

Abstract
Werner syndrome (WS) is characterized by premature onset of age-associated disorders and predisposition to cancer. The WS protein, WRN, encodes 3′ → 5′ DNA helicase and 3′ → 5′ DNA exonuclease activities, and is implicated in the maintenance of genomic stability. Translesion (TLS) DNA polymerases (Pols) insert nucleotides opposite replication-blocking DNA lesions and presumably prevent replication fork stalling/collapse. Here, we present in vitro and in vivo data that demonstrate functional interaction between WRN and the TLS Pols, Polη, Polκ, and Polι. In vitro, WRN stimulates the extension activity of TLS Pols on lesion-free and lesion-containing DNA templates, and alleviates pausing at stalling lesions. Stimulation is mediated through an increase in the apparent Vmax of the polymerization reaction. Notably, by accelerating the rate of nucleotide incorporation, WRN increases mutagenesis by Polη. In vivo, WRN and Polη colocalize at replication-dependent foci in response to UVC irradiation. The functional interaction between WRN and TLS Pols may promote replication fork progression, at the expense of increased mutagenesis, and obviate the need to resolve stalled/collapsed forks by processes involving chromosomal rearrangements.
Keywords: DNA damage, mutagenesis
WRN is a DNA helicase-exonuclease that belongs to the RecQ family of DNA helicases (1). Loss-of-function mutations in WRN result in the premature aging disorder, Werner syndrome (WS). WS is distinguished by an early onset of age-associated conditions, including bilateral cataracts, type II diabetes, atherosclerosis, and osteoporosis, and by an elevated incidence of unusual cancers (2–4).
Primary cells from WS patients exhibit genomic instability and diminished replicative life span in culture (5). Instability is manifested at the chromosomal level by multiple nonclonal rearrangements termed variegated translocation mosaicism, and at the molecular level by large DNA deletions (6–8). WS cells are sensitive to DNA damaging agents including 4-nitroquinoline-1-oxide, cross-linking agents (such as mitomycin C and cisplatin), and camptothecin and hydroxyurea (2, 9). WS cells also display sensitivity to DNA methylating agents but only when repair systems that remove these lesions are compromised (10).
In vitro, WRN exhibits 3′ → 5′ DNA helicase and 3′ → 5′ DNA exonuclease activities. The weak processivity of WRN helicase is enhanced by human single-stranded DNA binding protein, replication protein A (11, 12). WRN exonuclease resembles DNA polymerase (Pol) proofreading exonucleases in its preference for 3′ recessed ends and single-terminal mismatches (13). However, unlike proofreading exonucleases, WRN does not hydrolyze single-stranded DNA (13). WRN preferentially unwinds and degrades noncanonical DNA structures, some of which resemble intermediates of replication and recombination. These include bubble DNA, forked DNA, D-loops, synthetic Holliday junctions, and quadruplex DNA (2, 3).
Although WRN binds many cellular proteins, it interacts functionally with only a limited number, many of which are implicated in DNA replication and recombination. It markedly stimulates the activities in vitro of FEN-1 and Polδ, integral components of the replication apparatus (14, 15), and interacts with the MRN complex (Mre11-Rad50-Nbs1) (16, 17) and BLM (the product of the Bloom syndrome gene) (18) that function in recombination processes. The DNA substrate specificity of WRN, its interaction with replication/recombination proteins, and the sensitivity of WS cells to replication blocking DNA lesions all implicate WRN in DNA damage tolerance processes.
Translesion (TLS) Pols are specialized Pols whose primary function is to insert nucleotides across DNA lesions that block progression of replicative Pols. Eukaryotes are endowed with several TLS Pols, each presumably responsible for the bypass of specific lesions or class of lesions. Human cells have four TLS Pols, REV1, Polη, Polκ, and Polι, that belong to the Y family, and a family B Pol, Polζ (19). In vivo and in vitro evidence implicate Polη in error-free bypass of UV-induced cyclobutane pyrimidine dimers (CPD) (20, 21) and Polκ in the bypass of benzo[a] pyrene adducts (22–24). It is assumed that, by enabling the bypass of DNA lesions, TLS Pols mitigate against stalling and concomitant collapse of the replication complex. Inactivating mutations in POLH (encoding Polη) result in the UV sensitive, cancer-prone disorder, Xeroderma pigmentosum variant, XPV (25–27).
TLS Pols differ from replicative Pols in the following characteristics. (i) TLS Pols exhibit limited processivity. (ii) They lack proofreading exonucleolytic activity that enhances the fidelity of DNA synthesis by replicative Pols (19, 28). (iii) Although the bypass of some lesions is believed to be error-free (for example, bypass of CPD by Polη), TLS Pols are error-prone across normal bases. (iv) Active sites of TLS Pols can accommodate large bulky groups adducted to template bases. (v) The Y-family of TLS Pols possess a unique structural motif termed the “little finger domain” that presumably contributes to bypass activity.
