

Abstract
In this work the spectroscopic properties of the special low-energy absorption bands of the outer antenna complexes of higher plant Photosystem I have been investigated by means of low-temperature absorption, fluorescence, and fluorescence line-narrowing experiments. It was found that the red-most absorption bands of Lhca3, Lhca4, and Lhca1–4 peak, respectively, at 704, 708, and 709 nm and are responsible for 725-, 733-, and 732-nm fluorescence emission bands. These bands are more red shifted compared to “normal” chlorophyll a (Chl a) bands present in light-harvesting complexes. The low-energy forms are characterized by a very large bandwidth (400–450 cm−1), which is the result of both large homogeneous and inhomogeneous broadening. The observed optical reorganization energy is untypical for Chl a and resembles more that of BChl a antenna systems. The large broadening and the changes in optical reorganization energy are explained by a mixing of an Lhca excitonic state with a charge transfer state. Such a charge transfer state can be stabilized by the polar residues around Chl 1025. It is shown that the optical reorganization energy is changing through the inhomogeneous distribution of the red-most absorption band, with the pigments contributing to the red part of the distribution showing higher values. A second red emission form in Lhca4 was detected at 705 nm and originates from a broad absorption band peaking at 690 nm. This fluorescence emission is present also in the Lhca4-N-47H mutant, which lacks the 733-nm emission band.
INTRODUCTION
Photosystem I (PSI) is a plastocyanin/ferredoxin oxidoreductase. It is a multiprotein complex located in the stroma lamellae of higher plants and it can be divided into two moieties: the core complex which contains all the cofactors of the electron transport chain, 100 chlorophylls (Chls) and 22 β-carotene molecules, and the outer antenna system, composed of four Lhca complexes, which coordinates Chl a, Chl b, lutein, violaxanthin, and β-carotene (1).
A remarkable spectroscopic characteristic of PSI is the presence of Chls, which absorb at energy lower than the primary donor (red forms) (2,3). The transfer of excitation energy from these low-energy forms to the reaction center, where the energy is used for charge separation, is a thermally activated process (4,5). In higher plants most of the red forms are associated to the outer antenna system, composed of four Lhca complexes arranged on one side of the core (6,7). The genes coding for these proteins (Lhca) belong to the light-harvesting complex (Lhc) multigenic family, which also contains all the antenna complexes of Photosystem II (PSII; Lhcb) (8). From the resolution of the structures of PSI-light-harvesting complex I (LHCI) (7) and light-harvesting complex II (LHCII) (9), it can be inferred that these complexes share a high structural homology and have very similar Chl organization. Despite the high similarity, the spectroscopic properties of the complexes differ strongly: although all Lhcb complexes show low-temperature emission maxima at 680 nm (10), in the case of Lhca complexes the situation is different, with Lhca1 and Lhca2 showing the red-shifted emission at 702 nm, Lhca3 at 725 nm, and Lhca4 at 733 nm (11–14). Moreover the fluorescence band shape of the long wavelength chlorophyll state has a large full width at half-maximum (FWHM) at room temperature (e.g., 55 nm for Lhca4), which represents the broadest Qy Chl fluorescence band ever reported (15). These differences indicate that small changes in the structural organization of the complex can have a strong influence on the spectroscopic properties.
The origin of the low-energy absorption forms of Lhca complexes has been investigated by mutation analysis and refolding in vitro (16–19). It has been suggested that the red forms in all complexes represent the low-energy band of an excitonic interaction involving Chls in sites 1015 and 1025 (nomenclature from Ben-Shem (7)). It was also demonstrated that asparagine as a ligand for Chl 1015 in Lhca3 and Lhca4 is necessary to produce the strong absorption shift (20). Although the detection of the red forms is easy in fluorescence at low temperature, because these Chls represent an “energy sink” for the system and their contribution is strongly enhanced in the fluorescence spectra, detailed information about the absorption band responsible for these emissions is still lacking, mainly due to the lack of structure of the red absorption tail. Most data about the spectroscopic characteristics of the low-energy absorption bands have so far been obtained by analyzing a preparation containing all four Lhca complexes in a dimeric state (21–23). It has been proposed that the 733 nm emission originates from an absorption band centered at 710–711 nm (21). This band shows very different characteristics with respect to the “normal” absorption band of a Chl in a protein environment: large homogeneous broadening and Stokes shift, and a very large value for the electron-phonon coupling. It was proposed that the red excited states can be interpreted as excimer states (24).
It has also been shown that there is a strong correlation between the presence of red fluorescence and the fluorescence lifetime: the complexes with the reddest emission show shortened lifetimes, suggesting that the low-energy forms can act as quenchers (25).
Recently, the origin of the 702-nm fluorescence emission band of Lhca2 has been determined by a combined approach of mutation analysis and spectroscopy (18). The absorption form responsible for this emission shows a maximum at 690 nm and represents the low-energy band of the excitonic interaction between Chl 1015 and 1025. A value of 150 cm−1 for the interaction energy has been proposed (18).
The aim of this work is to determine the absorption bands that give rise to the red emission in Lhca3, Lhca4, and the Lhca1-4 dimer and the physicochemical characteristics of their low-energy absorption forms. To check a recent suggestion that a second red form is present in the Lhca4 complex (26), we also analyzed a mutant of Lhca4 (N-47H), which completely lacks the 733 nm emission.
MATERIALS AND METHODS
Sample preparation
Lhca polypeptides were reconstituted and purified as in Croce et al. (13) and Castelletti et al. (14). The pigment content of the complexes was determined by high performance liquid chromatography and fitting of the acetone extracts (13). The pigment content of the complexes was identical to what was reported before, except for the Lhca1–4 dimer, which was showing a Chl a/b ratio of 2.5 instead of 3.0. The samples were diluted in a buffer containing 10 mM Hepes (pH 7.5), 0.03% β-DM (n-dodecyl-β-D-maltoside) and were supplemented with 66% (w/w) glycerol for low-temperature measurements.
Spectroscopy
The optical density of the samples was 0.05 cm−1 at the maximum in the Qy region for most of the fluorescence emission spectra and around 1 cm−1 when the red-most band was investigated. The measurements were performed in acryl cuvettes with a path length of 1 cm. For cooling of the samples, a Utreks (Ukraine) cryosystem was used. Absorption spectra at 4 K were obtained using a home-built spectrophotometer. Fluorescence emission spectra upon nonselective and selective excitation were measured with a cooled charge-coupled device camera (Chromex Chromcam 1, Albuquerque, NM) equipped with a 1/2-m spectrograph (Chromex 500 IS). The spectral resolution for detection was 0.16 nm. Detection of the fluorescence was at 90° with respect to the direction of the excitation beam.
