

Abstract
Cell fusion mediated by Epstein-Barr virus requires three conserved glycoproteins, gB and gHgL, but activation is cell type specific. B cell fusion requires interaction between MHC-class II and a fourth virus glycoprotein, gp42, which complexes non-covalently with gHgL. Epithelial cell fusion requires interaction between gHgL and a novel epithelial cell coreceptor and is blocked by excess gp42. We show here that gp42 interacts directly with gH and that point mutations in the region of gH recognized by an antibody that differentially inhibits epithelial and B cell fusion significantly impact both the core fusion machinery and cell specific events. Substitution of alanine for glycine at residue 594 completely abrogates fusion with either B cells or epithelial cells. Substitution of alanine for glutamic acid at residue 595 reduces fusion with epithelial cells, greatly enhances fusion with B cells and allows low levels of B cell fusion even in the absence of gL.
Keywords: Epstein-Barr virus, glycoprotein gH, glycoprotein gp42, fusion, mutagenesis
Introduction
Although the process by which any herpesvirus mediates fusion with the cell membrane is poorly understood there is an emerging consensus that, for each, it involves a temporal cascade of events that requires participation of three conserved glycoproteins, gB, and gHgL (Spear and Longnecker, 2003). This fusion apparatus, sometimes referred to as the core fusion machinery, can be triggered into an active state either as a result of an interaction of one of the proteins with a cell surface molecule that functions as an entry mediator, or as a result of a similar interaction mediated by an additional accessory protein. Epstein-Barr virus (EBV) fusion can be triggered in either way, depending on the cell type involved. Fusion with B lymphocytes is triggered by an interaction with the major histocompatability class II protein (MHC-class II) mediated by an additional virus glycoprotein, gp42, which forms a non-covalently linked complex with gHgL (Li et al., 1997; Wang and Hutt-Fletcher, 1998). Fusion with epithelial cells is triggered by a direct interaction of gHgL and an as yet unidentified gHgL receptor, gHgLR (Borza et al., 2004; Wang et al., 1998). Whether this latter interaction involves residues of gH or a domain comprising residues of both gH and gL is unclear since, like all gH homologs, EBV gH is incompletely processed and transported in the absence of gL (Li, Turk, and Hutt-Fletcher, 1995; Yaswen et al., 1993). The EBV virion contains both gHgL and gHgLgp42 complexes as they are mutually exclusive for B cell and epithelial cell entry. Only three part gHgLgp42 complexes can mediate B cell infection and only two part complexes that lack gp42 can mediate epithelial cell infection (Wang et al., 1998). The presence of gp42 blocks the essential interaction with gHgLR (Borza et al., 2004).
We recently hypothesized that if the core fusion machinery of EBV for both B cells and epithelial cells is comprised of gB and gHgL and differs only in the way it is triggered, it might be possible to distinguish regions of gHgL that are capable of supporting fusion with an epithelial cell but not a B cell, or vice versa. We made a series of two amino acid insertions between residues 501 to 628 of gH, to which we had mapped the reactivity of a monoclonal antibody (Mab) that blocked epithelial cell but not B cell entry. Of these, mutant M10, in which a serine and an arginine residue were inserted after residue 592, and mutant M11, in which the same amino acids were inserted after residue 607, had opposing properties. In a cell-based fusion assay M10 could mediate fusion with an epithelial cell but not a B cell whereas M11 mediated no fusion with an epithelial cell but mediated considerably more fusion with a B cell than could the wild type protein (Wu, Borza, and Hutt-Fletcher, 2005). To explore further the importance of this region of gH to fusion with either cell type we made a series of more conservative alanine substitutions around residues 592 and 607. We report here that individual residues between 592 and 597 significantly impact both the core fusion machinery and cell specific events and that some changes in gH can at least ameliorate its need for coexpression with gL.
Results
Expression of proteins with alanine substitutions
A total of nine point mutants were made in which alanine was substituted for residues between positions 592 and 607 of a Flag tagged gH cloned in the pCAGGS vector (Fig. 1). The substitutions clustered around the sites of insertions previously used to generate insertion mutants M10 and M11 (Wu, Borza, and Hutt-Fletcher, 2005). When transfected into AGS epithelial cells each point mutant expressed a protein that could be recognized in indirect immunofluorescence assays of permeabilized cells by Mab anti-Flag and by three Mabs to gH or gHgL, CL59, CL40 and E1D1 (data not shown). None of the EBV specific Mabs react with EBV proteins in Western blots of native gels (unpublished) and it is assumed that each recognizes a conformational epitope. Surface expression of each mutant was determined by flow cytometry with Mab anti-Flag and compared with that of the wild type pCAGGS-Flag-gH. All mutants were expressed at or close to wild type levels at the cell surface (Fig. 2).
Fig. 1.
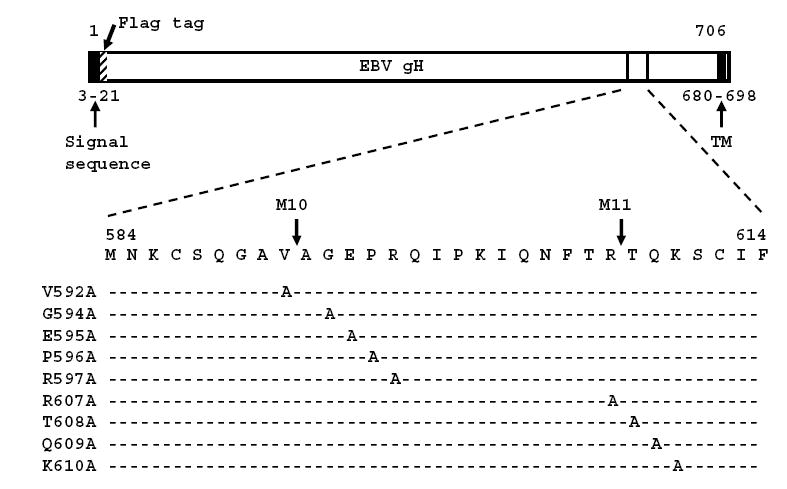
Linear map of EBV gH indicating the positions of the predicted signal sequence, the predicted transmembrane domain and the Flag tag. The sequence of amino acids surrounding the insertion mutants M10 and M11 and the positions of the alanine substitutions are indicated.