We reported (15) that WRN interacts functionally with eukaryotic Polδ but does not affect DNA synthesis by eukaryotic Pols α, β, or ε, the Klenow fragment of E. coli PolI, murine Moloney leukemia virus reverse transcriptase, or the thermostable Thermus aquaticus (Taq) Pol. Here, we demonstrate that WRN stimulates the polymerase and lesion bypass activity of human TLS Pols, Polη, Polκ, and Polι. Further, by accelerating the rate of DNA polymerization, WRN increases mutagenesis by Polη. Importantly, the in vitro functional cooperation between WRN and Polη can also be observed in vivo. We show that WRN and Polη colocalize in distinct nuclear foci after UV irradiation. These findings suggest a new role for WRN in DNA damage tolerance.
Results
WRN Stimulates DNA Synthesis by Bypass Pols.
Translesion Pols synthesize DNA across replication-blocking lesions and prevent stalling/collapse of the replication fork. Although the precise role of WRN is unclear at present, it has been implicated in processes that maintain the integrity of the replication fork. Because both TLS polymerases and WRN are involved in DNA damage tolerance pathways, we inquired whether they interact functionally with each other.
Because TLS Pols exhibit low processivity, we monitored the effect of WRN on extension, by human Polη, Polκ, and Polι, of a 28-nt DNA primer hybridized to a 36-nt long DNA template. Under conditions that limit primer extension by TLS Pols, WRN exerted a dramatic stimulatory effect on DNA synthesis by these enzymes (Fig. 1A). For example, whereas Polη elongated only 1% of the primer in the absence of WRN, 97% of the primer was extended in reactions with WRN. Notably, 20% of the reaction product corresponded to the length of the DNA template. Under identical reaction conditions, stimulation by WRN was not observed with human Polε and was very weak with human Polα (10% partially extended primer) versus 98% fully extended primer with Polη, demonstrating a marked preference for TLS Pols (Fig. 1B). Similar stimulatory effects of WRN on TLS Pols were observed with several different DNA substrates, including those requiring synthesis of up to 30 nt [see Fig. 3 and supporting information (SI) Fig. 7]. Assuming monomeric molecular weights and 100% activity of each enzyme preparation, stimulation was observed over a range of WRN concentrations, including a WRN:TLS Pol molar ratio as low as 6:1 (Fig. 2). However, reports that WRN exists as a multimer (29) suggest that the effective stimulatory concentration of WRN could be considerably lower. Interestingly, we have also observed the converse effect, namely, stimulation of the enzymatic activities of WRN by TLS Pols. However, this effect required a considerably large (>100-fold) excess of polymerase to detect by a gel-based assay (SI Fig. 8).
Fig. 1.

WRN stimulates the activity of Y-family TLS Pols. (A) A 5′-end-labeled 28-nt DNA primer, hybridized to a 36-nt DNA template (oligonucleotides 1 and 2; SI Table 2), was extended by Polη (0.375 fmol), Polκ (1 fmol), or Polι (0.4 fmol) in the absence (−) or presence (+) of WRN (30 fmol) at 37°C for 10 min. Reaction aliquots were electrophoresed through 14% polyacrylamide-urea gels; extension products were visualized and quantified by PhosphorImager analysis. (B) Extension of the 28-nt DNA primer was carried out as described in A except that human Polα and Polε were assayed in parallel with Polη. S, (−) enzyme; W, WRN alone.
Fig. 3.

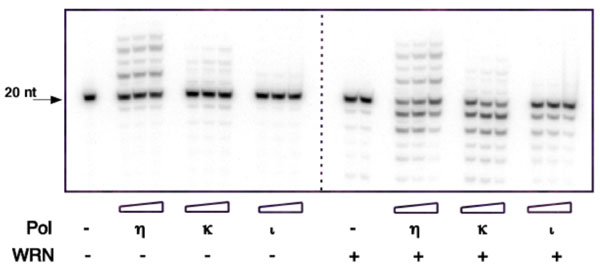
WRN stimulates lesion bypass activity of TLS Pol. (A) A 14-nt primer (oligo 8), hybridized to a DNA template with a site-specific cyclobutane pyrimidine dimer (oligo 10), was extended by 0.75 fmol Polη, 4 fmol Polκ, or 0.4 fmol Polι in the absence or presence of a fixed amount of WRN (30 fmol) as described. A lesion-free DNA template (oligo 9) served as a control. (B) A 20-nt DNA primer (oligo 11) was annealed either to a DNA template containing a single BPDE-dG adduct (oligo 13) or lacking the lesion (oligo 12) and extended by either Polκ (4 fmol, lanes 1 and 2; 16 fmol, lanes 3 and 4) or Polη (1.5 fmol, lanes 1 and 2; 6 fmol, lanes 3 and 4) with or without WRN (40 fmol) as indicated. Extension reactions were as described except that the dNTP concentration was increased to 0.2 mM. (C) A 28-nt primer (oligo 1), annealed to a 36-nt DNA template (oligo 2) containing a site-specific abasic site analog (AB), O6-methylguanine (O6mG), O4-methylthymine (O4mT), 1, N6-etheno adenine (eA), or 8-oxoguanine (8-oxdG), was extended by Polη (0.375 fmol) or Polκ (2 fmol) in the absence (−) or presence (+) of WRN (30 fmol). Sequences of DNA templates are indicated at the left of each panel.