For temperature-dependent fluorescence, a 150-W tungsten halogen lamp in combination with two filters was used for broadband excitation. The infrared output of the lamp was blocked by a cutoff filter, which was made of heat-resistant glass and blocks light above 800 nm. In addition, an interference filter was used, transmitting light at 420, 475, or 506 nm (each with a FWHM of 20 nm). For fluorescence line-narrowing spectroscopy, a continuous wave dye laser (Coherent (Santa Clara, CA) CR-599) with 4-dicyanomethylene-2-methyl-6-p-dimethylaminostyryl-4H-pyran dye, pumped by an argon ion laser (Coherent Innova 310) was used as an excitation source. The spectral bandwidth of the laser was <0.1 nm; the excitation power was regulated by neutral density filters and was kept below 150 μW to minimize hole-burning effects. Polarized fluorescence data were obtained by using polarizers in the excitation and emission beams. Fluorescence emission spectra were recorded with polarization direction oriented parallel, at magic angle or perpendicular, relative to the polarization of the excitation light. The anisotropy of the emission induced by the excitation light is defined as
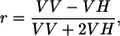 |
where VV and VH are the fluorescence intensities of the vertically and horizontally polarized emissions, respectively, upon excitation with vertically polarized light. Fluorescence anisotropy was corrected for the polarization-dependent efficiency of the monochromator.
RESULTS
Absorption
The absorption spectra of Lhca3 and Lhca4 monomeric complexes and the Lhca1-4 dimer at 4 K are reported in Fig. 1 together with their second derivatives (multiplied by −10). The absorption spectrum of the Lhca4-N-47H mutant, which lacks the red-most forms (20), is also shown. In Table 1 the absorption forms deduced from the second derivative analysis are presented.
FIGURE 1.

Absorption spectra at 4 K of the four complexes together with their second derivative spectra multiplied by −10.
TABLE 1.
Second derivative components of the absorption spectra of the complexes in nm
| Chl b | Chl b | Chl b | Chl a | Chl a | Chl a | Chl a | Chl a | Chl a | |
|---|---|---|---|---|---|---|---|---|---|
| Lhca3 | 649.5 | 656 | 665 | 668.5 | 674 | 680.5 | 688 | 704–6 | |
| Lhca4 | 644.5 | 653.5 | 662 | 671 | 674.5 | 680.5 | 689 | >700 | |
| Lhca4N-47H | 644.5 | 653.5 | 662 | 675 | 680.5 | ||||
| dim | 644.5 | 653 | 662 | 674.5 | 679.5 | 688 | 708 |
The absorption maxima are at 680 nm for Lhca3, 675 nm for Lhca4 and Lhca4-N-47H, and 677.5 nm for the Lhca1-4 dimer. The main Chl b form peaks at 644.5 nm in Lhca4, Lhca4-N-47H, and Lhca1–4 and at 650 nm in Lhca3. An additional Chl b contribution at 653 nm is visible in Lhca4, Lhca4-N-47H, and the Lhca1-4 dimer. The Chl a absorption region is dominated by forms at 674 and 680 nm, which are present in all complexes. The three wild-type (WT) complexes show absorption at wavelengths longer than 700 nm, but due to the lack of spectral structure in this region, it was not possible to clearly determine the maximum and width of the low-energy band. The low-energy absorption is not present in Lhca4-N-47H for which the absorption above 700 nm is only 0.2% of the absorption in the Qy region. For the WT Lhca3, Lhca4, and Lhca1-4 complexes this value is 7.4%, 5.5%, and 5.4%, respectively.
Fluorescence
The fluorescence emission spectra at 4 K of the WT complexes are reported in Fig. 2 A where they are normalized to their maxima. Lhca3 and Lhca4 show maxima at 725 and 733.5 nm, respectively, and in both samples a contribution around 690 nm is detectable, possibly representing the emission of some of the “bulk Chls” (the Chls which contribute to the main absorption band of the complexes around 670–680 nm), which do not transfer energy to the red forms, suggesting the presence of heterogeneous populations. The emission spectrum of the Lhca1-4 dimer is similar to that of Lhca4, although it is narrower and the contribution at shorter wavelengths is strongly reduced as compared to the monomeric complexes.
FIGURE 2.

(A) Emission spectra of Lhca3, Lhca4, and Lhca1–4 complexes at 4 K upon excitation at 506 nm. The spectra were normalized to the maximum. (B) Second derivative of the spectra in panel A. (C) Emission spectra of Lhca4-N-47H complex at different temperatures (4 K, 40 K, 77 K, 100 K, and 130 K), with 420 nm excitation.
To detect other possible emission forms, second derivative analysis was performed on the emission spectra (Fig. 2 B). Lhca3 shows mainly one red component at 725 nm, corresponding to the maximum of the spectrum. In the case of Lhca4 and Lhca1–4 the second derivative shows, in addition to the minimum at 732 nm, minima at 705 nm and at 725 nm. A blue component at 687–690 nm is present in all complexes.
The emission spectra of Lhca4-N-47H at different temperatures are reported in Fig. 2 C. Two components can be observed at 687 and 704 nm, and they are easily distinguishable in the spectrum at 130 K, whereas upon lowering the temperature a red shift of the blue-most component is observed, making it more difficult to resolve the two emissions. The relative intensity of the two forms changes very little with temperature, indicating that there is no energy transfer between the Chls responsible for these two emissions, which thus implies heterogeneity of the sample.
Site-selected fluorescence
To investigate the nature of the red emission of the complexes, the fluorescence of the samples was analyzed at 4 K upon selective laser excitation in the red tail of the absorption spectrum. Similar experiments on the PSI core complex of Synechocystis PCC 6803 and the PSI-LHCI complex from spinach were reported by Gobets et al. (27), on PSI cores of Synechococcus elongatus by Palsson et al. (28), on the native LHCI preparation by Ihalainen et al. (21), and on the PSI-LHCI complex from Chlamydomonas reinhardtii by Gibasiewicz et al. (29).