Fig. 2. Surface expression of point mutants measured by flow cytometry.
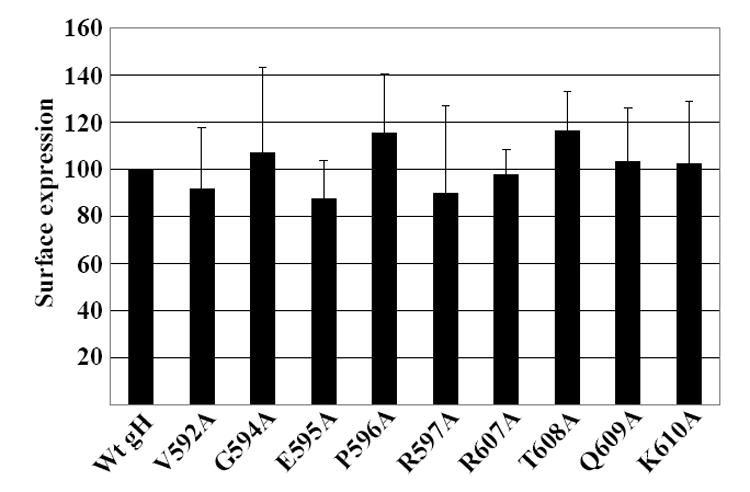
AGS cells were transfected with plasmids expressing gB and gL together with wild type gH or mutant gH and stained with Mab anti-Flag and fluorescein-conjugated sheep anti-mouse antibody. The mean level of fluorescence for cells transfected with gB, gL and wild type gH and stained with fluorescein-conjugated sheep anti-mouse antibody alone was subtracted from the mean levels of cells transfected with wild type or mutant gH. The remaining value for wild type gH was set at 100 and the remaining values for each mutant were expressed as a percent of this value. Vertical lines equal the standard deviation of 3 experiments
Comparison of the ability of wild type gH and point mutants to mediate cell-to-cell fusion of epithelial cells
Each of the point mutants was next compared with pCAGGS-Flag-gH for the ability to mediate cell-to-cell fusion when transfected into AGS epithelial cells together with pCAGGS-gL and pCAGGS-gB. pCAGGS-Flag-gH had previously been shown to support fusion at a level equivalent to a wild-type untagged version of gH in the same vector (Wu, Borza, and Hutt-Fletcher, 2005). The original insertion after residue 607 which had been made in M11 had made a protein that failed to mediate fusion with epithelial cells. All but one of the point mutants clustered around this site, however, now mediated wild type or very close to wild type levels of fusion. Only mutant K610A was slightly impaired mediating fusion at about 80% of wild type levels (Fig. 3). The original insertion after residue 592 which had been made in M10 had produced a protein that was better able to mediate epithelial cell fusion, though still not at wild type levels. Of the point mutants around this site P596A and R597A now mediated fusion at wild type levels and fusion mediated by V592A was reduced to 80% of this. However, E595A mediated only sixty percent of the level of fusion mediated by wild type Flag gH and G594A mediated essentially no fusion at all.
Fig 3. Fusion of AGS cells supported by point mutants.
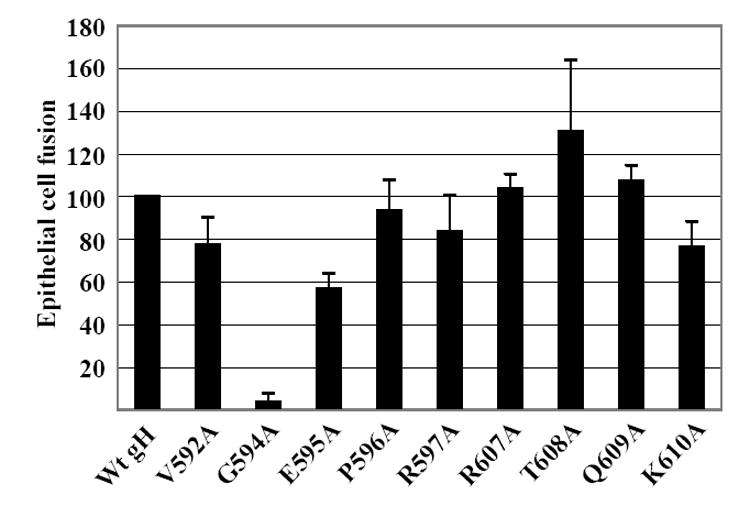
AGS cells were transfected with plasmids expressing gB and gL together with wild type gH, mutant gH or empty vector. After 24 h cells were stained with Mab CL55 to gB and fusion events were counted by fluorescence microscopy. Numbers of cells containing four or more nuclei were considered as having undergone fusion and the percent of cells that had undergone fusion with each mutant is expressed as a percentage of those that had undergone fusion with wild type gH. In the absence of gH no fusion events were seen. Vertical lines equal the standard deviation of 6 experiments.