Fig. 2.

TLS polymerase stimulation is specific to WRN. Extension of the 28/36 P/T DNA substrate (Fig. 1) by Polη (0.375 fmol) was monitored in the absence (−) or presence of increasing, equimolar amounts (3–30 fmol) of hWRN, hBLM or E. coli RecQ. S, (−) enzyme.
The Interaction of TLS Pols Is Specific to WRN.
To address the specificity of the functional interaction between TLS Pols and WRN, we carried out several experiments. First, heat inactivation of WRN abolished the stimulatory effect, indicating a requirement for a structured protein component. Second, stimulation was not a result of added salt. Third, stimulation was observed with four independent preparations of homogeneous wild-type WRN and two different preparations of Polη. Lastly and most importantly, stimulation of TLS Pol activity was not observed with other RecQ helicases. Equimolar amounts of E. coli RecQ and human BLM failed to detectably stimulate the activities of either Polη (Fig. 2) or Polκ (data not shown). Interestingly, helicase and exonuclease-deficient WRN mutants stimulated Polη to a similar extent as the wild-type protein (SI Fig. 9). These data suggest that, at least with a simple DNA primer-template, the helicase or exonuclease activity of WRN is not required for stimulating DNA synthesis by Polη.
WRN Stimulates Lesion Bypass.
Because the designated function of TLS Pols is lesion bypass, we assayed bypass activity across site-specific DNA lesions in the absence or presence of WRN. Addition of WRN to limiting amounts of Polη stimulated extension activity on a CPD-containing template, comparable with that observed on an identical DNA template lacking the T–T dimer; i.e., 8% extension (−) WRN versus 95% (+) WRN (Fig. 3A). In contrast, limiting amounts of Polκ and Polι were unable to synthesize DNA across and beyond the CPD lesion even in the presence of WRN, although stimulation of synthesis up to the site of the lesion and on the lesion-free DNA template was readily apparent (Fig. 3A).
Benzo[a]pyrene-diol-epoxide guanine (BPDE-dG) is a bulky replication-blocking lesion. In accord with published reports, we observed insertion of nucleotides opposite BPDE-dG and minimal DNA synthesis beyond the lesion by Polκ and Polη (Fig. 3B; Polκ and Polη, lane 3). Addition of WRN stimulated extension; the most significant effect was manifested on synthesis of products extending 2 nt past the lesion and up to the length of the template strand. The increase in lesion bypass synthesis with, versus without, WRN was at least 5- and 4-fold with Polκ and Polη, respectively (Fig. 3B; Polκ and Polη, lane 4). Robust stimulation of Polκ and Polη was observed with the identical lesion-free DNA substrate (Fig. 3B; Polκ and Polη; compare lanes 1 and 2).
WRN also enhanced extension on DNA templates containing site-specific methyl adducts O6-methylguanine and O4-methylthymine, the oxidative lesion, 8-oxoguanine, an abasic site or 1, N6-ethenoadenine lesion (Fig. 3C). However, the extent of bypass (i.e., synthesis across and beyond the lesion) varied with the type of lesion. For example, the ≈35% bypass of 8-oxoguanine by Polη + WRN was similar to that seen in the absence of any lesion (37%). In contrast, no detectable bypass of the 1, N6-ethenoadenine lesion was observed with either Polη or Polκ, even in the presence of WRN. On the other hand, measurable bypass, ≈7% with Polη and as much as 20% with Polκ, of the methyl adducts was promoted by WRN (Fig. 3C).
Mechanism of TLS Pol Stimulation by WRN.
To understand the basis of the stimulatory effect of WRN, we monitored reaction kinetics in the absence or presence of WRN. Polη and Polκ by themselves extended only between 1- 2% of the primer after 30 s, 2 min, and 6 min at 37°C. In contrast, in the presence of WRN, both polymerases extended 10%, 46%, and 82% of the primer, respectively, in the same time periods (SI Fig. 10). We also determined the apparent Vmax and Km of Polη for the initiating nucleotide, dTTP, keeping the amounts of DNA primer-template and Polη constant. Whereas the apparent Km for dTTP was not appreciably different in the absence or presence of WRN, there was a reproducible increase (2.5- to 3-fold) in the Vmax with WRN (Table 1), further supporting the observation that WRN increases reaction rates.
