

SUMMARY
Formation of the mitotic cleavage furrow is dependent upon both microtubules and activity of the small GTPase RhoA. GEF-H1 is a microtubule-regulated exchange factor that couples microtubule dynamics to RhoA activation. GEF-H1 localized to the mitotic apparatus in HeLa cells, particularly at the tips of cortical microtubules and the midbody, and perturbation of GEF-H1 function induced mitotic aberrations, including asymmetric furrowing, membrane blebbing, and impaired cytokinesis. The mitotic kinases Aurora A/B and Cdk1/Cyclin B phosphorylate GEF-H1, thereby inhibiting GEF-H1 catalytic activity. Dephosphorylation of GEF-H1 occurs just prior to cytokinesis, accompanied by GEF-H1-dependent GTP-loading on RhoA. Using a live cell biosensor, we demonstrate distinct roles for GEF-H1 and Ect2 in regulating Rho activity in the cleavage furrow, with GEF-H1 catalyzing Rho activation in response to Ect2-dependent localization and initiation of cell cleavage. Our results identify a GEF-H1-dependent mechanism to modulate localized RhoA activation during cytokinesis under the control of mitotic kinases.
INTRODUCTION
GEF-H1 and its mouse homologue Lfc represent novel members of the Dbl family of guanine nucleotide exchange factors (GEFs) with RhoA-specific enzymatic activity (Krendel et al., 2002; Ren et al., 1998). Subcellular localization analysis demonstrated that GEF-H1 is associated with microtubules (MTs), and that MT depolymerization leads to GEF-H1 activation, accompanied by a RhoA-dependent reorganization of the actin cytoskeleton (Krendel et al., 2002). Thus, GEF-H1 regulation by MTs provides a novel mechanism to link RhoA-controlled cellular activities to MT depolymerization. Because GEF-H1 activity is dependent on the polymerization state of MTs, we speculated that the extensive MT rearrangements that occur during mitosis might affect GEF-H1 and, thereby, RhoA signaling.
The small GTPase RhoA plays a central role in regulating the formation of the acto-myosin cleavage furrow (CF) during cell division. When RhoA is depleted genetically or through biochemical means, formation of the CF is prevented (Dean et al., 2005; Drechsel et al., 1997; Kishi et al., 1993; O’Connell et al., 1999). Moreover, the localization of RhoA activity to a precisely bounded zone seems to be critical for formation of the contractile apparatus necessary for cell cleavage (Bement et al., 2005). CF formation thus results from the local activation of RhoA in many cell types (Piekny et al., 2005). However, in Rat-1 (Yoshizaki et al., 2004) and Rat-2 (Bakal et al., 2005) cells, inhibition of RhoA does not prevent cytokinesis, but appears to exert effects on early mitotic spindle formation. Thus, the requirements for RhoA during cytokinesis appear to vary from cell type to cell type.
Studies manipulating the position or the integrity of the mitotic spindle have indicated that spindle MTs send CF initiation signals to the cell cortex to regulate the assembly and function of the contractile ring (Alsop and Zhang, 2004; Hamilton and Snyder, 1983; Rappaport, 1997). In particular, astral MTs emanating from the spindle poles, and antiparallel MT bundles between separating chromosomes (i.e. the midzone spindle) have crucial roles in activating cortical regions, most likely by controlling localized activation of RhoA at the cell equator (Bringmann, 2005; Canman et al., 2003; D’Avino et al., 2005; Dechant and Glotzer, 2003; Glotzer, 2004; Mandato et al., 2000; Strickland et al., 2005). However, the MT-regulated signals and mechanistic principles underlying RhoA activation during cytokinesis are poorly understood.
Since small GTPase activation requires the action of specific GEFs, it is conceivable that MT signaling may impinge on Rho-selective GEFs. Ect2 represents such an activator for Rho GTPases that has been implicated in cell division (Tatsumoto et al., 1999). There is accumulating evidence that the primary role of Ect2 is to ensure the localized concentration of RhoA at the CF (Kamijo et al., 2006 Nishimura, 2006 #1145; Yuce et al., 2005). However, whether Ect2 catalytic activity is required to activate RhoA during cytokinesis in mammalian cells remains unclear.
