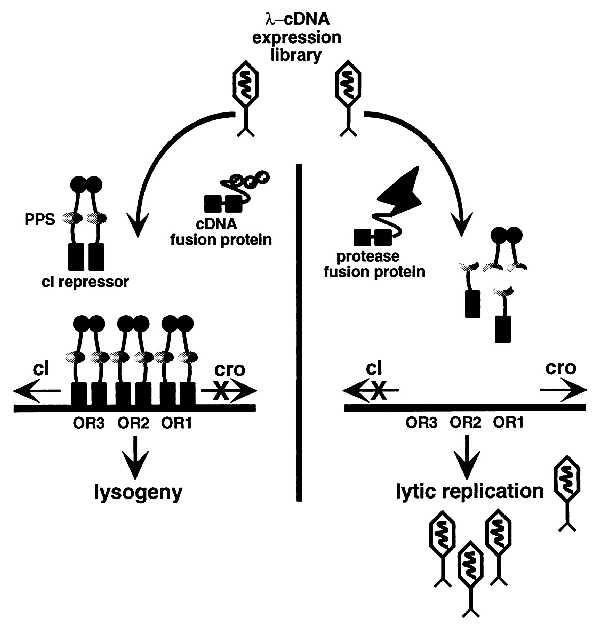
A genetic system for the isolation and characterization of site-specific proteases. (Left) The infecting phage encodes a random β-gal:cDNA fusion protein that has no effect on the steady-state levels of the target repressor containing the proteolytic processing site (PPS). (Right) The infecting phage encodes the appropriate site-specific protease that cleaves the recombinant repressor, ultimately resulting in lytic replication.
Of the known site-specific proteases, viral-encoded enzymes have been the most extensively characterized. Therefore, the parameters of a model system were defined by utilizing the protease encoded by HIV. This enzyme is required for the processing of the viral polyprotein, and its activity and specificity have been studied in vitro and in vivo (22–25). In addition, the HIV protease is the target of novel therapeutics and uniquely illustrates the potential applications of this system in the characterization of site-specific proteases and in the development of anti-protease drugs.
MATERIALS AND METHODS
Construction of Recombinant Repressors.
A unique BssH2 site was created in the cI linker domain of p280AP by site-directed mutagenesis (pcI.Bss). Prm (Promoter for Repressor Maintenance) [TAGATATTTATCCCTTGCGGTGATAGATT (21)] and mutant Prm sequences [2S: GATATATTTATCCCTTGCGGTGATAGATT (26); 2X: TAAATATTTATCCCTTGCGGTAATAGATT (27)] were generated by PCR of λ DNA with the appropriate Prm or mutant Prm oligonucleotides. cI.HIV and cI.HIVmt contain HIV proteolytic cleavage site 1 [amino acids 129–136 of the HIV-1 GAG-POL polyprotein (28, 29) or mutant site 1 (30), respectively]. pcI.HIV-cro and pcI.HIVmt-cro were constructed in pAlterEX-2 (Promega) by substitution of the appropriate repressor for the wild-type (WT) cI in the λ MunI fragment that encodes cro and cI.
HIV Protease and Recombinant Phage.
The HIV-1 protease gene was isolated from pVK3 [HIV-1 strain Hxb2 (31)] by PCR, inserted into pBSK− to generate the βgal-HIV protease fusion protein (pHIVprotease), and verified by nucleotide sequencing. Plasmids for cotransformation were produced by insertion of repressors expressed from the Prm promoter into pAlterEX-2. The HIV-1 protease gene from pHIVprotease was ligated to EcoRI-digested lambda ZapII and packaged in vitro. Phage stocks were prepared from the correct (λ-HIVp) and inverted (λ-HIVinv) insert orientation phage.
Western Blots.
Samples were resolved in 10–20% gradient SDS/polyacrylamide gels, transferred to nitrocellulose or poly(vinylidene difluoride) membranes, and blocked in PBS/0.2% Tween-20 (PBST) with 5% milk. Primary antibodies were preincubated in PBST/5% milk/12.5% bacterial lysate for 10 min before use. Duplicate Western blots were processed for chemiluminescent detection and for quantitative chemifluorescent imaging (Storm, Molecular Dynamics). For coexpression experiments, E. coli JM109 cells were cotransformed with pAlterEx-2 repressor plasmids and pHIVprotease or pHIVinv. Cultures were harvested at 0 and 3 hr postinduction with 1.0 mM isopropyl β-d-thiogalactopyranoside (IPTG) and lysed in SDS/PAGE sample buffer, and equivalent amounts of total cell protein were subjected to Western blot analyses as described above.