Table 1.
Kinetic constants of Polη for incorporation of the initiating nucleotide
| WRN | Km, μM | Vmax, % extension min−1 |
|---|---|---|
| (−) | 1.5 ± 1.0 | 22 ± 7.2 |
| (+) | 2.0 ± 0.8 | 59 ± 8.5 |
The apparent Km and Vmax values (mean ± SD) were calculated from Hanes–Woolf plots of measurements from three independent experiments carried out with DNA primer-template formed by annealing oligonucleotides 8 and 9.
WRN Increases the Amount of Errors Generated by Polη.
Three sets of experiments were carried out to determine whether WRN influences the fidelity of Polη. First, we carried out polymerase reactions in the presence of single dNTPs. Polη misincorporated and misextended to a large extent, particularly in reactions containing either dGTP or dATP (Fig. 4A). WRN enhanced the extent of misincorporation and misextension but did not alter lengths of extension products. The accuracy of synthesis was also monitored in reactions lacking one of four dNTPs. Polη exhibited a high error frequency in reactions lacking either dGTP, dATP, or dCTP; addition of WRN stimulated the incorporation and extension of noncomplementary nucleotides, most notably in reactions lacking dTTP (Fig. 4B).
Fig. 4.

WRN increases the extent of nucleotide misinsertion and misextension by Polη. Polη (1.5 fmol) was incubated with 0.1 pmol of a 16/30 P/T (oligo 14/oligo 9) (A) or a 14/30 P/T (oligo 8/oligo 9) (B) and either a single dNTP (A) or three of four dNTPs (B). Extension reactions were carried out at 37°C for 10 min ± 15 fmol WRN. N, reactions with all four dNTPs; S, DNA P/T (−) polymerase. Sequences of DNA templates are indicated at the left of each panel.
Finally, we measured the error frequency of Polη during extension of a gapped M13mp2 DNA substrate containing the lacZ α-complementation target sequence. Using a 3-fold molar excess (12 fmol) of Polη over that of DNA, the frequency of mutant plaques generated by Polη alone was ≈3%. When the Polη:DNA molar ratio was increased to 50:1, we observed the reported mutation frequency of ≈30% (30). Addition of stoichiometric amounts of WRN (12 fmol) resulted in a reproducible 2-fold increase in the frequency of mutant plaques relative to Polη. Sequence analysis of DNA from the mutant plaques revealed mutational hot spots; a preponderance of T → C substitutions and insertions at template dT and dA residues, and deletions thereof, was observed. The positions of frequent substitutions, insertions, and deletions were not altered by the addition of WRN. Moreover, the types of sequence changes in the target DNA were independent of WRN (SI Fig. 11). However, the number of mutations within the first 100 nucleotides of each gapped DNA molecule synthesized by Polη was higher with WRN than without (Fig. 5). For example, the colorless plaques generated by Polη alone had predominately one, two, or three substitutions in the lacZ sequence, whereas those with Polη + WRN displayed, in addition, four and even as many as six mutations per clone. A nonparametric Wilcoxon rank-sum test analysis of these data demonstrated that this difference was statistically significant (P = 0.006).
Fig. 5.

WRN increases mutagenesis by Polη. A 407-nt gap within the lacZ α-complementation sequence of bacteriophage M13mp2 was copied by Polη (12 fmol) ± an equimolar amount of exonuclease-deficient WRN. Reaction aliquots were transformed and plated on minimal medium containing X-gal. DNA isolated from colorless or light blue plaques was sequenced to score alterations in the target region. The frequencies at which DNA from mutant plaques displayed single or multiple mutations within the first 100 nucleotides of the gap in reactions lacking (open bars) or containing WRN (filled bars), are presented.
WRN Colocalizes with Polη After UV Damage.
We investigated whether the in vitro functional collaboration between WRN and Polη can be observed in vivo. To do so, we cotransfected HeLa cells with EGFP-WRN and DsRed-Polη and monitored their cellular localization before and after UVC exposure. Both WRN and Polη distributed homogeneously in the nucleus in the absence of UV irradiation. However, in response to UVC, Polη formed discrete, DNA replication-dependent nuclear foci, as reported (31). WRN, too, formed distinct nuclear foci; importantly, on average, 94% of WRN foci colocalized with those of Polη, as revealed by the abundance of yellow foci after a merger of the individual images (Fig. 6A). Polη and WRN form foci independent of each other; UV-induced DsRed-Polη foci were visible in two independent WRN−/− cell lines (AG11395 and WV1) and likewise, GFP-WRN formed foci in XP7TA cells lacking Polη (data not shown). WRN constructs lacking the N-terminal exonuclease domain or, with the missense K577M mutation that eliminates helicase activity, formed foci that colocalized with those of Polη (Fig. 6B). On the other hand, WRN constructs encoding either the helicase and ribonuclease D C-terminal domain or the C-terminal domain failed to form foci in response to UVC irradiation.