Cdk1/Cyclin B and Aurora kinases have emerged as major regulators of mitotic progression through their ability to phosphorylate numerous critical mitotic modulators (Ferrari, 2006; Nigg, 1995; Nigg, 2001). Several lines of evidence have implicated mitotic kinases in the control of RhoA activity during cell division. Cdk1/Cyclin B modulates the activity of Ect2 during cytokinesis (Hara et al., 2006; Niiya et al., 2006), while phosphorylation by Aurora B is required to confer RhoA-specific GAP-activity to MgcRacGAP in the same context (Minoshima et al., 2003). In the present work we show that GEF-H1 is phosphorylated during mitosis by both Aurora A/B and Cdk1/Cyclin B, and that phosphorylation of GEF-H1 during M-phase coincides with reduced catalytic activity. GEF-H1 dephosphorylation occurs at the onset of cytokinesis, and is accompanied by GEF-H1-dependent GTP-loading on RhoA. Perturbation of endogenous GEF-H1 by both gene knock down and overexpression of inhibitory mutants caused impairment of HeLa cell cytokinesis. Examination of RhoA activation dynamics in HeLa cells stably expressing a FRET-based RhoA biosensor indicate an important regulatory role for GEF-H1 in the activation of RhoA during cytokinesis.
RESULTS
GEF-H1 subcellular distribution correlates with CF formation
Using affinity purified antibodies specific for GEF-H1 (Zenke et al., 2004), we observed that GEF-H1 accumulates at the mitotic spindle during cell division (Fig. S1, A). Detailed analysis of the subcellular distribution of GEF-H1 during mitosis in asynchronous HeLa cells revealed that a substantial fraction of GEF-H1 localizes to the spindle apparatus throughout all stages of mitosis (Fig. 1, A). During telophase, a subset of GEF-H1 immunoreactivity was enriched in a narrow equatorial band comprised of aligned short segments at sites of CF formation, most likely representing overlapping plus ends of equatorial astral MTs (Fig. 1, A; telophase I). With the furrow advancing inwards, GEF-H1 segments became more condensed into a ring-like structure encompassing the midzone (Fig. 1, A; telophase II). Interestingly, our GEF-H1 antibodies did not stain the central spindle, suggesting that GEF-H1 distribution is restricted to astral and kinetochore MTs (see also Fig. 1, B). Further examination using laser scanning confocal microscopy showed that GEF-H1 immunoreactivity is enriched at the tips of spindle MTs (Fig. 1, B; tubulin panels and Fig. S1, C,D). We noticed that MT tip-localized GEF-H1 was more efficiently detected by a phosphorylation-specific GEF-H1 antibody (Fig. 1, B; pGEF-H1 panels) than by a non-phospho GEF-H1 antibody (Fig. S1, C), possibly due to differences in epitope accessibility upon phosphorylation (see below). To visualize GEF-H1 distribution relative to centromeres, we stained metaphase and telophase cells with human CREST serum, which detects the centromere protein CENP-A (Fig. 1, B; CREST panels). In metaphase cells GEF-H1-positive dots at MTs aligned juxtaposed to the centromeres. Later in telophase, when the chromosomes moved pole wards, GEF-H1 dots became compacted into a single band in the midzone region. Double labeling experiments revealed that GEF-H1 and actin form a double-ring structure (Fig. 1, C; merge channel) composed of an internal GEF-H1 immunoreactive circle (Fig. 1, C; green channel) associated with the tubulin cytoskeleton, and aligned with the inner face of the contractile acto-myosin ring (Fig. 1, C; red channel). Together, these results indicate that GEF-H1 localizes to structures critical for CF formation and ingression, i.e. cortical MTs and the midbody region.