Selections.
E. coli strains containing p2X-cI.HIV or p2X-cI.HIVmt were transformed with pcI.HIV-cro or pcI.HIVmt-cro, respectively. The resulting strains were infected with λ-HIVp or λ-HIVinv for 15 min, washed with 10 mM MgSO4, and resuspended in Luria–Bertani medium (LB)/12.5 μg/ml tetracycline/0.2% maltose/10 mM MgSO4/0.1 mM IPTG. At 3–5 hr postinfection, aliquots of the cultures were coplated with E. coli XL-1 Blue cells in top agar/12.5 μg/ml tetracycline/0.1 mM IPTG for 6 hr. Additional cycles of selective growth were done by resuspending the infected cells with a fresh aliquot of pcI.HIV-cro or pcI.HIVmt-cro cells and continued incubation as described. After two or three selective cycles, phage from representative plaques were eluted and the orientation of the protease gene was determined by restriction analysis.
λ-cDNA Expression Libraries.
In the course of these studies, it was observed that many commercially available libraries contain a population of irrepressible phage that results in high background. The frequencies of irrepressible phage in several libraries were determined by infecting cells that express the WT repressor from a constitutive promoter (p2X-cI). An aliquot of the selected λ-cDNA library (Corpus striatum #936213, Stratagene) was amplified to create a high titer stock with less than one irrepressible phage in 5.5 × 107 pfu. E. coli cells containing pcI.HIV-cro were infected with a mixture of l-HIVp and the amplified library (1:105, respectively). After one selective growth cycle, the resulting phage pool was amplified by infection of E. coli XL-1 Blue cells, and 1 × 108 pfu of this amplified pool was subjected to a second selective growth cycle. The resulting phage population was analyzed as describe above.
Protease Inhibitors.
Indinavir sulfate (crixivan, Merck) and saquinavir mesylate (invirase, Roche) were obtained from the National Institutes of Health Clinical Pharmacy. The inhibitors were included in the LB and the top agar at the indicated concentrations.
RESULTS AND DISCUSSION
Function of Target cI Repressors.
As shown in Fig. 2A, the λ cI repressor consists of several well characterized domains (21). To construct a recombinant repressor for the insertion of target proteolytic processing sites, the recA-mediated cleavage site in the linker domain of cI was altered (A-G to A-R), resulting in the creation of a unique restriction site (cI.Bss). The function of cI.Bss was tested by expression in E. coli under the control of several promoters [Prm and various Prm down-mutants (21, 26, 27)] producing strains that expressed various steady-state levels of the repressor. Infection with λ resulted in significant repression of phage replication in all strains as compared with cells that express the wild-type cI (WT.cI), indicating that the recombinant repressor was stable and functional.
Figure 2.
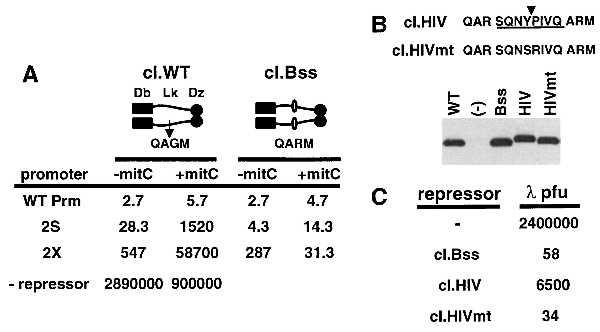
Construction and function of target repressors. The λ cI repressor consists of a DNA-binding domain (Db), a flexible linker (Lk), and a dimerization domain (Dz). The site of recA-mediated cleavage was altered (A-G to A-R) to create a unique site for the insertion of target proteolytic processing sites (cI.Bss). (A) Cells expressing cI.WT or cI.Bss from the indicated promoters were infected with λ in the absence or presence of mitomycin C (32). After 15 min, the cells were washed, resuspended in LB/0.2% maltose/10 mM MgSO4, and incubated for 5 hr. The resulting phage supernatants were titered as described. The progeny virus (pfu/μl) is shown for each repressor strain in comparison to an infection of cells that do not express a repressor. (B) HIV target repressors contain the GAG-POL site 1 (cI.HIV) or a mutant site (cI.HIVmt). A Western blot of cells expressing the WT and recombinant repressors from the Prm promoter using anti-cI sera. (C) Repressor function was determined by infection of strains expressing the repressors at subsaturation levels (2X promoter). (-), cells that do not express repressor.