Fig. 6.

WRN colocalizes with Polη after UVC irradiation. (A) HeLa cells expressing EGFP-WRN and DsRed-Polη were irradiated with 20 J/m2 UVC light, +UVC. After 6 h incubation in growth media, cells were fixed and examined for foci formation. DNA was stained with DAPI; −UVC, unirradiated controls. (B) WRN mutants used for transfection. K577M, helicase-defective; ΔExo, deletion of exonuclease domain; HRDC (helicase and ribonuclease D C-terminal domain), amino acids 1021–1432; C-ter (C-terminal) amino acids 1229–1432. HeLa cells, transfected with one of these EGFP-WRN constructs and DsRed-Polη, were irradiated and processed as described.
Discussion
We present evidence here for functional cooperation between WRN and the Y-family TLS Pols, Pols η, κ and ι. We show that, in vitro, WRN stimulates DNA synthesis by these TLS Pols on normal (Fig. 1) and lesion-containing (Fig. 3) DNA templates in a sequence-independent manner. In so doing, WRN increases mutagenesis by Polη (Fig. 5). In vivo, WRN colocalizes with Polη in response to UV irradiation (Fig. 6).
The interaction between WRN and TLS Pols is specific to WRN. Neither the prototype E. coli RecQ nor the homologous human BLM helicase detectably stimulates polymerase activity (Fig. 2). Although mutations in WRN and BLM result in genomic instability disorders, WS and BS cells are sensitive to similar DNA damaging agents, and WRN and BLM share common DNA substrates and interacting proteins, cooperation with Pols appears to be thus far limited to WRN (2, 3, 32). The WRN–TLS Pol interaction is also not common to all Pols. We have shown that WRN stimulates the polymerase activity of Polδ and enables it to traverse replication-blocking tetraplex DNA structures but has minimal or undetectable effects on Pols α and ε (Fig. 1B and refs. 15 and 33). By enhancing the activities of Polδ and TLS Pols, and by alleviating pausing at replication-blocking DNA structures and lesions, WRN may promote efficient progression of DNA synthesis and prevent fork stalling.
Like WRN, proliferating cell nuclear antigen (PCNA) has also been shown to stimulate the activity of Y-family TLS Pols (19). However, the mechanism of action of PCNA appears to differ from that of WRN. Whereas PCNA stimulates DNA polymerization by decreasing the apparent Km for nucleotides (34–36), WRN increases the rate of nucleotide incorporation by increasing the apparent Vmax of Polη (Table 1 and SI Fig. 10). Further, stimulation by WRN occurs in the absence of PCNA and does not require the PCNA binding domain of Polη as demonstrated by similar extents of stimulation of wild-type and a truncated Polη protein lacking the C-terminal PCNA binding motif (A.S.K.-L., unpublished data). Further, the processivity of TLS Pols on a 14/46 primer/template (SI Fig. 7) did not differ in the absence or presence of WRN (data not shown). It is unclear if the interaction between WRN and TLS Pols is direct or is mediated by interactions with DNA. Gel mobility shift assays to demonstrate a putative supershifted complex of DNA with WRN and Polη have been unsuccessful, suggesting that such a complex may be transient and unstable (data not shown). It is possible that the interaction between WRN and TLS Pols triggers a conformational change in the polymerase to increase rates of polymerization and frequency of mutagenesis.
Several striking findings emerge from this study. First, WRN stimulates DNA synthesis and alleviates TLS Pol stalling on lesion-containing DNA templates (Fig. 3). Second, and in accord, WRN and Polη cooperate in vivo in response to UV irradiation (Fig. 6). Third, WRN increases mutagenesis by Polη on unmodified DNA templates (Fig. 5).
In addition to stimulating the extension activity of TLS Pols on unmodified DNA templates, WRN enhances extension on DNA templates containing lesions (Fig. 3). A compelling observation is that synthesis by Polη on a CPD-containing DNA template is stimulated as strongly as on an otherwise identical, lesion-free template (Fig. 3A). Not only does WRN stimulate primer extension, it also alleviates pausing/stalling of Polκ and Polη at lesions that slow DNA synthesis. For example, despite the strong pause site observed at the nucleotide immediately preceding O6-methylguanine and O4-methylthymine, WRN facilitates extension beyond the lesions; bypass products account for as much as 20% of the extension products synthesized by Polκ (Fig. 3C). Likewise, although Polκ and Polη stall at the bulky, polycyclic BPDE-dG adduct, addition of WRN enhances extension beyond the lesion to generate full-length products (Fig. 3B). However, some lesions cannot be bypassed even in the presence of WRN, e.g., 1, N6-ethenoadenine that blocks extension by Polη and Polκ, and CPD that blocks synthesis by Polκ and Polι. Apparently, WRN can facilitate synthesis past only those lesions that TLS Pols can bypass, albeit inefficiently, on their own. Nonetheless, in vivo, if the cellular burden of stalling lesions is sufficiently high to render the amounts of TLS Pols rate limiting on bypass, the capacity of WRN to increase the efficiency of TLS Pols could become crucial for cell survival. By accelerating DNA polymerization and facilitating the bypass activity of TLS Pols, WRN may prevent replication fork demise in encounters with stalling or blocking lesions.