Comparison of the ability of wild type gH and point mutants to mediate cell-to-cell fusion with target B cells
A similar analysis was done of the ability of point mutants to mediate fusion with B cells. Plasmids expressing EBV proteins together with a plasmid expressing the luciferase gene under control of a T7 promoter were transfected into CHO-K1 cells, which, unlike AGS cells, do not fuse with each other (McShane and Longnecker, 2004). The transfected cells were overlayed with Daudi cells expressing the T7 polymerase and fusion was measured in terms of an increase in luciferase activity. Of the point mutants clustered around the original mutant M11, which had mediated greater than wild type levels of B cell fusion, two, T608A and K610A, now behaved as wild type protein (Fig 4). Two, Q609A and R607A, both of which had mediated wild type levels of epithelial cell fusion, had reduced ability to mediate B cell fusion. Q609A was at 80% and R607A was at 60% of wild type levels. The point mutants clustered around the site of the insertion mutant M10, which had been poorly fusogenic for B cells, showed greater divergence. Mutant G594A was just as unable to support B cell fusion as it had been to support fusion of epithelial cells. Mutant V592A, which had mediated epithelial cell fusion at 80% of wild type levels, mediated only 60% of wild type fusion for B cells. Mutant R597A, which had been able to mediate wild type levels of fusion with epithelial cells, mediated B cell fusion at less than 40% of wild type levels. In contrast, mutant E595A, which, despite its wild type expression levels, had mediated only 60% of wild type levels of fusion of epithelial cells mediated fusion with B cells at levels more than five times those of wild type gH.
Fig 4.
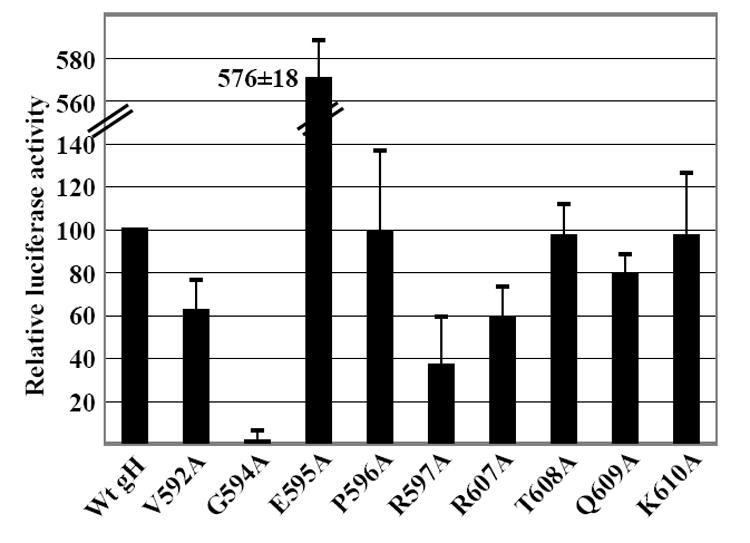
Fusion of Daudi B cells supported by point mutants and measured as relative luciferase activity. CHO-K1 cells were transfected with plasmids expressing gB and gL together with wild type gH, mutant gH or empty vector and a plasmid expressing luciferase under control of the T7 promoter. Transfected cells were overlayed with Daudi 29 cells expressing T7 RNA polymerase. Luciferase activity in the presence of empty vector was subtracted from activity in the presence of wild type or mutant gH. The remaining value for wild type gH was set at 100 and luciferase activity in the presence of each mutant was expressed as a percentage of this value. Vertical lines indicate the standard deviation of 8 experiments.
Complex formation by point mutants
Point mutant G594A, which mediated very little fusion with epithelial cells and no fusion at all with B cells, reacted with the Mab E1D1, which only recognizes gH that is complexed with gL (Li, Turk, and Hutt-Fletcher, 1995), (Wu and Hutt-Fletcher, unpublished). However, it remained possible that its inability to mediate B cell fusion reflected an inability to complex with gp42. To examine this possibility the point mutant was reconstructed in pTM1-EBV gH for higher levels of expression. Cells were transfected with wild type or mutated pTM1-Flag gH together with pTM1 gL and pTM1 gp42, radiolabeled and immunoprecipitated with Mab F-2-1 to gp42. Both gH constructs (the only protein of the gHgLgp42 complex that contains methionine residues for labeling beside the start residue) could be immunoprecipitated with F-2-1, confirming that the mutant retained the ability to form a three part gHgLgp42 complex (Fig. 5). The association with gL was also reconfirmed by immunoprecipitation with an antibody to gL.
Fig. 5. Interaction of wild type gH and G495A with gL and gp42.
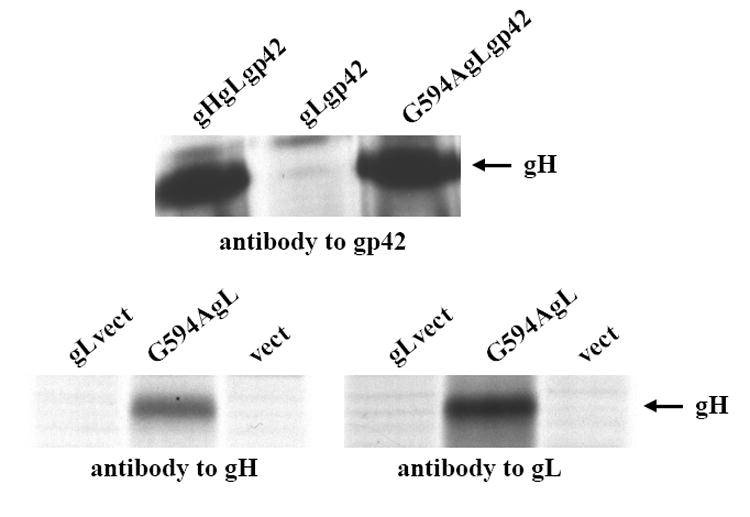
SDS-PAGE analysis of proteins immunoprecipitated by antibody to gp42, antibody to gL or antibody to gH from CV1 cells labeled with [35S] methionine and transfected as indicated with pTM1-EBV gH, pTM1-EBV gL and pTM1-EBV gp42, pTM1-EBV gL and pTM1-EBV gp42, pTM1-EBV G594A, pTM1-EBV gL and pTM1-EBV gp42, pTM1 gL and pTM1 empty vector (vect), pTM1 gL and pTM1-EBV G594A or pTM1 alone. The band indicated with an arrow is gH.