Interestingly, in the presence of mitomycin C, a gratuitous inducer of the recA-mediated cleavage of the phage repressor (32), only strains expressing the WT repressor from the mutant promoters (2S and 2X) allowed significant phage replication (Fig. 2A). This indicated that it is critical to express the repressor in a manner that efficiently represses nonspecific phage while still allowing for the replication of a phage encoding a protease of modest activity. Finally, the addition of mitomycin C to cells that express the recombinant Bss repressor had no effect, confirming that the altered repressor is not a target for recA-mediated cleavage.
For the construction of HIV protease-specific target repressors, processing site 1 from the viral GAG-POL polyprotein (28, 29) or a control mutant site (30) was inserted into the Bss repressor (cI.HIV and cI.HIVmt, respectively). These repressors were expressed at steady-state levels equivalent to WT.cI and cI.Bss (Fig. 2B) and efficiently repressed the infecting phage (Fig. 2C), even when expressed from the 2X promoter at subsaturating levels. Although the repression mediated by cI.HIV was less efficient than the controls, it was significant in comparison to cells that did not express a repressor.
The target specificity of the HIV repressors was determined by coexpression of these repressors or the control cI.Bss with a β-galactosidase (β-gal)–HIV protease fusion (HIV protease, Fig. 3A). As illustrated (Fig. 3B), expression of the HIV protease resulted in nearly complete cleavage of cI.HIV (80% reduction in steady-state level) while having no effect on the control cI.HIVmt or Bss repressors.
Figure 3.
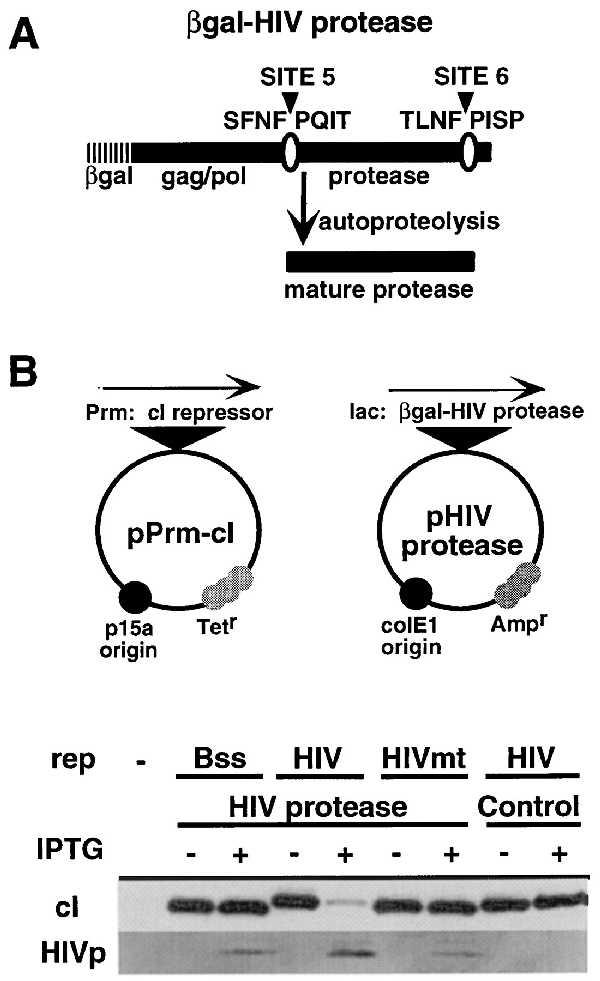
The HIV protease specifically reduces the steady-state levels of cI.HIV. (A) The β-gal–HIV protease fusion contains amino acids 444–605 of HIV-1 GAG-POL polyprotein (31) fused in-frame to the amino-terminal 38 aa of β-gal. Autocatalytic cleavage at sites 5 and 6 releases the mature viral protease (28, 29). The control contains the identical sequences inserted in the noncoding orientation (HIVinv). (B) The cI repressors were coexpressed with the HIV protease fusion under the control of the lac promoter. Expression of the protease was induced with IPTG for 3 hr, and equivalent amounts of total cell protein were blotted with anti-cI and anti-HIV protease sera. Control, HIV protease gene inserted in the antisense orientation. The steady-state levels of the HIV protease in cells which express cI.HIV was consistently higher than in cI.HIVmt cells, possibly because of the stabilization or the reduced toxicity of the protease in the presence of the appropriate target substrate.