Consistent with this hypothesis, we have shown that WRN and Polη colocalize in vivo in response to UV irradiation (Fig. 6A). Although Polη has been reported to form UV-induced nuclear foci that colocalize with PCNA and incorporated BrdU (31), the effect on WRN localization has not been previously demonstrated. We show here that WRN also redistributes, exhibiting uniform nuclear staining in unirradiated cells and forming UV-induced foci that show 94% coincidence with replication foci containing Polη. These observations imply that both enzymes relocate to the same replication sites when ongoing synthesis is impeded by DNA lesions. Colocalization of WRN foci with those of Polη is independent of the helicase and exonuclease domains of WRN (Fig. 6B), in accord with our finding that stimulation of TLS Pol activity in vitro does not require these enzymatic activities (SI Fig. 9).
Another significant finding is that WRN modulates the extent of mutagenesis by Polη; specifically, WRN elevates the mutation frequency of Polη without altering its mutation spectrum. In stimulating the polymerase activity of Polη, WRN increases the incorporation and extension of both correct and incorrect nucleotides. This conclusion is supported by data from two experimental approaches. First, WRN increases the extent of misincorporation and misextension by Polη when reactions are carried out either with single dNTPs, or in reaction mixtures lacking one of the four dNTPs (Fig. 4 A and B), the latter presumably mimicking conditions of in vivo nucleotide pool imbalance. Second, in an in vitro lacZ forward mutation assay, stoichiometric amounts of WRN increase the mutation frequency of Polη and the total number of mutations per mutant clone (Fig. 5). Although WRN increases mutagenesis, the distribution and types of substitutions generated by Polη do not differ in the absence or presence of WRN. Interestingly, Polη may be one of the candidate low-fidelity TLS Pols operative in somatic hypermutation at the Ig variable regions (37, 38). The ability of WRN to elevate the mutation frequency of Polη could, therefore, be important in the generation of diversity at the Ig gene locus.
Our findings of in vitro and in vivo functional collaborations between WRN and TLS Pols reveal a previously unrecognized aspect of WRN function at the replication fork. They suggest that, by enhancing polymerization by error-prone Y-family TLS Pols, WRN may direct cells to make mutations and continue replication, rather than stall and risk reconstitution of collapsed forks by processes that can cause chromosomal rearrangements or cell death. The defects of WS cells include a prolonged S-phase, a reduced rate of DNA replication, and a decreased frequency of replication initiation (39–42). Absence of the interaction of WRN and TLS Pols could slow the progression of DNA synthesis particularly when the replication fork encounters DNA lesions. As an alternative to translesion synthesis, cells can tolerate DNA lesions by homologous recombination pathways. Data suggest that WS cells are defective in faithfully executing homologous recombination (43, 44). Inefficient translesion synthesis, together with the recombination defect, could explain, at least in part, the delayed progression of DNA replication and the prevalence of chromosomal rearrangements in WS cells. Although WRN has been implicated in the generation of chromosomal rearrangements and large deletions, in vivo frequencies of spontaneous and damage-induced base substitution and small insertion/deletion mutations in WS cells have thus far not been determined. WS patients present a wide range of pathology including an increased incidence of cancers and age-associated disorders. The functional interaction between WRN and TLS Pols could contribute to these pathologies.
Methods
Materials.
Human WRN proteins, wild-type, exonuclease-deficient (D82A, E84A), and helicase-deficient (K577M), were purified as described in ref. 45; concentrations of each ranged from 10 to 25 ng/μl. Homogeneous preparations of human Y-family TLS Pols, Polη, Polκ, and Polι (20–50 ng/μl) were purchased from Enzymax (Lexington, KY). Details of oligonucleotides used in the studies are in SI Table 2.
Primer Extension Reactions.
5′-γ-32P-end-labeled DNA primers were annealed with a 2-fold molar excess of corresponding DNA templates (46). Polymerase reactions (10 μl) were carried out in 40 mM Tris·HCl buffer (pH 7.9)/5 mM MgCl2/10 mM DTT/60 mM KCl/2.5% glycerol/100 μM each of dATP, dTTP, dGTP/dCTP/10 nM DNA primer-template, and indicated amounts of Pols and WRN. Reactions were incubated at 37°C for 10 min, and aliquots were electrophoresed through 14% polyacrylamide–8.0 M urea denaturing gels. Extension products were visualized and quantified by PhosphorImager analysis (GE Healthcare, Piscataway, NJ).