E595A, in contrast to G594A, had a significantly enhanced ability to mediate B cell fusion. The ability of E595A to mediate complex formation was thus also examined more closely by immunoprecipitation with antibodies to gL and gp42. Immunoprecipitation with antibody to gL confirmed its expected interaction with gL (Fig 6). Previous work with disrupted virus had suggested that incorporation of gp42 into a gHgLgp42 complex might result from an interaction with gL (Li, Turk, and Hutt-Fletcher, 1995), although such an interaction had not proven possible to confirm when the two proteins were expressed together in isolation (unpublished). In the course of this new analysis, however, we found that gp42 interacted directly with either wild type gH or E595A in the absence of gL.
Fig 6. Complex formation by wild type gH and E595A.
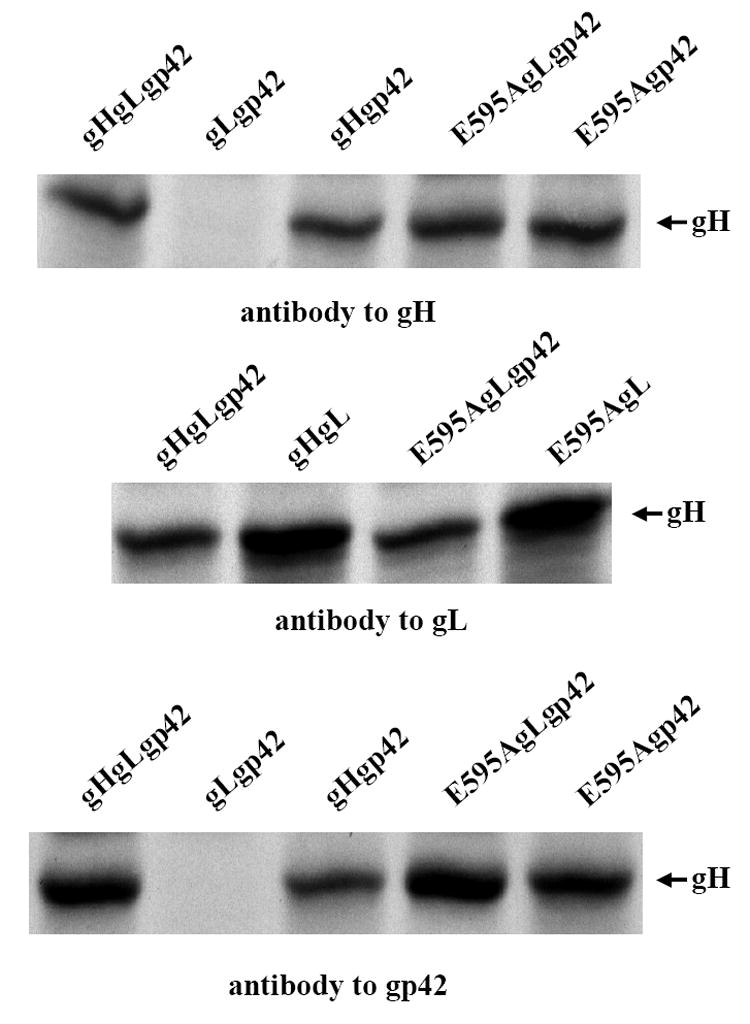
SDS-PAGE analysis of proteins immunoprecipitated by antibody to gH, antibody to gL or antibody to gp42 from CV1 cells labeled with [35S] methionine and transfected as indicated with pTM1-EBV gH, pTM1-EBV gL and pTM1-EBV gp42, pTM1-EBV gH and pTM1-EBV gp42, pTM1-EBV E595A, pTM1-EBV gL and pTM1-EBV gp42 or pTM1-EBV E595A and pTM1-EBV gp42. The band indicated with an arrow is gH.
Surface expression of gp42 in the presence of gH and E595A
The finding that gp42 interacts directly with gH suggested that one possible explanation for the enhanced ability of E595A to mediated B cell fusion might be that its interaction differed in a way that resulted in expression of higher levels of gp42 at the cell surface. Previous work had suggested that transport of gp42 occurs independently of gH (Li, Turk, and Hutt-Fletcher, 1995), but this had been determined on a qualitative and not a quantitative basis. A Mab antibody to gp42 was therefore used to make a flow cytometric analysis of the effects of gH and E595A on the absolute levels of expression of gp42 at the cell surface. Expression of gp42 with gB, gL and E595A was actually slightly lower in than in the presence of gB, gL and wild type gH (Fig 7). Absence of gH reduced gp42 expression further. Absence of gL which would be expected to significantly inhibit transport of gH to the cell surface, had a similar effect.
Fig 7.