Selective Growth of a Site-Specific Protease Encoding Phage.
The selective degradation of cI.HIV demonstrates that this system exhibits a high level of specificity; an important consideration for the isolation of the appropriate site-specific protease. However, as previously indicated, the target repressor must be expressed in a manner that efficiently represses nonspecific phage but that allows for the replicative isolation of a phage encoding a protease of modest expression or activity. To accomplish this, the λ cI-cro regulatory circuit was utilized because this dual-gene system amplifies a relatively small decrease in the steady-state level of cI by induction of cro-mediated repression of cI transcription (21). Adapting the λ cI-cro circuit would theoretically allow the system to respond to relatively low levels of specific proteolytic activity, thereby significantly increasing the sensitivity of the screen.
As shown in Fig. 4A, E. coli cells were cotransformed with plasmids encoding the cI.HIV-cro and the β-gal–HIV protease fusion. Upon infection of this strain, phage replication was as efficient as in cells that did not express cI. In contrast, replication was efficiently repressed in control cells that did not express the protease, strongly suggesting that the cI.HIV-cro system would allow selective replication of a phage encoding the HIV protease. Therefore, E. coli strains containing the HIV repressor-cro constructs were infected with a recombinant phage encoding the β-gal–HIV protease (λ-HIVp) or a control antisense phage (λ-HIVinv). In these experiments, λ-HIVp replicated up to 7,000-fold more efficiently than λ-HIVinv in cells expressing cI.HIV (Fig. 4B). In addition, λ-HIVp replication was specific for the cI.HIV strain because both phages were repressed in cells expressing cI.HIVmt. The selection of λ-HIVp was further enhanced (up to 74,000-fold) by successive infection of cells expressing cI.HIV, clearly indicating that the system is capable of isolating a site-specific protease from a library pool. Furthermore, as the activity of the HIV protease [1.7 × 104 s−1M−1 (25)] is significantly lower than that of other characterized site-specific enzymes including CPP32 [2.3 x 106 s−1M−1 (33)], lep [2.3 × 106 s−1M−1 (34)], subtilisin [2.6 × 105 s−1M−1 (35)], and Kex2 [2.5 × 107 s−1M−1 (36)], this system has the potential and sensitivity to isolate novel enzymes of limited activity.
Figure 4.