In Fig. 3 B a selection of the emission spectra of Lhca4 after different excitations is presented. The shape of the emission spectrum changes with the excitation wavelength. In addition to the main red emission peaking at 725–750 nm, two other emission forms can be detected in the spectra: i) around 688–690 nm, whose relative amplitude is maximal for excitation around 680 nm; and ii) around 705 nm, which becomes prominent for excitation at 690 nm. Similar results were obtained for Lhca1–4 (Fig. 3 C), whereas Lhca3 shows only the 690 nm emission in addition to the red-most one. These data suggest that the 688–690 nm emission originates from bulk Chls, which are not transferring energy to the red-most forms, and the 705 nm emission originates from pigments in Lhca4 absorbing around 690 nm, which are at 4 K (at least partially) uncoupled from the red-most emitting forms.
FIGURE 3.

Selection of the emission spectra of (A) Lhca3, (B) Lhca4, (C) Lhca1–4, and (D) Lhca4-N-47H at 4 K for selective laser excitations in the 680–720 nm range. The sharp peak is due to the elastic scattering of the excitation light. The spectra were normalized to the maximum.
In the case of the Lhca4 N-47H (Fig. 3 D), excitation of the bulk Chls leads to the emission of both forms (688–690 and 705), in agreement with the presence of two subpopulations one of which possesses the red-most form (emission at 704 nm). For excitation wavelengths ≥690 nm only the subfraction emitting at 704 nm becomes excited.
In Fig. 4 the various fluorescence spectra of Lhca4 upon excitation at different wavelengths in the far-red region of the absorption spectrum are shown. The spectra are shifted in such a way that the excitation is set to 0 cm−1. We conclude that the shape and width of the emission spectra are identical for excitations above 720 nm, an indication that maximal selectivity has been reached. Moreover, for far-red excitation the gap between excitation and emission maximum, which represents the energy gap between the zero-phonon line and its phonon wing (i.e., the optical reorganization energy), is very large and has a value around 350 cm−1 (see Discussion). Similar results were obtained for Lhca3 and Lhca1–4, for which values of 280 cm−1 and 325 cm−1 were found. It is worth noticing that this last value is identical to the one which can be calculated from fluorescence line narrowing (FLN) experiments for the native LHCI preparation (24). Smaller values were found for the red forms of Synechocystis (115 cm−1), Synechococcus (200 cm−1), and PSI-LHCI from spinach (240 cm−1) (27,28). This value is 150 cm−1 for Lhca4-N-47H, in which case the spectra are identical for excitation above 690 nm.
FIGURE 4.

Emission spectra of Lhca4 for excitation at 716 nm, 720 nm, 724 nm, and 730 nm. The spectra are shifted in a way that the excitation wavenumbers correspond to 0 cm−1. The spectra were normalized to the maximum.
In Fig. 5 the dependence of the fluorescence emission maximum (λem) on the excitation wavelength (λex) is shown for the three WT complexes. It is clear that the λem strongly depends on λex. For all WT complexes, the λem is blue shifted compared to that of the nonselectively excited emission if λex is between 690 and 705 nm. Excitation at wavelengths longer than 705 nm results in a gradual red shift of λem. The blue shift of λem while λex is moved to the red between 690 and 705 nm is due to preferential excitation in the blue wing of the inhomogeneous distribution of the red forms, which is selectively contributing to the emission (27,30). This blue shift is more pronounced for Lhca4 and the Lhca1–4 dimer, which exhibit a close to 7-nm maximal blue shift. Upon 705–708 nm excitation, the λem/λex relation becomes linear, with a slope (calculated as cm−1 per cm−1) of 0.67 for Lhca4 and 0.7 for Lhca3 and 0.53 for the dimer, an indication of a large contribution of the inhomogeneous width to the total width of the red-most band for all samples (see Discussion). These values are in agreement with the value obtained from the analysis of an LHCI preparation which contains all four Lhca complexes (21). For the excitation in the very red edge of the band the slope becomes 1, thus indicating that we are exciting the zero-phonon lines directly and that the maximum selectivity has been reached. Also in the case of the Lhca4-N-47H complex the emission is shifting to the red with the excitation, and for excitation above 688 nm the slope is 1.05 (data not shown).
FIGURE 5.

Emission and absorption characteristics at 4 K of the red tail of (A) Lhca3, (B) Lhca4, and (C) Lhca1–4 complexes. (Solid squares) dependence of the emission maximum on the excitation wavelength. (Open circles) values of the anisotropy of the emission detected at the emission maximum (e.g., 725 nm for Lhca3, 733 nm for Lhca4, and 732.5 nm for Lhca1–4 dimer). (Line) red tail of the absorption spectrum at 4 K. The straight line represents the value of the emission maximum for nonselective excitation.
The site-selected measurement also allows us to obtain an estimation of the absorption maximum of the red-most form. Following Gobets et al. (27) the absorption maximum of the emitting band corresponds to the excitation value at which the emission maximum has the same value as observed for nonselective excitation (see Gobets et al. (27) for details). Accordingly, the maximum of the red-most absorption forms of Lhca3, Lhca4, and Lhca1–4 was determined to be 705 nm, 708 nm, and 709 nm, respectively.
Anisotropy measurements
To get information about the relative orientation of the absorbing and emitting dipoles, polarized fluorescence measurements were performed and the anisotropy at the maximum of the emission was calculated for each excitation wavelength. The results are presented in Fig. 5 (open circles). The anisotropy values reach a maximum (0.32) between 705 nm and 710 nm excitation for all samples (note that a value of 0.32 for a low-temperature experiment is already very high and close to 0.4; the deviation from 0.4 could be due to experimental imperfections like birefringence in the cryostat and cuvette windows). There are two possible interpretations: i), all excitations with λ > 705 nm are trapped on the red-most form(s); and ii), the absorption in this region is due to two or more Chls connected by energy transfer with the same orientations of their Qy transitions. Although the second possibility cannot be completely ruled out, it is hard to imagine that the low absorption intensity above 705 nm contains the contribution of two Chls, which in addition should have the same orientation. Moreover, the absorption spectrum of the N-47H mutant of Lhca4, which completely lacks the 732 nm emission, is almost zero at 705 nm. This mutant coordinates the same number of Chls as the WT, and it has been shown that the mutation only changes the position of a single Chl, 1051, thus suggesting that the absorption above 705 nm originates from only one state (20). For the Lhca4-N-47H mutant the maximum is reached for excitation at 694 nm.