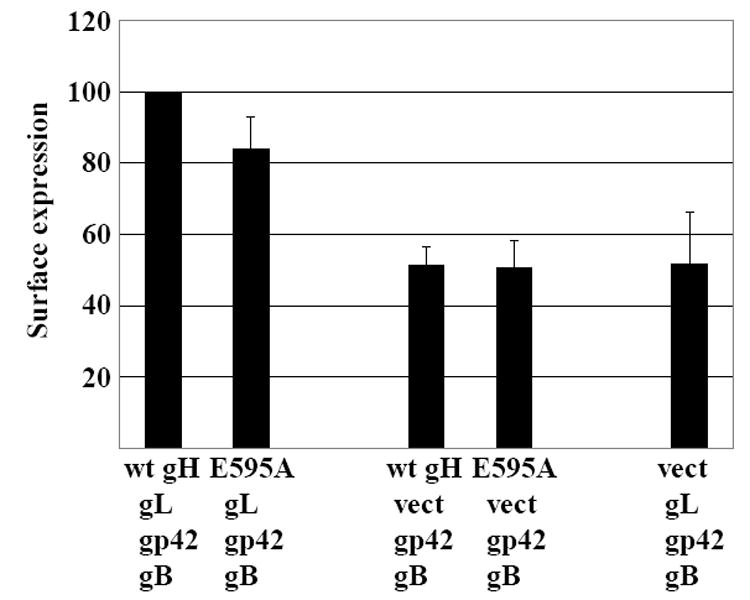
Effect of wild type gH, E595A and gL on surface expression of gp42 measured by flow cytometry. AGS cells were transfected with plasmids expressing gB and gp42 together with combinations of gL, gH, E595A or empty vector (vect) as indicated and stained with Mab to gp42 and fluorescein-conjugated sheep anti-mouse antibody. The mean level of fluorescence for cells transfected with gB, gL gH and gp42 and stained with fluorescein-conjugated sheep anti-mouse antibody alone was subtracted from the values obtained for each combination. The remaining value for expression of gp42 with gB, gL and wild type gH was set at 100 and the values for each of the other combinations were expressed as a percent of this value. Vertical lines indicate the standard deviation of 3 experiments.
Surface expression of gH and E595A and fusion mediated in the absence of gL
When previous analyses of gH expression at the cell surface in the absence of gL had been performed no Mabs specific for gH had been available, and the gH constructs had not been tagged. The conclusion that transport to the cell surface was interrupted had been based on observations that gH expressed alone does not carry the endoglycosidase H resistant N-linked sugars that are characteristic of processing of native gH in the Golgi apparatus (Yaswen et al., 1993) and that iodination of cell surface proteins can only detect its expression at the cell surface in the presence of gL (Li, Turk, and Hutt-Fletcher, 1995; Yaswen et al., 1993). Since our gH constructs were now Flag-tagged, we also reevaluated cell surface expression of gH as well by flow cytometry using the Mab anti-Flag. Although only very small amounts of both wild type gH and E595A could be detected on the surface of the cell in the absence of gL the signal was, for both, greater than that seen in the absence of primary antibody (Fig. 8).
Fig 8.
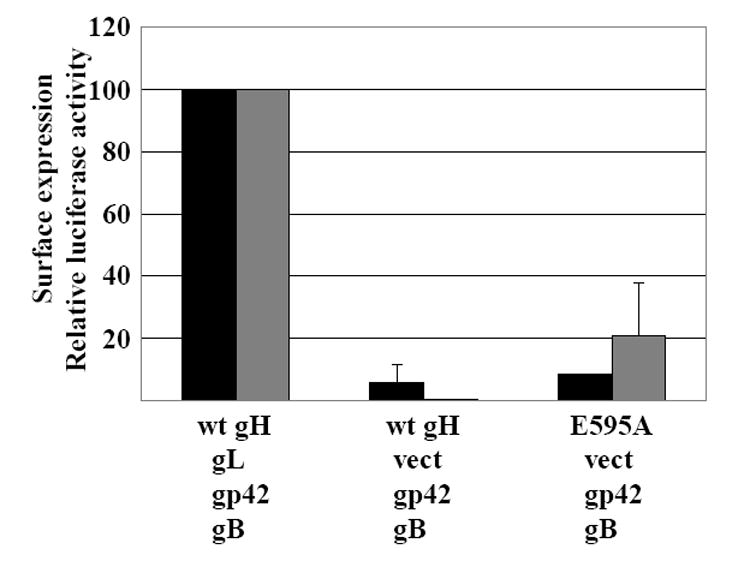
Surface expression of wild type gH and E595A in the absence of gL and the ability to mediate fusion of Daudi B cells. Black bars: AGS cells were transfected with combinations of plasmids expressing gH or E595A, gB, gp42 and gL or empty vector (vect) as indicated, stained with Mab anti-Flag and fluorescein-conjugated sheep anti-mouse antibody and analyzed by flow cytometry. The mean level of fluorescence for cells transfected with gB, gL gH and gp42 and stained with fluorescein-conjugated sheep anti-mouse antibody alone was subtracted from the values obtained for each combination. The remaining value for cells transfected with plasmids expressing gH, gB, gp42 and gL was set at 100 and values for other combinations were expressed as a percent of this value. Vertical lines equal the standard deviation of 3 experiments. Grey bars: CHO-K1 cells were transfected with a plasmid expressing luciferase under control of the T7 promoter and the indicated combinations of plasmids expressing gB, gL, gp42, gH, and E595A. Transfected cells were overlayed with Daudi 29 cells expressing T7 RNA polymerase. Luciferase activity in the presence of gB, gL, gp42 and empty vector was subtracted from activity in the presence of gH or E595A. The remaining value for gB, gL, gp42 and gH was set at 100 and luciferase activity in the presence of other combinations was expressed as a percentage of this value. Vertical lines indicate the standard deviation of 4 experiments.