Fig. 1. Subcellular localization of endogenous GEF-H1 during mitosis.
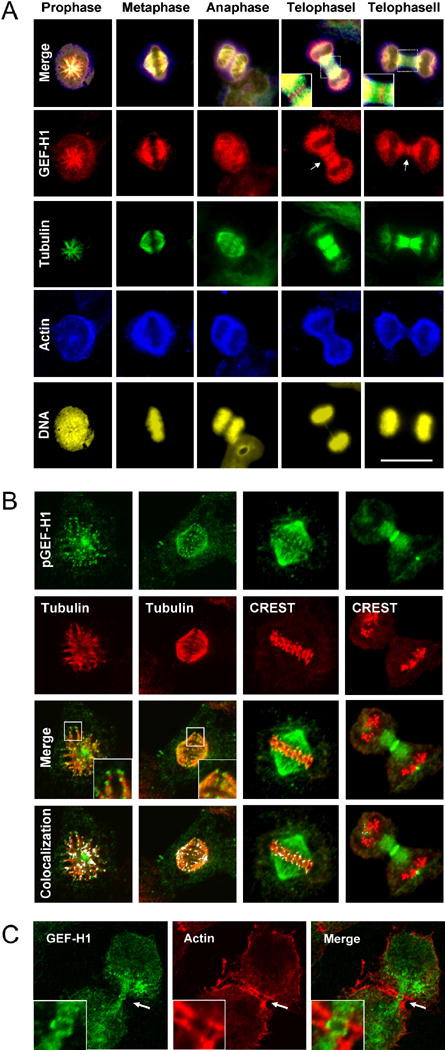
(A)GEF-H1 is associated with the spindle apparatus throughout mitosis. Asynchronous HeLa cells were fixed with methanol/acetone and quadruple-stained for endogenous GEF-H1 (red channel), tubulin (green channel), actin (blue channel) and DNA (yellow). Scale bar represents 20 μm.
(B) GEF-H1 localizes to the tips of cortical MTs. Asynchronous HeLa cells were extracted for soluble tubulin, fixed as above and stained using anti-pGEF-H1 (green), anti-tubulin (red) antibodies or CREST serum. Boxed regions are shown in higher magnification. Confocal micrographs (single slices of 0.54 μm thickness) were analyzed for colocalized data points (displayed as white overlays in the colocalization panel).
(C) During cytokinesis, GEF-H1 forms an equatorial ring encompassed by contractile actin structures. Confocal microscopy of mitotic HeLa cells stained with anti-GEF-H1 (green) and anti-actin (red) antibodies. Indicated regions (white arrows) are shown in higher magnification in the inserts.
Perturbation of GEF-H1 function causes defects in cytokinesis
If GEF-H1 is an important regulator of contractile ring formation and cytokinesis, then perturbation of endogenous GEF-H1 function should increase the extent of multinucleation as an indicator of failed cytokinesis. Indeed, siRNA-mediated depletion of endogenous GEF-H1 significantly increased the formation of multinucleated cells (18.9 % ± 6.4%) compared with control (CTRL) siRNA-transfected cells (8.4 % ± 3.7 %) (Fig. 2, A). As a positive control for multinucleation we also depleted cells of Ect2: similar to previously published studies (Kim et al., 2005), the number of nuclei was markedly increased in Ect2-siRNA treated cells (55% ± 5.3%, see also Supp. Material).
Fig. 2. Multinucleation caused by GEF-H1 perturbation.