Description of the absorption spectra
To get further insight into the low-energy bands, the absorption spectra at 4 K of the three complexes were fitted in terms of Gaussians, using as starting parameters the values obtained by second derivative analysis. Due to a lack of structure in the red absorption tail, several fits of similar quality can be found for this region. However, the fitting has to satisfy three criteria, which come from the fluorescence measurements: 1) The maxima of the red-most absorption band should be in the range suggested by the site-selective fluorescence measurements (e.g., 704–705 nm for Lhca3, 708–709 nm for Lhca4, and 709 nm for the Lhca1–4 dimer). 2) The red-most band should show absorption at the wavelengths where the red shifting of excitation induces a blue shift of the emission peak (e.g., 690 nm for Lhca1–4), demonstrating that at these wavelengths we start exciting the red-most absorption form. 3) A band absorbing up to 705 nm should be present in the description to satisfy the constraints from the anisotropy data (only above 705 nm there is exclusive excitation of the red-most band, below this wavelength the lower anisotropy values indicate excitation energy transfer to the red-most pigments).
The Gaussian fits which obey these three restrictions are presented in Fig. 6 for the three WT samples. The red-most forms of Lhca3, Lhca4, and Lhca1–4 dimer are located around 705 nm, 708 nm, and 709 nm, respectively (see Table 2 for the characteristics of the bands). The error is larger in the case of Lhca4 due to the fact that its spectrum is completely lacking structure in the red tail. The amplitude of the bands is similar for Lhca4 and Lhca1–4 dimer (6.1%–6.3% of the absorption in the Qy region) and it is higher for Lhca3 (9.7%). In all cases the red-most bands exhibit a very large bandwidth.
FIGURE 6.

Gaussian analysis of the 4 K absorption spectra of (A) Lhca3, (B) Lhca4, (C) Lhca1–4, and (D) Lhca4-N-47H. Note that the fitting is mainly meant to extract the characteristics of the red bands, using the restrictions as explained in the text.
TABLE 2.
Characteristics of the red-most absorption bands of red-emitting Lhca proteins
| Sample | Max (nm) | Amplitude (%) | Γtot (cm−1) | Γhom (cm−1) | Γinh (cm−1) |
|---|---|---|---|---|---|
| Lhca3 | 704.8 ± 0.4 | 9.7 | 415 ± 8 | 228 | 347 |
| Lhca4 | 707.8 ± 2.2 | 6.3 | 468 ± 34 | 297 | 362 |
| Lhca1–4 | 709.3 ± 0.4 | 6.1 | 424 ± 11 | 290 | 309 |
The errors of the fit are reported.
In addition to the red-most band, in all complexes a second form, peaking at 689–691 nm was required to describe the red absorption tail. This band is rather broad (around 15 nm) and it accounts for 6%–7% of the absorption in the monomers and 4% in the dimer. However the errors for this band are quite large due to the lack of structure in this spectral region. The fitting of the absorption spectrum of Lhca4-N-47H is presented in Fig. 6 D. The fitting still requires a broad band with maximum around 690 nm, although its intensity (3% of the absorption in the Qy region) is smaller compared to the WT complex.
DISCUSSION
Site-selection fluorescence measurements and Gaussian analysis suggest that the absorption maxima of the red-most band is around 705 nm for Lhca3 and 708 and 709, respectively, for Lhca4 and the Lhca1–4 dimer at 4 K. The associated bandwidths are far larger than what is typically found for antenna Chls in a protein environment (e.g., (31,32)), though most BChl a antenna systems have bandwidths comparable to the ones observed here (e.g., the “deconvoluted” monomer band of B820 has a width of 400 cm−1 at 77 K (33)). One difference between BChl a and Chl a is the much higher difference dipole moment that is observed for BChl a (2.6 D) (34) as compared to Chl a (0.6 D) (35). Since it is in first approximation the coupling between the difference dipole moment and the environment (protein, solvent) that gives rise to spectral broadening, it is not surprising that BChl a in general has stronger electron-phonon coupling than Chl a.
Generally, the absorption spectrum of a pigment in a protein coordination site consists of a very narrow zero-phonon line (ZPL), which corresponds to the pure electronic transition, and a blue-shifted broad phonon band, which is associated with low-frequency vibrations of the protein matrix, coupled to the electronic transition. The energy distance between the ZPL and its phonon wing is approximately identical to a parameter known as optical reorganization energy (Sν) where S is the strength of the electron-phonon coupling (or Huang-Rhys factor) and v is the mean frequency of the protein matrix phonons (36). This broadening is often referred to as homogeneous broadening, and it is temperature dependent. The spectral broadening for an ensemble of pigments coordinated to a protein also involves a second factor, the inhomogeneous broadening. This is due to the fact that the protein is not a rigid matrix. Single-molecule fluorescence experiments allow one to observe the spectral fluctuations resulting from sudden changes in the realization of the disorder in real time (37). These (relatively slow) structural fluctuations in the protein modify the binding site of the pigments and generally lead to a Gaussian distribution of the frequency of the optical transition. This statistical distribution is usually considered to be temperature independent (38).
An estimation of the homogeneous (Γhom) and inhomogeneous (Γinh) bandwidth can be obtained from the plot of λem versus λex (29). The slope, in the wavelength range of the absorption of the red-most band, is approximated by 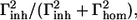 where
where 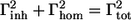 (Γtot is the FWHM of the absorption band assumed to be a Gaussian (see Gobets et al. (27) for details). The obtained values for the red-most band of the three complexes are reported in Table 2. All complexes show high values that are very similar to the values obtained for the red forms in the native LHCI preparation (24).
(Γtot is the FWHM of the absorption band assumed to be a Gaussian (see Gobets et al. (27) for details). The obtained values for the red-most band of the three complexes are reported in Table 2. All complexes show high values that are very similar to the values obtained for the red forms in the native LHCI preparation (24).
Homogeneous broadening
The Stokes shift (i.e., the energy separation between the absorption and the emission maximum of a pigment) is directly related to the homogeneous width of the band and thus to the optical reorganization energy (Stokes shift approximately equal to 2Sν). In general for bulk Chl associated to an antenna system the Stokes shift is around 2 nm (with S = 0.8 and ν = 20 cm−1). In the case of the red forms of Lhca3, Lhca4, and the Lhca1–4 dimer these values are 20 nm, 25 nm, and 23 nm, respectively (Table 3).
TABLE 3.
Characteristics of the red-most band of Lhca3, Lhca4, and the Lhca1–4 dimer
| Sample | Absorption (nm) | Emission (nm) | Sν (from gap ex vs. em) (cm−1) | Sν (from the Stokes shift) (cm−1) |
|---|---|---|---|---|
| Lhca3 | 704.8 | 725 | 280 | 200 |
| Lhca4 | 707.8 | 733 | 325 (350) | 240 |
| Lhca1–4 | 709.3 | 732.5 | 325 | 230 |
The gap between the excitation and the emission maximum upon excitation at the very red edge of the absorption spectra (region where the slope s = 1) provides the values for the reorganization energy. Sν from the Stokes shift was calculated using Stokes shift = 2Sν (see text).