Since small amounts of both E595A and wild type gH could be seen at the cell surface in the absence of gL and since B cell fusion supported by E595A was significantly higher than that supported by wild type gH, we also looked to see if any B cell fusion could be mediated by E595A, gp42 and gB in the absence of gL. When compared to wild type gH, E595A could mediate a modest but detectable amount of fusion in the absence of gL (Fig. 8). Although the variability was high in repeated experiments, the difference from wild type gH was significant (p 0.005). No similar fusion was seen in the absence of both gL and gp42 and no similar fusion could be detected with epithelial cells (not shown).
Discussion
The amino terminus of gH has been implicated as being critical to its role in support of cell fusion. Potentially alpha helical residues 377-397 of the gH homolog of herpes simplex virus have been proposed to represent an internal fusion peptide and similar motifs are found in equivalent positions in other herpesviruses (Gianni et al., 2005). Residues that are part of a heptad repeat sequence just downstream of this peptide are also essential to fusion (Gianni, Menotti, and Campadelli-Fiume, 2005). Peptides corresponding to a putative coiled-coil domain in human cytomegalovirus gH between residues 108-128 inhibit membrane fusion. Mutations in residues 52-79 of EBV gH which are also predicted, although perhaps less robustly, to be part of a coil-coiled domain, impact fusion with both B cells and epithelial cells (Omerovic, Lev, and Longnecker, 2005). However, residues close to the transmembrane domain of herpes simplex virus gH have also been shown to be essential (Galdiero et al., 1997) and our work is consistent with this.
The original insertion mutants M10 and M11 had reciprocal effects on B cell and epithelial cell fusion despite the fact that they were only 15 amino acids apart. This suggested that these sequences were in some way important to triggering fusion both via HLA class II and gHgLR. Since a soluble form of gp42 that binds to gHgL in trans also blocks the interaction of gHgL and gHgLR, this did not seem unreasonable. The single alanine substitutions made here are consistent with the previous observations in that they show that even closely adjacent residues can differentially affect the behavior of the protein. However, they did not entirely recapitulate the effects of the original insertions.
Single amino acid substitutions in the sequence around site of the M11 insertion had relatively little impact on fusion with either cell type, but the two that had modest but significant effects, R607A and K610A, reciprocally affected B cell and epithelial cell fusion. Adjacent single amino acid substitutions around the site of insertion of M10 had more pronounced effects. Alanine substitutions at residues 592 and 597 (V592A and R597A) both significantly impacted, though did not abrogate B cell fusion and had little or no effect on fusion with an epithelial cell. These mutations were in line with the original M10 which only mediated epithelial cell fusion. Substitution of an alanine for a glycine residue at position 594, however, essentially abrogated fusion with both cell types. Whether this means that this residue is important for a “core” event downstream of fusion or rather that it blocks triggering either via HLA class II or gHgLR cannot currently be determined. The protein at least retained the ability to interact with gp42, although it may have been impaired in translating a signal from the gp42 HLA class II interaction downstream. The substitution of alanine for glutamic acid at the immediately adjacent residue 595 produced a phenotype similar to that of the original M10 in its ability to mediate moderate levels of epithelial cell fusion, but differed dramatically in its acquisition not merely of the ability to mediate B cell fusion but to mediate it at levels significantly higher than the wild type protein.
Further exploration of the phenotype of the E595A mutant led to the previously unrecognized finding that gp42 can interact directly with gH in the absence of gL. We could, however, find no evidence for a substantial change in the ability of E595A to increase the levels of gp42 at the cell surface. This despite the fact that reduction of gH levels at the cell surface, either by omitting gL and impairing its transport or by omitting gH itself from the transfection mix, did reduce the surface levels of gp42 and implied that at least a portion of gp42 is either transported to or stabilized at the surface as a result of complex formation. It was a surprise to find that a very small but detectable amount of gH could be expressed at the cell surface in the absence of gL and that the small amount of E595A at the surface of the cell in the absence of gL could mediate detectable cell fusion. It did, however, suggest that the E595A mutation may produce a protein with a conformation least in part similar to that supported by coexpression of gL and that it perhaps has an enhanced ability to transmit or amplify an otherwise weak triggering signal from gp42.
Although there is general consensus that the three conserved homologs, gB, gH and gL make up the core fusion machinery of herpesviruses the order in which they are triggered into an active state is generally less clear and it has remained uncertain as to whether the fusion event itself involves one or more than one of the three. Peptides corresponding to predicted alpha helical coiled-coil domains in the gB and gH homologs of human cytomegalovirus can independently block virus entry, but these domains do not appear to function in an identical manner as coiled coil domains found in class I fusion proteins and inhibition presumably could reflect either the need for interaction between the two proteins or the involvement of both in the events proximal to fusion (Lopper and Compton, 2004). The interaction of glycoprotein gD with one of several entry mediators triggers fusion of herpes simplex virus and recent studies have suggested that this is followed by sequential interaction of gD, gB and then gHgL (Gianni, Forghieri, and Campadelli-Fiume, 2006). The order in which EBV proteins act may be different, however. A mutated form of EBV gB is able to mediate fusion in the absence of gHgL, suggesting that it is the final player in the process (McShane and Longnecker, 2004). The fact that gHgL are directly involved in triggering of fusion with epithelial cells, might suggest that they function upstream of gB. The work done here provides no kinetic data, but the observation that gp42 directly interacts with gH and that the E595A mutant can, together with gB, mediate at least small amounts of fusion in the absence of gL, is consistent with the suggestion that, for B cells at least, it is to gH that the initial triggering signal is transmitted.