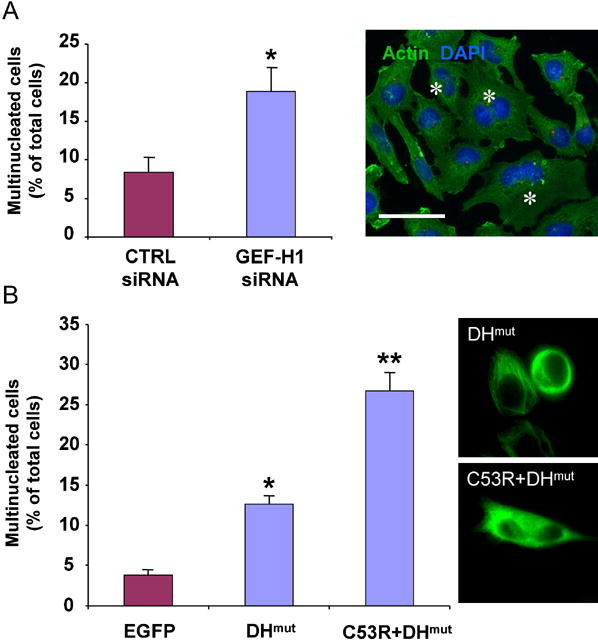
(A) Multinucleated cells were found at 48 h after transfection with GEF-H1 siRNA. Values shown are the means from four independent experiments in which over 1000 cells were counted per experiment; error bars indicate SEM. For statistical analysis, all data were evaluated by two-tailed Student’s t-test. Values significantly different from controls (p≤ 0.01) are marked with an asterisk. Cells harboring more than 1 nucleus are indicated by an asterisk in the micrograph. Scale bar represents 20 μm.
(B) Overexpression of GEF-H1 inhibitory mutants induced multinucleation. The number of the multinucleated cells as a percentage of the total cell population expressing the EGFP-tagged constructs was quantified in HeLa cells 48 h after transfection. A minimum of 100 cells from each of three independent experiments was scored for each construct. Values significantly different from EGFP-expressing cells are marked with one (p≤0.01) or two asterisks (p ≤0.001). Micrographs on the right illustrate expression patterns of the different constructs.
To corroborate the siRNA depletion data, we analyzed the consequences of overexpression of GEF-H1 inhibitory mutants on cell division. Fig. 2B shows that overexpression of a non-MT-associated, enzymatically inactive GEF-H1 mutant (EGFP-GEF-H1C53R+DHmut) had a greater effect on multinucleation than a MT-associated d/n EGFP-GEF-H1DHmut construct (26.9% ± 4.9 % vs 12.6 % ± 2.4 %, compared to 3.8 % ± 1.1% for EGFP alone-expressing cells). These findings are consistent with data showing that d/n GEF-H1 mutants defective in MT binding block endogenous RhoA activation more efficiently than do MT-bound forms, supporting the idea that endogenous GEF-H1 is most active when released from the tubulin cytoskeleton (Krendel et al., 2002; Zenke et al., 2004). Thus, both multinucleation caused by ablation of endogenous GEF-H1 and via overexpression of GEF-H1 inhibitory mutants suggest that perturbation of GEF-H1 impairs cytokinesis.
GEF-H1 depletion induces membrane instability and ectopic furrowing during cytokinesis
Prolonged synchronization with nocadazole (noc) was frequently associated with cell death in GEF-H1 siRNA-treated cells (Fig. S2, B). Under milder conditions in which the apoptotic phenotype was attenuated (see Methods), we still observed that more cells displayed cortical aberrations during early mitotic stages when exposed to GEF-H1 siRNA (Fig. 3, b), although the majority of cells progressed to later stages of mitosis, and chromosome alignment, segregation and furrow initiation appeared normal in both GEF-H1- and control-depleted cells (Fig. S8 and Supp. Movies 1-3). As the furrow started to constrict, about 2/3 of the progressing GEF-H1-depleted cells (i.e. 40.8 % of all cells) exhibited membrane aberrations, including excessive cortical blebbing and the formation of unstable ectopic furrows (Fig. 3, c-e; Supp. Movie 2). Interestingly, uncoordinated furrowing and plasma membrane instabilities did not necessarily culminate in cytokinesis failure. Instead, ectopic furrows regressed or joined with a robust central furrow, while other uncoordinated membrane activities along the cortex abrogated upon daughter cell separation (24.8 % ± 4.7%; instability, c panel).