An independent determination of the value of the optical reorganization energy was obtained from the site-selected fluorescence emission measurements, being the energy gap between the excitation and the emission wavelength, for excitation in the very red part of the absorption band (Fig. 4). Values between 280 and 325 cm−1 (Table 3) were obtained. The data indicate that, independent from the applied calculation method, the Sν value for Lhca4 and the Lhca1–4 dimer are very similar to each other. It is smaller for Lhca3, in agreement with the narrower bandwidth. However, it is also clear that the two methods lead to quantitatively different results (see Table 3). We note that a similar discrepancy was present also in the native LHCI preparation (24). At least two effects can contribute to this discrepancy. First of all it should be noted that the lifetime is progressively shorter for the redder forms (25), and therefore the quantum yield of the redder forms in the ensemble will be reduced. Thus the redder forms are less important in determining the average, bulk excited, emission spectrum, leading to a reduced Stokes shift. Second, we have to take into account that although the Sν value calculated from the Stokes shift represents an average value for the entire red band, the value obtained upon selectively exciting the zero-phonon lines refers to a low-energy subpopulation of the red band. If the value of Sν is not constant over the entire band, then neither is the Stokes shift. Large values for Stokes shifts are usually attributed to the presence of interacting dimers (39–41). In this case it appears that the pigments absorbing at the red edge of the inhomogeneous distribution possess a higher value for the optical reorganization energy than the pigments absorbing on the blue edge.
Exciton coupling and charge transfer states
It has been shown that the red-most band of Lhca complexes represents the low-energy band of an excitonic interaction involving the Chls in sites 1015 and 1025 (17–20). This interaction alone would neither explain the red shift (in LHCII, which has very similar pigment organization and in which the interaction energy is half of what is observed in Lhca4—120 cm−1 vs. 260 cm−1—the red-most transition is around 681 nm (42,43)) nor explain the large bandwidth observed for the red forms, because exciton coupling per se in Lhcs primarily gives rise to narrowing of absorption bands. This was, e.g., observed for the B820 subunit of LH1, in which a dimer of BChl shows clear signs of exchange narrowing (33). However, the formation of a dimeric species can also lead to broadening of the spectra, since bringing the monomers close together enhances the possibility of electron transfer processes between them. As a result charge transfer states become of importance. Because these charge transfer states are generally close in energy to the singlet-excited states of the dimer, they will tend to mix into the excited state (44,45). For a dimer of identical monomers in a symmetric environment this need not be too important. As long as the two possible intradimer charge transfer states are degenerate, their mixing into the excited states of the dimer will be balanced, and the excited states do not acquire a polar character (46). However, when this symmetry gets broken, the two CT-states need not be isoenergetic anymore, and as a consequence the unbalanced mixing of these states into the excited state will result in an excited-state dipole moment. The ground state is energetically too far away from the CT-states and will be left unchanged. Therefore the mixing of CT-states will result in a difference dipole moment (Δμ) between ground and excited state.
Since the difference dipole moment is responsible for the coupling between the optical transition and (polar) phonon modes of the environment, this will result in an increase in the exciton-phonon coupling. Stark experiments on the excitonic states of Chl a in LHCII have shown that in that case the LHCII excitonic states probably behave like monomeric Chl a being characterized by a rather small difference dipole moment (35), typically ∼0.5 D, whereas corresponding values for monomeric BChl a are 2–4 times larger (47). Since the Stokes shift caused by the difference dipole moment scales with (Δμ)2 (48,49), this is a significant difference. Therefore, for an excitonically coupled dimer of Chl as coupled to a charge transfer state, the acquired difference dipole moment can cause a significant change in its spectroscopic properties (line broadening, large Stokes shift), making it more similar to BChl a antenna systems. Note that in the Lhcas, as in the BChl a antenna systems, in addition to the broadening prominent red shifts also occur as a consequence of the coupling of the excitonic states to CT-states (50).
In Lhca3 and Lhca4 the environment of the two interacting Chls is indeed very different, with the Chl 1025 being located in a polar environment (ligand Glu with an Arg and a second Glu nearby), whereas no charged amino acids are located in the surrounding of the Chl 1015. Therefore it is highly likely that this dimer has acquired a charge transfer character, which explains the dramatic increase in the exciton-phonon coupling.
We found strong indications that the Stokes shift is not constant over the absorption band of the red forms but in fact increases upon redder excitation although it seems to reach a maximum around 720 nm and it is constant above this wavelength, which could indicate that the mixing of the excited state and the charge transfer state reaches a maximum around 720 nm. The changes in Stokes shift is partly explained within the context of the disordered dimer model (39), which predicts that the red tail of the inhomogeneous distribution contain dimers, which are relatively more localized and consequently have a stronger electron phonon coupling. By combining Redfield theory with the disordered exciton model, a very good description of the change in shape in the fluorescence spectrum from single LH2 and LH1 complexes as a function of the emission wavelength was obtained (51,52).
Since the spectral properties of the red states of Lhca differ so much from those of monomeric Chl a (as a consequence of the acquired difference dipole moment), we consider the possibility that the inhomogeneity over the band is also directly related to the difference dipole moment. Such a correlation is not part of the disordered dimer model. Then the variability over the band is natural given the fact that the dipole moment depends on the degree of asymmetry of the environment of the two Chl a molecules and on the distance between them. The same symmetry breaking (with respect to the energetics of the two Chl a sites) that produces states in the red tail of the distribution of the disordered dimer model would then be responsible for an increase in the difference dipole moment, resulting in a correlation between the spectral position within the band, the difference dipole moment, and thus the spectroscopic characteristics.
This correlation can be observed in our site-selection experiments. In the Lhca4-N-47H mutant and in the Lhca2 complex, both exhibiting a 702–705 nm emission (18), the energy of the emission maximum as a function of excitation frequency (energy) had a slope that exceeds unity, which could be explained by presuming that redder species from the ensemble had a relatively stronger exciton-phonon coupling. Note that the slope can be at most equal to one. For the red pigments of Lhca4, the most red-shifted member of this family of complexes, we obtained a slope of 1, which gives us no direct proof of disorder in the coupling strength. This could be an indication that the red pigments of Lhca4 are in a less flexible/variable environment than the ones in Lhca2 and Lhca4-N-47H. Considering that the red form of Lhca4 is at lower energy than the one of Lhca2 and Lhca4 N-47H, it can be suggested that the red-most forms have a well-defined configuration/structure, whereas the intermediate red forms are more in a variable environment.