Materials and methods
Cells
CV-1 monkey kidney cells were grown in Dulbecco’s modified Eagle’s medium. AGS, a human gastric carcinoma cell line and Chinese hamster ovary (CHO-K1) cells were grown in Ham’s F12 medium. Daudi 29 cells (a gift of Richard Longnecker, Northwestern University), B cells that stably express T7 RNA polymerase from the pOS2 vector (Whetter et al., 1994), were grown in RPMI medium. All culture media were obtained from Gibco-BRL Life Technologies (Grand Island NY) and supplemented with 10% heat inactivated fetal bovine serum.
Antibodies
Antibodies used were monoclonal antibodies (Mabs) E1D1 to gHgL, CL59 and CL40 to gH, CL55 to gB, F-2-1 to gp42 (Li, Turk, and Hutt-Fletcher, 1995) and M2 (Sigma-Aldrich, St Louis, MO) to the Flag epitope. A rabbit antibody, anti-gL, made to a peptide corresponding to residues 125 to 137 of gL was also used. All non-commercial antibodies were affinity purified on protein A-agarose.
Virus
Stocks of recombinant vaccinia virus expressing the T7 RNA polymerase (vvT7) (a gift of William Britt, University of Alabama, Birmingham) were grown in CV-1 cells infected at a multiplicity of infection of 0.01 and harvested by freeze-thawing and sonication of cells.
Plasmids
Plasmids pTM1-EBV gH and pTM1-EBV gL and pTM1-gp42 (Li, Turk, and Hutt-Fletcher, 1995) were made by cloning PCR amplified sequences into the pTM1 vector (Moss et al., 1990) which contains a T7 promoter, the encephalomyocarditis virus cap-independent translation signal, a multiple cloning site, and a T7 transcriptional terminator. Translation initiates at the Nco I site in the multiple cloning region. In pTM1-EBV gH this site is lost as a result of blunt end cloning. Plasmids pCAGGS-gH pCAGGS-gL, pCAGGS-gB and pCAGGS-gp42 (Wu, Borza, and Hutt-Fletcher, 2005) were made by cloning PCR amplified sequences into the pCAGGS/MCS vector (a gift of Martin Muggeridge, LSUHSC) for expression under control of the β-actin promoter in cooperation with the HCMV-IE enhancer (Niwa, Yamamura, and Miyazaki, 1991). A Flag tagged version of pCAGGS-gH, pCAGGS-Flag-gH (Wu, Borza, and Hutt-Fletcher, 2005), was made by insertion of an 8 amino acid Flag epitope between residues 22 and 23 of the EBV gH sequence, 4 residues after the predicted cleavage site of the putative signal sequence. Point mutations in pCAGGS-Flag-gH were made using the Quickchange II protocol (Stratagene, La Jolla, CA) in a 1.6 kb Bgl II-Pvu II fragment of gH encompassing sequences coding for residues 500 to 629 that had been cloned into the 2.5 kb pSP72 vector (Promega, Madison, WI) as previously described (Wu, Borza, and Hutt-Fletcher, 2005). The mutated Bgl II-Pvu II fragments were cloned back into pCAGGS-Flag-EBVgH for assay of fusion activity or into pTM1-EBV gH for biochemical analysis. The desired mutation was confirmed by sequencing of the fragments after return to pCAGGS-EBV Flag-gH.
Transfection-infection protocol
CV-1 cells were grown to 90% confluency in 100-mm-diameter Petri dishes and infected with vvT7 at a multiplicity of infection of 5. Thirty minutes later, the inoculum was removed, and the cells were washed twice in medium without serum and transfected with pTM1 plasmids. 10 μg of DNA was mixed with 30ul of Lipofectamine (Gibco/BRL) made to a total volume of 2.5 ml with serum-free medium.
Immunofluorescence
For internal staining, slides bearing air-dried acetone-fixed cells were incubated at 37°C for 30 min in a humidified atmosphere with Mabs, washed three times with phosphate-buffered saline for 5 min each, reincubated for 30 min with the appropriate dilution of fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse immunoglobulin (ICN/Cappel, Irvine, CA), washed three times and mounted in Mounting Medium (Kirkegaard Perry Laboratories, Gaithersburg, MD). For surface staining, unfixed cells were sequentially incubated with Mabs and FITC-conjugated anti-mouse immunoglobulin. Cells were washed three times between incubations and three times before mounting in Mounting Medium.
Levels of cell surface expression
The levels of expression of wild type and mutant gH constructs at the cell surface were measured by flow cytometric analysis of CHO-K1 cells transfected with pCAGGS-Flag-gH or gH mutants and pCAGGS-gL. 3.2 μg DNA was mixed with 4 μl Target transfectin F2 and 12 μl Target peptide enhancer (Targeting Systems, Santee, CA) in high glucose DMEM media. Twenty four hours later, transfected cells were washed with ice-cold phosphate-buffered saline containing 2% fetal bovine serum and serially reacted with MAb M2 (anti-Flag) and phycoerythrin conjugated anti-mouse immunoglobulin (Jackson ImmunoResearch Laboratories, West Grove, PA) with washing between each step.
AGS cell-cell fusion assay
AGS cells were seeded in two-well chamber slides and were transfected at 70-80% confluency for 4 hours with 0.25 μg pCAGGS-Flag-gH or 0.25 μg mutated pCAGGS-Flag-gH together with 0.25 μg pCAGGS-gL and 0.6 μg pCAGGS-gB. Plasmids were mixed with 1 μl Target transfectin F2 and 2 μl Target peptide enhancer in high glucose DMEM media. Twenty four hours post transfection cells were fixed with ice-cold acetone and stained with Mab to gB. Cells expressing gB and containing four or more nuclei were considered to have undergone fusion and were recorded as one fusion event. The extent of fusion was calculated as the number of fusion events as a percentage of the total number cells expressing gB. No fusion was seen in cells that did not stain with Mab to gB.