Fig. 3. Downregulation of GEF-H1 causes mitotic aberrations.
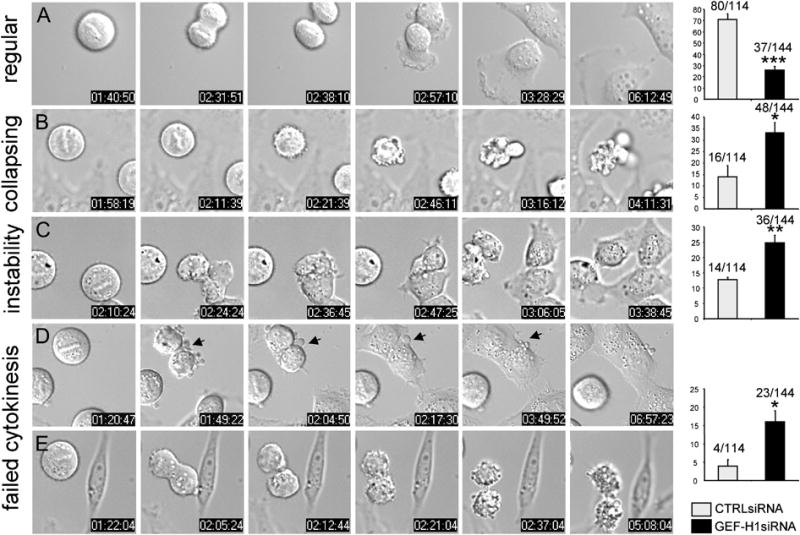
Distribution of mitotic phenotypes in GEF-H1 (GEF-H1 siRNA) or control siRNA (CTRL siRNA)-transfected cells. Classification of phenotypes: a) regular cytokinesis; b) collapsing: cortical hyperactivity during metaphase, finally collapsing; c) instability: membranous aberrations around the CF w/o impairment of cytokinesis; d) failed cytokinesis: membranous aberrations during cytokinesis that are not compensated and gave rise to cytokinesis failure. Values are given as percentage of total cells with error bars indicating ± SEM. Actual numbers are indicated. In four independent experiments, a total of 144 GEF-H1-depleted and 114 control-depleted cells were scored (*≤ 0.05; **≤ 0.005; *** ≤0.0005).
In about 16 % ± 6 % of GEF-H1-depleted cells, abnormal membrane activities could not be compensated and caused impairment of daughter cell abscission and separation, leading to the formation of multinucleated cells (20 out of 144 cells; cytokinesis failure, d panels; supplementary Movie 3) or total collapse of the daughter cells (3 out of 144 cells; e panels). In contrast, the vast majority of cells expressing control siRNA did not exhibit membrane instabilities, and passed normally through cell division (regular, a panel). Although some blebbing occurred in control cells, it was much less pronounced than observed upon GEF-H1 ablation, and was usually confined to the cell poles. Down-regulation of Ect2 induced severe cytokinetic defects, where 38.4 % ± 1 % (n=54 cells) of the cells analyzed were not able to perform cleavage (not shown). Multinucleation in this case, however, was not associated with cortical instabilities but solely with defects in CF formation and ingression (Fig. S3). Overall, these results indicate distinct roles for Ect2 and GEF-H1 in cytokinesis and suggest that Ect2 is essential for early events occurring during CF induction whereas GEF-H1 activity is important to coordinate cortical activities during CF ingression.