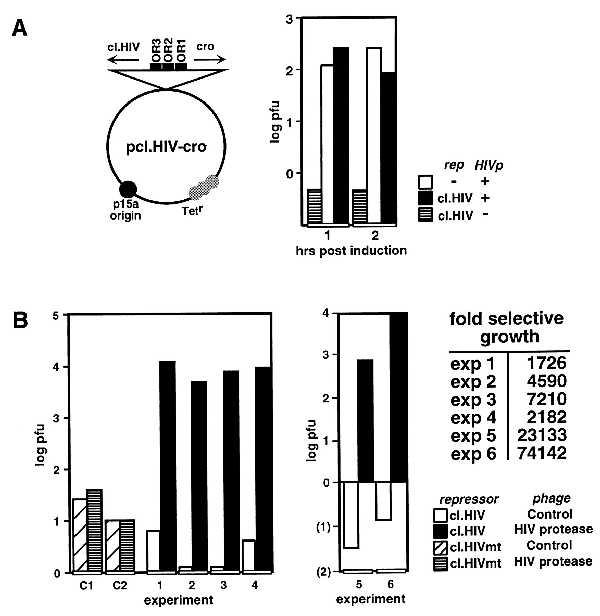
Selective growth of λ-HIVp in cells expressing cI.HIV. (A) The structure of the repressor-cro plasmid is illustrated. The expression of the cotransformed HIV protease was induced with IPTG (1 and 2 hr), and the cells were infected with λ. The graph illustrates the resulting phage titer/μl of culture supernatant. (B) Cells transformed with cI.HIV-cro or cI.HIVmt-cro cells were infected with λ-HIVp or λ-HIVinv for 5 hr. C1 and C2 are control infections of cells expressing cI.HIVmt. Experiments 1–4 and 5 and 6 are the resulting phage titers after one or three rounds of infection–selection in cI.HIV-cro cells, respectively. Fold-selective growth, pfu λ-HIVp/λ-HIVinv. As shown, selection in cI.HIV cells resulted in maintenance replication of λ-HIVp, whereas the replication of λ-HIVinv was inhibited more severely with each successive selection cycle.
Selective Isolation of a Site-Specific Protease from a Complex Pool.
To demonstrate the isolation of a rare phage encoding a site-specific enzyme, cells expressing cI.HIV were infected with a mixture of λ-HIVp and control λ-HIVinv (1:105, respectively). After successive rounds of selection, λ-HIVp represented 12.5% (two rounds, 12,500-fold selection) and 40% (three rounds, 40,000-fold selection) of the final phage population (Fig. 5). In a subsequent experiment, cells expressing cI.HIV were infected with a similar mixture of λ-HIVp and a λ human brain cDNA library to simulate the isolation of a site-specific protease from a complex cDNA library pool. After two rounds of selection, λ-HIVp constituted 8% of the resulting phage population (8,000-fold selection, data not shown).
Figure 5.
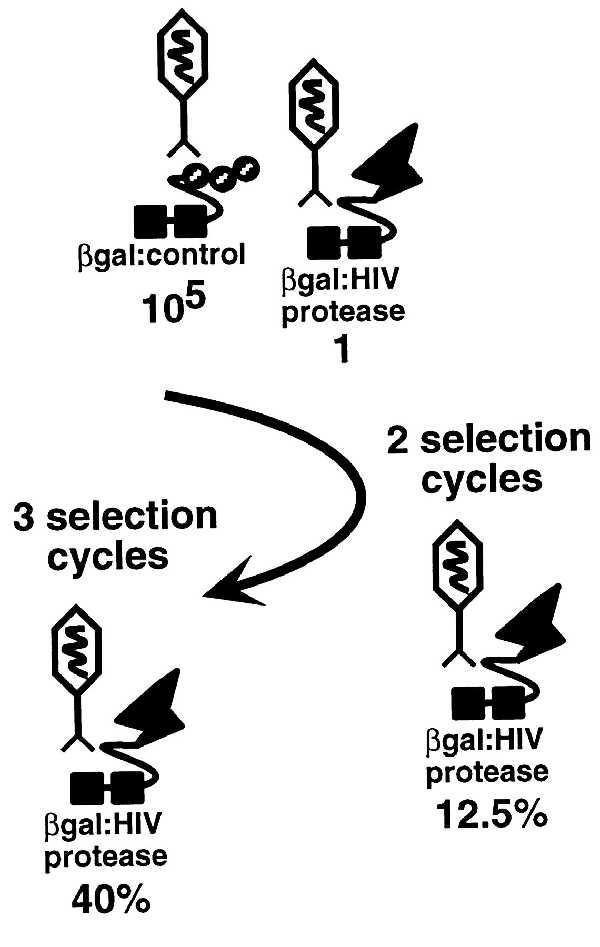
Selective isolation of λ-HIVp from a phage pool. Cells transformed with pcI.HIV-cro were infected with a mixture of λ-HIVp and control λ-HIVinv (1:105). After two and three selection cycles, the percent of λ-HIVp in the final population was determined.
The Isolation and Characterization of Site-Specific Proteases: Applications to Therapeutics.