Gap-Filling Reactions.
Bacteriophage M13mp2 DNA containing a 407-nt gap within the lacZ α-complementation target sequence was prepared and filled, as described in ref. 47. The gapped DNA substrate (4 fmol) was extended by Polη ± WRN (12 fmol each) at 37°C for 10 min. Reaction aliquots were transformed and plated on α-complementation host E. coli cells. The number of wild-type (dark blue) and mutant (light blue and white) plaques was scored. M13mp2 DNA was sequenced and analyzed for mutations with Sequencher software, Version 4.2 (Gene Codes, Ann Arbor, MI). Further details of the assay may be found in SI Methods.
Plasmids, Cells, and Conditions of UVC Irradiation.
cDNA encoding human WRN and POLH were amplified with primers containing XhoI and NotI restriction sites at the 5′ and 3′ termini, respectively. PCR products were sequenced and cloned into the corresponding sites of pEGFP-C1 or pDsRed-C1 (tetramer type) vector (Clontech Laboratories, Mountain View, CA), respectively. Construction of WRN mutants was as described in ref. 48. HeLa, XP7TA (POLH deficient), AG11395, and WV1 (WRN deficient) cells were cultured in DMEM containing 10% FCS. Cells were plated at a density of 1 × 105 cells per 3.5-cm dish and transfected 24 h later with plasmid DNA, using FuGene6 (Roche, Indianapolis, IN). Two days after transfection, cells were washed with Hanks's buffer and irradiated with 20 J/m2 UVC light. Cells were incubated in growth medium for an additional 6 h after irradiation.
Immunofluorescence Microscopy.
To observe localization of EGFP-WRN or DsRed-Polη, cells were washed twice in PBS and then fixed with methanol:acetone (1:1) at −20°C for 10 min. After fixing, cells were washed with buffer containing 50 mM Tris·HCl buffer (pH 7.5), 150 mM NaCl, and 0.05% Tween 20 for 5 min, and stained with DAPI. Confocal fluorescence images were captured with a FV500 laser-scanning microscope (Olympus, Tokyo, Japan). Percent colocalization was calculated by determining the number of WRN foci that colocalized with Polη foci divided by the total number of WRN foci generated after UVC irradiation. Determinations were from an average of 10 independent cells.
Supplementary Material
Acknowledgments
We thank Drs. S. Iwai (Osaka University, Osaka, Japan) and N. Geacintov (New York University, New York) for their generous gifts of DNA oligonucleotides containing site-specific CPD and BPDE-dG adducts, respectively; Drs. S. Kowalczykowski (University of California, Irvine, CA) and N. Maizels (University of Washington, Seattle, WA) for supplying E. coli RecQ and human BLM, respectively; Michael Schmitt and Ali Ozgenc for their assistance in preparing gapped M13mp2 DNA; Dr. Ted Gooley for statistical analysis of data from the forward mutation assay; Drs. Michael Fry and Ann Blank for critically reading the manuscript and providing insightful advice; and members of the University of Washington Werner Syndrome Program Project for helpful discussions. This work was supported by Genome Network Project Grant PO1 CA077852 (to L.A.L.) and the Grants-in-Aid for Scientific Research Grant 18012003 from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to A.Y.).
Abbreviations
- BPDE-dG
benzo[a]pyrene-diol-epoxide guanine
- CPD
cyclobutane pyrimidine dimer
- PCNA
proliferating cell nuclear antigen
- TLS
translesion
- Pol
DNA polymerase
- WS
Werner syndrome.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702513104/DC1.