GEF-H1 is phosphorylated early in mitosis by Aurora A kinase
Using phospho-specific antibodies raised against the pSer885 epitope, we observed high levels of endogenous phospho-GEF-H1 in lysates from noc-arrested mitotic HeLa cells (Fig. 4, A; upper panel), with concomitant increase in 14-3-3 binding (Fig. S5, B). In contrast, GEF-H1 phosphorylation and 14-3-3 binding was nearly undetectable in asynchronous HeLa cell lysates. When synchronized HeLa cells were allowed to progress through mitosis, a dramatic decrease in the amount of phosphorylated GEF-H1 was observed at 90 min after release, when the majority of cells entered telophase and underwent cytokinesis (Gohla et al., 2005) (Fig 4, A; lower panel). Dephosphorylation of GEF-H1 coincided with proteolysis of cyclin B1, which starts at meta-/anaphase and is required for M phase exit (Clute and Pines, 1999). Phosphorylation of GEF-H1 at Ser885 was no longer detectable 180 min after release from the noc block, when most of the cells were in G1 phase. These results demonstrate that GEF-H1 is phosphorylated early during mitosis and undergoes specific dephosphorylation prior to the final stages of cell division.
Fig. 4. Association of GEF-H1 with Aurora A.
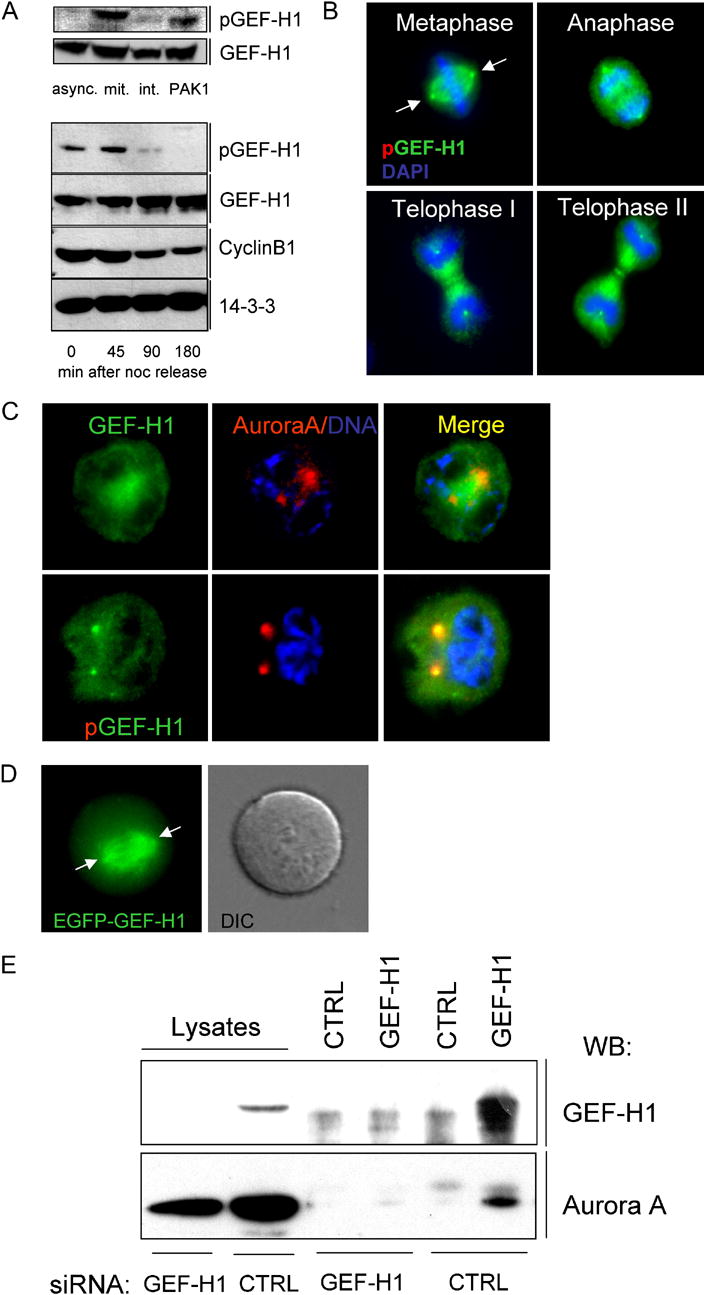
(A) Upper panel: GEF-H1 is phosphorylated at the onset of mitosis. HeLa cells were incubated with 100ng/ml noc for 16 h. Rounded mitotic cells, harvested by mechanical knock-off (mit.) and remaining adherent interphase cells (int.) were lysed and analyzed by immunoblotting with anti-pGEF-H1 or anti-GEF-H1 antibodies. Lysates from asynchronous (async.) or Pak1-transfected (PAK1; pSer885 control) HeLa cells were used as controls. Lower panel: Dephosphorylation of GEF-H1 during telophase/cytokinesis. HeLa cells were synchronized in mitosis by thymidine and noc, collected by knock-off and replated on poly-L-lysine-coated culture dishes. Lysates were analyzed by anti-pGEF-H1 or anti-GEF-H1 immunoblotting at the indicated time points. The degradation of Cyclin B was used as an additional marker for cell synchrony. 14-3-3 zeta controls were included to confirm comparable protein loading.