As previously noted, viral enzymes are significant both as models of basic cellular processes and as targets of antiviral therapeutics. For HIV, specific protease inhibitors have recently become important components of clinical treatments (37). As shown in Fig. 6, the described system may also be applicable to the rapid characterization of novel anti-protease drugs. Cells expressing cI.HIV were infected with λ-HIVp in the presence of various concentrations of HIV-specific protease inhibitors. Although these drugs had no effect on the growth of λ-HIVp in control cells, micromolar concentrations significantly inhibited the growth of this phage in cI.HIV cells. Notably, the inhibition of λ-HIVp growth correlated well with the effective clinical drug concentration range [indinavir sulfate (crixivan; Merck, publication 7979800); saquinavir mesylate (invirase; Roche, publication 25974580–0197C)]. In addition to further illustrating the specificity of this model system, these experiments demonstrate the potential of the screen for the rapid characterization of anti-protease drugs. More significantly, treatment with specific protease inhibitors has recently shown indications of the selection of drug-resistant virus isolates in the clinical population (38, 39). The potential ability to rapidly select and analyze drug-resistant enzymes in this system may aid in the prediction of important clinical isolates and provide targets for further drug development.
Figure 6.
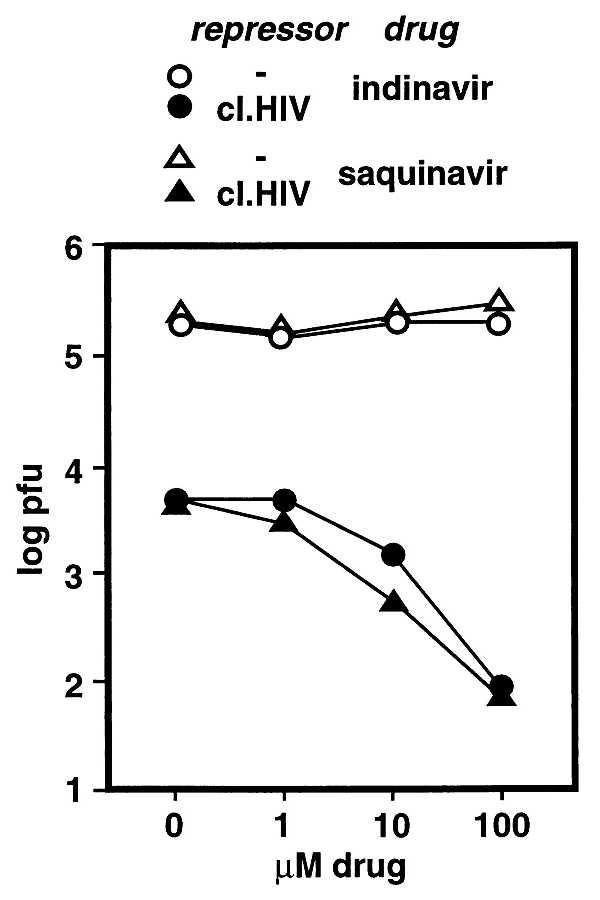
Specific inhibition of l-HIVp growth in the presence of HIV protease inhibitors. Cells transformed with pcI.HIV-cro or the control (-repressor) were infected with λ-HIVp in the absence or presence of various concentrations of specific HIV protease inhibitors. Five hours postinfection, the resulting phage was titered. Each data point is the average of two independent samples.
Site-specific proteolysis is an important regulatory mechanism in many basic biological processes (1–20). In addition, specific proteolysis is critical to the replication of many viruses and may also play a role in the development of other disease states such as Alzheimer’s (40–43) and hypocholesterolemia (9, 44–47). Although the characterization of site-specific proteases is of significant interest, many of the enzymes are unknown. A genetic screen therefore has been developed that provides a rapid methodology for the isolation and characterization of these enzymes.
It is important to note that the isolation of a novel enzyme utilizing this system depends on the representation of the cDNA in the library, the expression of catalytically active protein, and the recognition of the appropriate target site in the heterologous cI repressor substrate. However, as demonstrated by the selective isolation of a phage encoding the HIV-1 protease, the λ cI-cro dual regulatory circuit responds to a low level of specific proteolytic activity, allowing selective growth of the appropriate phage. Thus, this system clearly has significant potential to specifically amplify and isolate enzymes of low catalytic activity. In addition, adaptations of this system can be used to characterize known enzymes with respect to their activity, specificity, and structure.