Epithelial cell-B cell fusion assay
CHO-K1 cells were seeded in 6-well plates and were transfected at 70-80% confluency for 4 hours with mutant 0.8 μg pCAGGS-Flag-gH or mutated pCAGGS-Flag-gH together with 0.8 μg gL, 1.4 ug pCAGGS-gB and 1.2 μg pCAGGSgp42 as well as 0.8 μg pST7-Luc containing the T7 promoter upstream of the luciferase gene (Ferrer et al., 1999) (a gift of Martin Muggeridge). Each well of transfected cells was then overlayed with two million Daudi 29 cells that expressed T7 RNA polymerase. 24 hours later, cells were washed twice with phosphate buffered saline and lysed with 500 μl Passive Lysis Buffer (Promega). Luciferase substrate (100 μl) was added to 20 μl supernatant of lysate. Luminosity readings were obtained by using a TD-20/20 luminometer (Promega).
Radiolabeling and immunoprecipitation
CV-1 cells were been infected with vvT7 and transfected with pTM1 plasmids were labeled biosynthetically with 100 μCi of Pro-Mix (70% [35S]methionine, 30% [35S]cysteine) and 300 μCi of [35S]cysteine (>1,000 Ci/mmol; Amersham Biosciences, Arlington IL) per dish (approximately 107 cells) as previously described (Li, Turk, and Hutt-Fletcher, 1995). Labeled cells were solubilized in radioimmunoprecipitation buffer (50 mM Tris-HCl, pH 7.2, 0.15 M NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.1 mM phenylmethyl-sulfonylfluoride, and 100 U of Aprotinin per ml) and immunoprecipitated with antibody and protein A-Sepharose CL4B (Sigma). Immunoprecipitated proteins were washed, dissociated by boiling for 2 min in sample buffer with 2-mercaptoethanol, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 10% polyacrylamide cross-linked with 0.28% N,N’-diallyltartardiamide followed by fluorography.
Acknowledgments
This work was supported by Public Health Services grants AI20662 from the National Institute of Allergy and Infectious Diseases and DE016669 from the National Institute of Dental and Craniofacial Research. Support was also provided by Cobre grant P20-RR018724 from the National Center for Research Resources of the National Institutes of Health.
We thank Richard Longnecker, Northwestern University, for Daudi 29 cells and Martin Muggeridge, Louisiana State University Health Sciences Center, for the pCAGGS/MCS and pST7-Luc vectors and for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J Virol. 2004;78:5007–5014. doi: 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M, Kapoor TM, Strassmaier T, Weissenhorn W, Skehel JJ, Oprian D, Schreiber SL, Wiley DC, Harrison SC. Selection of gp41 mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nature Struct Biol. 1999;6:953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J Virol. 1997;71:2163–2170. doi: 10.1128/jvi.71.3.2163-2170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Forghieri C, Campadelli-Fiume G. The herpesvirus glycoproteins B and H-L are sequentially recruited to the receptor-bound gD to effect membrane fusion at virus entry. Proc Natl Acad Sci USA. 2006;103:14572–14577. doi: 10.1073/pnas.0606127103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gianni T, Martelli PL, Casadio R, Campadelli-Fiume G. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha-helix with attributed of an internal fusion peptide, positionally conserved in the herpesviridae family. J Virol. 2005;79:2931–2940. doi: 10.1128/JVI.79.5.2931-2940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Menotti L, Campadelli-Fiume G. A heptad repeat in herpes simplex 1 gH, located downstream of the α-helix with attributes of a fusion peptide, is critical for virus entry and fusion. J Virol. 2005;79:7042–7049. doi: 10.1128/JVI.79.11.7042-7049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol. 1997;71(6):4657–4662. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Turk SM, Hutt-Fletcher LM. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J Virol. 1995;69:3987–3994. doi: 10.1128/jvi.69.7.3987-3994.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopper M, Compton T. Coiled-coil domains in glycoproteins B and H are involved in human cytomegalovirus membrane fusion. J Virol. 2004;78:8333–8341. doi: 10.1128/JVI.78.15.8333-8341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane MP, Longnecker R. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci USA. 2004;101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B, Elroy-Stein O, Mizukami T, Alexander WA, Fuerst TR. New mammalian expression vectors. Nature. 1990;348:91–92. doi: 10.1038/348091a0. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression tansfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Omerovic J, Lev L, Longnecker R. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with B cells and epithelial cells. J Virol. 2005;79:12408–12415. doi: 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG, Longnecker R. Herpesvirus entry: an update. J Virol. 2003;77:10179–10185. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hutt-Fletcher LM. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J Virol. 1998;72:158–163. doi: 10.1128/jvi.72.1.158-163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kenyon WJ, Li QX, Mullberg J, Hutt-Fletcher LM. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol. 1998;72:5552–5558. doi: 10.1128/jvi.72.7.5552-5558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetter LE, Day SP, Brown EA, Elroy-Stein O, Lemon SM. Analysis of hepatitis A virus translation in a T7 polymerase-expressing cell line. Arch Virol. 1994;(Suppl 9):291–298. doi: 10.1007/978-3-7091-9326-6_29. [DOI] [PubMed] [Google Scholar]
- Wu L, Borza CM, Hutt-Fletcher LM. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J Virol. 2005;79:10923–10930. doi: 10.1128/JVI.79.17.10923-10930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen LR, Stephens EB, Davenport LC, Hutt-Fletcher LM. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology. 1993;195:387–396. doi: 10.1006/viro.1993.1388. [DOI] [PubMed] [Google Scholar]