Thus we propose that for the Lhca complexes the excitonic state is mixed with a charge transfer state, giving a high value for the reorganization energy and thus for the Stokes shift. The same suggestion was made for the red forms in the PSI core of Synechocystis (41,53) and Synechococcus (28) on the basis of fluorescence line-narrowing experiments. Using Stark spectroscopy it was later shown that indeed the red states are strongly mixed with charge transfer states (53). Furthermore, the fact that the red forms of LHCI exhibit a large frequency shift under pressure lends additional support for the proposed mixing of excitonic and charge transfer states in the peripheral Lhcs of PSI (24).
A second red emission form in Lhca4
It has recently been proposed that besides the red-most emission band in Lhca4, there is a second emission form peaking at 713 nm in Lhca4 at room temperature, which partly transfers its energy to the red-most one. This energy transfer is not accompanied by any depolarization which was explained by a parallel orientation of the corresponding transition dipole moments (26). In our experiments at 4 K we did not find any evidence for an emission band at 713 nm in Lhca4, but a band was observed at 705 nm. Also in the Lhca4-N-47H mutant, which is lacking the red-most band, only a band at 704 nm was observed. The fluorescence-polarized experiments show that this form is still transferring to the red-most one, but the transition dipole moment of the two forms are differently oriented.
Stoichiometry of the red forms
An estimation of the oscillator strength of the red-most band can be obtained from the Gaussian analysis. Considering a stoichiometry of 10 Chls per polypeptides for monomeric Lhca3 and Lhca4 and of 21 Chls per Lhca1–4 dimer, the analysis indicates that the 705-nm absorption form of Lhca3 corresponds to the absorption of almost 1 Chl molecule. A lower value of 0.6 Chl was obtained for the 708-nm absorption form of Lhca4. In the case of the dimer, the amplitude of the red-most band corresponds to the absorption of 1.2 Chls. In the native preparation containing all Lhca complexes, the red absorption tail represents 5% of the absorption, which should correspond to the absorption of 2–2.5 Chl molecules. This is in agreement with the data on the recombinant proteins with an oscillator strength of 1.2 Chl associated to the Lhca1–4 dimer and 1 associated to Lhca3.
The data indicate that the intensity of the red-most band of the Lhca1–4 dimer is double compared to the Lhca4 monomer. The difference absorption spectrum Lhca1–4 minus the sum of Lhca1 and Lhca4 is reported in Fig. 7. The spectrum shows positive amplitude with a maximum at 710.5 nm and a bandwidth of 430 cm−1. Both the absorption maximum and the bandwidth are in agreement with the data obtained by FLN and Gaussian deconvolution. The origin of the difference between the dimer and the sum of the monomers is not clear; the characteristics of the band in Lhca4 and Lhca1–4 are very similar, thus suggesting a similar origin. Moreover, the dimer of Lhca4-N-47H with Lhca1 WT does not show red forms (T. Morosinotto, R. Bassi, R. Croce, unpublished results), thus indicating that this increase is not due to a new form which arises from protein-protein interaction or to a change in the environment of one Lhca1 chromophore. Another possibility is that due to the monomer-monomer interaction there is a change in the orientation of one of the Chls responsible for the red form in the monomer, which would change the distribution of the excitation over the two exciton bands. The fact that there is a negative band in the difference spectrum at 682 nm, where the high-energy band of the excitonic interaction is located (20), supports this interpretation.
FIGURE 7.

Absorption difference spectrum between the Lhca1–4 dimer and the sum of the spectra of Lhca1 and Lhca4 monomers (solid line). The dashed lines represent the Gaussian fitting of the spectrum. The spectra were normalized to the Chl content before calculating the difference.
Acknowledgments
We thank Herbert van Amerongen for helpful discussions and for critically reading the manuscript.
This work was supported by the Photosystem I Consortium Research Training Network of the European Union, grant HPRN-CT-2002-00248, “Ministero dell'Università e della Ricerca” Progetto FIRB No. RBNE01LACT, and the Provincia Autonoma di Trento, progetto SAMBAx2.
Editor: Robert Callender.
References
- 1.Nelson, N., and C. F. Yocum. 2006. Structure and function of Photosystems I and II. Annu. Rev. Plant Biol. 57:521–565. [DOI] [PubMed] [Google Scholar]
- 2.Butler, W. L. 1978. Energy distribution in the photochemical apparatus of photosynthesis. Annu. Rev. Plant Physiol. 29:345–378. [Google Scholar]
- 3.Gobets, B., and R. van Grondelle. 2001. Energy transfer and trapping in Photosystem I. Biochim. Biophys. Acta. 1057:80–99. [DOI] [PubMed] [Google Scholar]
- 4.Jennings, R. C., G. Zucchelli, R. Croce, and F. M. Garlaschi. 2003. The photochemical trapping rate from red spectral states in PSI-LHCI is determined by thermal activation of energy transfer to bulk chlorophylls. Biochim. Biophys. Acta. 1557:91–98. [DOI] [PubMed] [Google Scholar]
- 5.Palsson, L. O., C. Flemming, B. Gobets, R. van Grondelle, J. P. Dekker, and E. Schlodder. 1998. Energy transfer and charge separation in Photosystem I P700 oxidation upon selective excitation of the long-wavelength antenna chlorophylls of Synechococcus elongatus. Biophys. J. 74:2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boekema, E. J., P. E. Jensen, E. Schlodder, J. F. van Breemen, H. van Roon, H. V. Scheller, and J. P. Dekker. 2001. Green plant Photosystem I binds light-harvesting complex I on one side of the complex. Biochemistry. 40:1029–1036. [DOI] [PubMed] [Google Scholar]