Acknowledgments
We thank A. Hochschild for p280AP; J. Roberts for cI antisera; B. Korant for HIV-1 protease antisera; Pat Earl for pVK3; K. Siller for acquisition of HIV protease inhibitors; S. Gottesman for expert advice; J. Sisler for oligonucleotide synthesis; B. Moss, J. Vogel, and members of the Laboratory of Viral Diseases for helpful discussions; and B. Moss, J. Vogel, A. McBride, L. D’Adamio, J. Yewdell, A. Sears, and J. L. C. McKnight for critical reading of this manuscript. This work was supported by the Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, and the NIH Intramural AIDS Targeted Antiviral Program.
ABBREVIATIONS
- Prm
Promoter for Repressor Maintenance
- IPTG
isopropyl β-d-thiogalactopyranoside
- LB
Luria–Bertani medium
- β-gal
β-galactosidase
- WT
wild type
References
- 1.Lee J J, Ekker S C, von Kessler D P, Porter J A, Sun B I, Beachy P A. Science. 1994;266:1528–1537. doi: 10.1126/science.7985023. [DOI] [PubMed] [Google Scholar]
- 2.Coughlin S R. Proc Natl Acad Sci USA. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hollenberg M D. Trends Pharmacol Sci. 1996;17:3–6. doi: 10.1016/0165-6147(96)81562-8. [DOI] [PubMed] [Google Scholar]
- 4.Artavanis-Tsakonas S, Matsuno K, Fortini M E. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 5.Goodburn S, King P. Biochem Soc Trans. 1996;25:498–502. doi: 10.1042/bst0250498. [DOI] [PubMed] [Google Scholar]
- 6.Whiteside S T, King P, Goodbourn S. J Biol Chem. 1994;269:27059–27065. [PubMed] [Google Scholar]
- 7.Orejas M, Espeso E A, Tilburn J, Sarkar S, Arst H N, Penalva M A. Genes Dev. 1995;9:1622–1632. doi: 10.1101/gad.9.13.1622. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmeister A E M, Londono-Vallejo A, Harry E, Stragier P, Losick R. Cell. 1995;83:219–226. doi: 10.1016/0092-8674(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Sato R, Brown M S, Hua X, Goldstein J L. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 10.Porter A G, Ng P, Janicke R U. BioEssays. 1997;19:501–507. doi: 10.1002/bies.950190609. [DOI] [PubMed] [Google Scholar]
- 11.Lazebnik Y A, Kaufman S H, Desnoyers S, Poirier G G, Earnshaw W C. Nature (London) 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 12.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 13.Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 14.Steller H. Science. 1995;267:1445. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 15.Stein D. Curr Biol. 1995;5:1360–1363. doi: 10.1016/s0960-9822(95)00270-3. [DOI] [PubMed] [Google Scholar]
- 16.Song Z, McCall K, Steller H. Science. 1997;275:536–540. doi: 10.1126/science.275.5299.536. [DOI] [PubMed] [Google Scholar]
- 17.Smith C L, Giordano H, Schwartz M, DeLotto R. Development. 1995;121:4127–4135. doi: 10.1242/dev.121.12.4127. [DOI] [PubMed] [Google Scholar]
- 18.Murugasu-Oei B, Rodrigues V, Yang X, Chia W. Genes Dev. 1995;9:139–154. doi: 10.1101/gad.9.2.139. [DOI] [PubMed] [Google Scholar]
- 19.Smith C L, Delotto R. Nature (London) 1994;368:548–551. doi: 10.1038/368548a0. [DOI] [PubMed] [Google Scholar]
- 20.Fields B N, Knipe D M, Howley P M. Fields Virology. New York: Lippincott–Raven; 1996. [Google Scholar]
- 21.Ptashne M. A Genetic Switch. Cambridge, MA: Cell Press; 1986. [Google Scholar]