References
- 1.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, et al. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 2.Hickson ID. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 3.Ozgenc A, Loeb LA. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Orren DK. Front Biosci. 2006;11:2657–2671. doi: 10.2741/1999. [DOI] [PubMed] [Google Scholar]
- 5.Martin GM, Sprague CA, Epstein CJ. Lab Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- 6.Salk D, Au K, Hoehn H, Martin GM. Cytogenet Cell Genet. 1981;30:92–107. doi: 10.1159/000131596. [DOI] [PubMed] [Google Scholar]
- 7.Gebhart E, Bauer R, Raub U, Schinzel M, Ruprecht KW, Jonas JB. Hum Genet. 1988;80:135–139. doi: 10.1007/BF00702855. [DOI] [PubMed] [Google Scholar]
- 8.Fukuchi K, Martin GM, Monnat RJ., Jr Proc Natl Acad Sci USA. 1989;86:5893–5897. doi: 10.1073/pnas.86.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JW, Harrigan J, Opresko PL, Bohr VA. Mech Ageing Dev. 2005;126:79–86. doi: 10.1016/j.mad.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Blank A, Bobola MS, Gold B, Varadarajan S, D Kolstoe D, Meade EH, Rabinovitch PS, Loeb LA, Silber JR. DNA Repair. 2004;3:629–638. doi: 10.1016/j.dnarep.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Shen J.-C., Gray MD, Oshima J, Loeb LA. Nucleic Acids Res. 1998;26:2879–2885. doi: 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brosh RM, Jr, Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, Bohr VA. J Biol Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- 13.Kamath-Loeb AS, Shen J-C, Loeb LA, Fry M. J Biol Chem. 1998;273:34145–34150. doi: 10.1074/jbc.273.51.34145. [DOI] [PubMed] [Google Scholar]
- 14.Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar J, Opresko PL, Piotrowski J, Dianov I, Dianov GL, Bohr VA. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamath-Loeb AS, Johansson E, Burgers PMJ, Loeb LA. Proc Natl Acad Sci USA. 2000;97:4603–4608. doi: 10.1073/pnas.97.9.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, Carney JP, Bohr VA. J Biol Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- 17.Pichierri P, Franchitto A. Bioessays. 2004;26:306–313. doi: 10.1002/bies.10411. [DOI] [PubMed] [Google Scholar]
- 18.von Kobbe C, Karmakar P, Dawut L, Opresko P, Zeng X, Brosh RM, Jr, Hickson ID, Bohr VA. J Biol Chem. 2002;277:22035–22044. doi: 10.1074/jbc.M200914200. [DOI] [PubMed] [Google Scholar]
- 19.Prakash S, Johnson RE, Prakash L. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 20.Johnson RE, Washington MT, Prakash S, Prakash L. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 21.Masutani C, Kusumoto R, Iwai S, Hanaoka F. EMBO J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor JS, Geacintov NE, Wang Z. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki N, Ohashi E, Kolbanovskiy A, Geacintov NE, Grollman AP, Ohmori H, Shibutani S. Biochemistry. 2002;41:6100–6106. doi: 10.1021/bi020049c. [DOI] [PubMed] [Google Scholar]
- 24.Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, Livneh Z. J Biol Chem. 2004;279:53298–53305. doi: 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- 25.Cleaver JE. J Invest Dermatol. 1972;58:124–128. doi: 10.1111/1523-1747.ep12538913. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RE, Kondratick CM, Prakash S, Prakash L. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 27.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 28.Friedberg EC. Nat Rev Mol Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- 29.Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. J Mol Biol. 2001;312:335–346. doi: 10.1006/jmbi.2001.4937. [DOI] [PubMed] [Google Scholar]
- 31.Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Genes Dev. 2001;15:158–172. doi: 10.1101/gad.187501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opresko PL, Cheng WH, Bohr VA. J Biol Chem. 2004;279:18099–18102. doi: 10.1074/jbc.R300034200. [DOI] [PubMed] [Google Scholar]
- 33.Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PMJ, Fry M. J Biol Chem. 2001;276:16439–16446. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 34.Haracska L, Johnson RE, Unk I, Phillips B, Hurwitz J, Prakash L, Prakash S. Mol Cell Biol. 2001;21:7199–7206. doi: 10.1128/MCB.21.21.7199-7206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haracska L, Johnson RE, Unk I, Phillips BB, Hurwitz J, Prakash L, Prakash S. Proc Natl Acad Sci USA. 2001;98:14256–14261. doi: 10.1073/pnas.261560798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haracska L, Unk I, Johnson RE, Phillips BB, Hurwitz J, Prakash L, Prakash S. Mol Cell Biol. 2002;22:784–791. doi: 10.1128/MCB.22.3.784-791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 38.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. Nat Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 39.Fujiwara Y, Higashikawa T, Tatsumi M. J Cell Physiol. 1977;92:365–374. doi: 10.1002/jcp.1040920305. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi F, Hanaoka F, Goto M, Akaoka I, Hori T, Yamada M, Miyamoto T. Hum Genet. 1982;60:365–368. doi: 10.1007/BF00569220. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi F, Hanaoka F, Goto M, Yamada M, Miyamoto T. Exp Gerontol. 1982;17:473–480. doi: 10.1016/s0531-5565(82)80009-0. [DOI] [PubMed] [Google Scholar]
- 42.Poot M, Hoehn H, Runger TM, Martin GM. Exp Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- 43.Prince PR, Emond MJ, Monnat RJ., Jr Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen J-C, Gray MD, Oshima J, Kamath-Loeb AS, Fry M, Loeb LA. J Biol Chem. 1998;273:34139–34144. doi: 10.1074/jbc.273.51.34139. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Reference Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 1989. [Google Scholar]
- 47.Bebenek K, Kunkel TA. Methods Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]
- 48.Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, Yasui A. J Cell Sci. 2005;118:4153–4162. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}