- 7.Ben Shem, A., F. Frolow, and N. Nelson. 2003. Crystal structure of plant Photosystem I. Nature. 426:630–635. [DOI] [PubMed] [Google Scholar]
- 8.Jansson, S. 1994. The light-harvesting chlorophyll a/b-binding proteins. Biochim. Biophys. Acta. 1184:1–19. [DOI] [PubMed] [Google Scholar]
- 9.Liu, Z., H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An, and W. Chang. 2004. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature. 428:287–292. [DOI] [PubMed] [Google Scholar]
- 10.Nussberger, S., J. P. Dekker, W. Kuhlbrandt, B. M. van Bolhuis, R. van Grondelle, and H. van Amerongen. 1994. Spectroscopic characterization of three different monomeric forms of the main chlorophyll a/b binding protein from chloroplast membranes. Biochemistry. 33:14775–14783. [DOI] [PubMed] [Google Scholar]
- 11.Schmid, V. H. R., K. V. Cammarata, B. U. Bruns, and G. W. Schmidt. 1997. In vitro reconstitution of the Photosystem I light-harvesting complex LHCI-730: heterodimerization is required for antenna pigment organization. Proc. Natl. Acad. Sci. USA. 94:7667–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmid, V. H. R., S. Potthast, M. Wiener, V. Bergauer, H. Paulsen, and S. Storf. 2002. Pigment binding of Photosystem I light-harvesting proteins. J. Biol. Chem. 277:37307–37314. [DOI] [PubMed] [Google Scholar]
- 13.Croce, R., T. Morosinotto, S. Castelletti, J. Breton, and R. Bassi. 2002. The Lhca antenna complexes of higher plants Photosystem I. Biochim. Biophys. Acta. 1556:29–40. [DOI] [PubMed] [Google Scholar]
- 14.Castelletti, S., T. Morosinotto, B. Robert, S. Caffarri, R. Bassi, and R. Croce. 2003. Recombinant Lhca2 and Lhca3 subunits of the Photosystem I antenna system. Biochemistry. 42:4226–4234. [DOI] [PubMed] [Google Scholar]
- 15.Jennings, R. C., F. M. Garlaschi, E. Engelmann, and G. Zucchelli. 2003. The room temperature emission band shape of the lowest energy chlorophyll spectral form of LHCI. FEBS Lett. 547:107–110. [DOI] [PubMed] [Google Scholar]
- 16.Morosinotto, T., S. Castelletti, J. Breton, R. Bassi, and R. Croce. 2002. Mutation analysis of Lhca1 antenna complex—low energy absorption forms originate from pigment-pigment interactions. J. Biol. Chem. 277:36253–36261. [DOI] [PubMed] [Google Scholar]
- 17.Morosinotto, T., M. Mozzo, R. Bassi, and R. Croce. 2005. Pigment-pigment interactions in Lhca4 antenna complex of higher plants Photosystem I. J. Biol. Chem. 280:20612–20619. [DOI] [PubMed] [Google Scholar]
- 18.Croce, R., T. Morosinotto, J. A. Ihalainen, A. Chojnicka, J. Breton, J. P. Dekker, R. van Grondelle, and R. Bassi. 2004. Origin of the 701-nm fluorescence emission of the Lhca2 subunit of higher plant Photosystem I. J. Biol. Chem. 279:48543–48549. [DOI] [PubMed] [Google Scholar]
- 19.Mozzo, M., T. Morosinotto, R. Bassi, and R. Croce. 2006. Probing the structure of Lhca3 by mutation analysis. Biochim. Biophys. Acta. 1757:1607–1613. [DOI] [PubMed] [Google Scholar]
- 20.Morosinotto, T., J. Breton, R. Bassi, and R. Croce. 2003. The nature of a chlorophyll ligand in Lhca proteins determines the far red fluorescence emission typical of Photosystem I. J. Biol. Chem. 278:49223–49229. [DOI] [PubMed] [Google Scholar]
- 21.Ihalainen, J. A., B. Gobets, K. Sznee, M. Brazzoli, R. Croce, R. Bassi, R. van Grondelle, J. E. I. Korppi-Tommola, and J. P. Dekker. 2000. Evidence for two spectroscopically different dimers of light-harvesting complex I from green plants. Biochemistry. 39:8625–8631. [DOI] [PubMed] [Google Scholar]
- 22.Croce, R., G. Zucchelli, F. M. Garlaschi, and R. C. Jennings. 1998. A thermal broadening study of the antenna chlorophylls in PSI-200, LHCI, and PSI core. Biochemistry. 37:17355–17360. [DOI] [PubMed] [Google Scholar]
- 23.Gobets, B., J. T. M. Kennis, J. A. Ihalainen, M. Brazzoli, R. Croce, L. H. M. van Stokkum, R. Bassi, J. P. Dekker, H. van Amerongen, G. R. Fleming, and R. van Grondelle. 2001. Excitation energy transfer in dimeric light harvesting complex I: a combined streak-camera/fluorescence upconversion study. J. Phys. Chem. B. 105:10132–10139. [Google Scholar]
- 24.Ihalainen, J. A., M. Ratsep, P. E. Jensen, H. V. Scheller, R. Croce, R. Bassi, J. E. I. Korppi-Tommola, and A. Freiberg. 2003. Red spectral forms of chlorophylls in green plant PSI—a site-selective and high-pressure spectroscopy study. J. Phys. Chem. B. 107:9086–9093. [Google Scholar]
- 25.Ihalainen, J. A., R. Croce, T. Morosinotto, I. H. M. van Stokkum, R. Bassi, J. P. Dekker, and R. van Grondelle. 2005. Excitation decay pathways of Lhca proteins: a time-resolved fluorescence study. J. Phys. Chem. B. 109:21150–21158. [DOI] [PubMed] [Google Scholar]
- 26.Zucchelli, G., T. Morosinotto, F. M. Garlaschi, R. Bassi, and R. C. Jennings. 2005. The low energy emitting states of the Lhca4 subunit of higher plant Photosystem I. FEBS Lett. 579:2071–2076. [DOI] [PubMed] [Google Scholar]
- 27.Gobets, B., H. van Amerongen, R. Monshouwer, J. Kruip, M. Rogner, R. van Grondelle, and J. P. Dekker. 1994. Polarized site-selected fluorescence spectroscopy of isolated Photosystem I particles. Biochim. Biophys. Acta. 1188:75–85. [Google Scholar]
- 28.Palsson, L. O., J. P. Dekker, E. Schlodder, R. Monshouwer, and R. van Grondelle. 1996. Polarized site-selective fluorescence spectroscopy of the long-wavelength emitting chlorophylls in isolated Photosystem I particles of Synechococcus elongatus Photosynth. Res. 48:239–246. [DOI] [PubMed] [Google Scholar]