- 22.Tomasselli A G, Heinrikson R L. Methods Enzymol. 1994;241:279–301. doi: 10.1016/0076-6879(94)41069-0. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan A H, Manchester M, Everitt L, Swanstrom R. Methods Enzymol. 1994;241:58–69. doi: 10.1016/0076-6879(94)41059-3. [DOI] [PubMed] [Google Scholar]
- 24.Carter C, Zybarth G. Methods Enzymol. 1994;241:227–253. doi: 10.1016/0076-6879(94)41067-4. [DOI] [PubMed] [Google Scholar]
- 25.Schock H B, Garsky V M, Kuo L C. J Biol Chem. 1996;271:31957–31963. doi: 10.1074/jbc.271.50.31957. [DOI] [PubMed] [Google Scholar]
- 26.Youderian P, Bouvier S, Susskind M M. Cell. 1982;30:843–853. doi: 10.1016/0092-8674(82)90289-6. [DOI] [PubMed] [Google Scholar]
- 27.Rosen E D, Gussin G N. Virology. 1979;98:393–410. doi: 10.1016/0042-6822(79)90562-2. [DOI] [PubMed] [Google Scholar]
- 28.Stebbins J, Debouck C. Methods Enzymol. 1994;241:3–16. doi: 10.1016/0076-6879(94)41055-0. [DOI] [PubMed] [Google Scholar]
- 29.Rizzo C J, Korant B D. Methods Enzymol. 1994;241:16–29. doi: 10.1016/0076-6879(94)41056-9. [DOI] [PubMed] [Google Scholar]
- 30.Gottlinger H G, Sodroski J G, Haseltine W A. Proc Natl Acad Sci USA. 1989;86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratner L, Fisher A, Jagodzinski L L, Mitsuya H, Liou R S, Gallo R C, Wong-Stall F. AIDS Res Hum Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- 32.Hendrix R W, Roberts J W, Stahl F W, Weisberg R A. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1983. [Google Scholar]
- 33.Margolin N, Raybuck S A, Wilson K P, Chen W, Fox T, Gu Y, Livingston D J. J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 34.Tschantz W R, Paetzel M, Cao G, Suciu D, Inouye M, Dalbey R E. Biochemistry. 1995;34:3935–3941. doi: 10.1021/bi00012a010. [DOI] [PubMed] [Google Scholar]
- 35.Ballinger M D, Tom J, Wells J A. Biochemistry. 1996;35:13579–13585. doi: 10.1021/bi961543h. [DOI] [PubMed] [Google Scholar]
- 36.Rockwell N C, Wang G T, Krafft G A, Fuller R S. Biochemistry. 1997;36:1912–1917. doi: 10.1021/bi961779l. [DOI] [PubMed] [Google Scholar]
- 37.McDonald C K, Kuritzkes D R. Arch Intern Med. 1997;157:951–959. [PubMed] [Google Scholar]
- 38.Moyle G, Gazzard B. Drugs. 1996;51:701–712. doi: 10.2165/00003495-199651050-00001. [DOI] [PubMed] [Google Scholar]
- 39.Jacobsen H, Hanggi M, Ott M, Duncan I, Owen S, Andreoni M, Vella S, Mous J. J Infect Dis. 1996;173:1379–1387. doi: 10.1093/infdis/173.6.1379. [DOI] [PubMed] [Google Scholar]
- 40.Haas C. Neuron. 1997;18:687–690. doi: 10.1016/s0896-6273(00)80309-8. [DOI] [PubMed] [Google Scholar]
- 41.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 42.Vito P, Lacana E, D’Adamio L. Science. 1996;271:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- 43.Wolozin B, Iwasaki K, Vito P, Ganjei J K, Lacana E, Sunderland T, Zhao B, Kusiak J W, Wasco W, D’Adamio L. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 44.Brown M S, Goldstein J L. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 45.Sakai J, Duncan E A, Rawson R B, Hua X, Brown M S, Goldstein J L. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 46.Hua X, Sakai J, Brown M S, Goldstein J L. J Biol Chem. 1996;271:10379–10384. doi: 10.1074/jbc.271.17.10379. [DOI] [PubMed] [Google Scholar]
- 47.Duncan E A, Brown M S, Goldstein J L, Sakai J. J Biol Chem. 1997;272:12778–12785. doi: 10.1074/jbc.272.19.12778. [DOI] [PubMed] [Google Scholar]