- 29.Gibasiewicz, K., A. Szrajner, J. A. Ihalainen, M. Germano, J. P. Dekker, and R. van Grondelle. 2005. Characterization of low-energy chlorophylls in the PSI-LHCI supercomplex from Chlamydomonas reinhardtii. A site-selective fluorescence study. J. Phys. Chem. B. 109:21180–21186. [DOI] [PubMed] [Google Scholar]
- 30.den Hartog, F. T. H., J. P. Dekker, R. van Grondelle, and S. Volker. 1998. Spectral distributions of “trap” pigments in the RC, CP47, and CP47-RC complexes of Photosystem II at low temperature: a fluorescence line-narrowing and hole-burning study. J. Phys. Chem. B. 102:11007–11016. [Google Scholar]
- 31.Jankowiak, R., J. M. Hayes, and G. J. Small. 1993. Spectral hole-burning spectroscopy in amorphous molecular solids and proteins. Chem. Rev. 93:1471–1502. [Google Scholar]
- 32.Zucchelli, G., R. C. Jennings, F. M. Garlaschi, G. Cinque, R. Bassi, and O. Cremonesi. 2002. The calculated in vitro and in vivo chlorophyll a absorption bandshape. Biophys. J. 82:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Mourik, F., C. J. R. Van der Oord, K. J. Visscher, P. S. Parkes-Loach, P. A. Loach, R. W. Visschers, and R. van Grondelle. 1991. Exciton interactions in the light-harvesting antenna of photosynthetic bacteria studied with triplet- singlet spectroscopy and singlet-triplet annihilation on the B820 subunit form of rhodospirillum-rubrum. Biochim. Biophys. Acta. 1059:111–119. [Google Scholar]
- 34.Lockhart, D. J., and S. G. Boxer. 1988. Stark effect spectroscopy of Rhodobacter sphaeroides and Rhodopseudomonas virids reaction centers. Proc. Natl. Acad. Sci. USA. 85:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palacios, M. A., R. N. Frese, C. C. Gradinaru, I. H. van Stokkum, L. L. Premvardhan, P. Horton, A. V. Ruban, R. van Grondelle, and H. van Amerongen. 2003. Stark spectroscopy of the light-harvesting complex II in different oligomerisation states. Biochim. Biophys. Acta. 1605:83–95. [DOI] [PubMed] [Google Scholar]
- 36.Rebane, K. K., and K. K. Rebane. 1970. Impurity Spectra of Solids. Elementary Theory of Vibrational Structure. Plenum Press, New York. 1–253.
- 37.Rutkauskas, D., V. Novoderezkhin, R. J. Cogdell, and R. van Grondelle. 2004. Fluorescence spectral fluctuations of single LH2 complexes from Rhodopseudomonas acidophila strain 10050. Biochemistry. 43:4431–4438. [DOI] [PubMed] [Google Scholar]
- 38.Van Amerongen, H., L. Valkunas, and R. van Grondelle. 2000. Photosynthetic Excitons. World Scientific Publishing, Hackensack, NJ.
- 39.Small, G. J. 1995. On the validity of the standard model for primary charge separation in the bacterial reaction center. Chem. Phys. 197:239–257. [Google Scholar]
- 40.Pullerits, T., F. van Mourik, R. Monshouwer, R. W. Visschers, and R. van Grondelle. 1994. Electron-phonon coupling in the B820 subunit form of LH1 studied by temperature dependence of optical spectra. J. Lumin. 58:168–171. [Google Scholar]
- 41.Ratsep, M., T. W. Johnson, P. R. Chitnis, and G. J. Small. 2000. The red-absorbing chlorophyll a antenna states of Photosystem I: a hole-burning study of Synechocystis sp PCC 6803 and its mutants. J. Phys. Chem. B. 104:836–847. [Google Scholar]
- 42.van Amerongen, H., and R. van Grondelle. 2001. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 105:604–617. [Google Scholar]
- 43.Novoderezhkin, V. I., M. A. Palacios, H. van Amerongen, and R. van Grondelle. 2005. Excitation dynamics in the LHCII complex of higher plants: modeling based on the 2.72 angstrom crystal structure. J. Phys. Chem. B. 109:10493–10504. [DOI] [PubMed] [Google Scholar]
- 44.Lathrop, E. J. P., and R. A. Friesner. 1994. Vibronic mixing in the strong electronic coupling limit. Spectroscopic effects of forbidden transitions. J. Phys. Chem. 98:3050–3055. [Google Scholar]
- 45.Renger, T. 2004. Theory of optical spectra involving charge transfer states: dynamic localization predicts a temperature dependent optical band shift. Phys. Rev. Lett. 93:188101. [DOI] [PubMed] [Google Scholar]
- 46.Zhou, H. L., and S. G. Boxer. 1997. Charge resonance effects on electronic absorption line shapes: application to the heterodimer absorption of bacterial photosynthetic reaction centers. J. Phys. Chem. B. 101:5759–5766. [Google Scholar]
- 47.Gottfried, D. S., J. W. Stocker, and S. G. Boxer. 1991. Stark effect spectroscopy of bacteriochlorophyll in light-harvesting complexes from photosynthetic bacteria. Biochim. Biophys. Acta. 1059:63–75. [Google Scholar]
- 48.Bakhshiev, N. G. 1972. Spectroscopy of Intermolecular Interactions. Nauka, Leningrad.
- 49.Vanderzwan, G., and J. T. Hynes. 1985. Time-dependent fluorescence solvent shifts, dielectric friction, and nonequilibrium solvation in polar-solvents. J. Phys. Chem. 89:4181–4188. [Google Scholar]
- 50.Warshel, A., and W. W. Parson. 1987. Spectroscopic properties of photosynthetic reaction centers. 1. Theory. J. Am. Chem. Soc. 109:6143–6152. [Google Scholar]
- 51.Rutkauskas, D., V. Novoderezhkin, A. Gall, J. Olsen, R. J. Cogdell, C. N. Hunter, and R. van Grondelle. 2006. Spectral trends in the fluorescence of single bacterial light-harvesting complexes: experiments and modified redfield simulations. Biophys. J. 90:2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novoderezhkin, V. I., D. Rutkauskas, and R. van Grondelle. 2006. Dynamics of the emission spectrum of a single LH2 complex: interplay of slow and fast nuclear motions. Biophys. J. 90:2890–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frese, R. N., M. A. Palacios, A. Azzizi, I. H. van Stokkum, J. Kruip, M. Rogner, N. V. Karapetyan, E. Schlodder, R. van Grondelle, and J. P. Dekker. 2002. Electric field effects on red chlorophylls, β-carotenes and P700 in cyanobacterial Photosystem I complexes. Biochim. Biophys. Acta. 1554:180–191. [DOI] [PubMed] [Google Scholar]