(B) Immunolocalization of Ser885-phosphorylated GEF-H1 in mitotic cells. Asynchronous HeLa cells were fixed and stained for GEF-H1 (red channel), tubulin (green channel) and DNA (DAPI) as described in Fig.1. Scale bar represents 20 μm.
(C) pGEF-H1 colocalizes with Aurora A in centrosomes. HeLa cells in prophase were co-stained for Aurora A (red channel) and either GEF-H1 or pGEF-H1 (green channels). DNA was detected by DAPI staining. Yellow color (merged image) indicates co-localization of pGEF-H1 with Aurora A in centrosomes.
(D) Centrosome localization of EGFP-GEF-H1. HeLa cells were microinjected with plasmid DNA encoding EGFP-GEF-H1 and analyzed by time-lapse video fluorescence microscopy. Arrows indicate the position of the centrosomes.
(E) GEF-H1 interacts with Aurora A in vivo. Immunoprecipitation of GEF-H1-depleted (GEF-H1 siRNA) or control-depleted (CTRL siRNA) mitotic lysates with either anti-GEF-H1 antibodies or control IgG after DSP crosslinking. The resulting immunoprecipitates were isolated, separated on SDS-PAGE, and immunoblotted with anti-Aurora A antibody. Lysate lanes shown represent ten percent of input lysate.
Examination of the subcellular distribution of Ser885-phosphorylated GEF-H1 in mitotic cells showed that immunoreactivity specifically accumulated at the polar regions of the spindle apparatus (Fig. 4, B and Fig. S4, A). This suggested that GEF-H1 might be phosphorylated by kinases associated with centrosomal structures such as Aurora A (Andrews et al., 2003). As shown in Fig. 4C, phospho-GEF-H1indeed accumulated in Aurora A positive double spots which correspond to the duplicated centrosomes. In contrast, these structures were poorly stained using non-phospho-GEF-H1 antibody, perhaps due to steric hindrance of antibody access. We therefore verified the presence of GEF-H1 in centrosomes after EGFP-GEF-H1 plasmid microinjection into thymidinesynchronized HeLa cells (Fig. 4, D). Since phospho-GEF-H1 antibodies and EGFP-GEF-H1 minimally stained centrosomes of interphase cells (not shown), this suggests that GEF-H1 is recruited to centrosomes during early mitotic events, perhaps coincident with phosphorylation by Aurora A.
We next tested whether endogenous Aurora A kinase and GEF-H1 form a complex in live cells. Immunoprecipitation of GEF-H1 resulted in co-precipitation of Aurora A kinase (Fig. 4, E), while no co-precipitation was observed from GEF-H1-depleted cell lysates or with control IgG. In addition, Ser885 phosphorylation of GEF-H1 in vivo was abolished by blocking the activities of the centrosomal kinases Aurora A and the Cdk1/Cyclin B complex but not by PKA or GSK3 β (Fig. 5) indicating that GEF-H1 phosphorylation during mitosis involves the mitotic kinases Aurora A/B and Cdk1/Cyclin B. We also found that inhibition of Aurora B, a paralogue of Aurora A that localizes to centromeres, abolished GEF-H1 phosphorylation later in mitosis (see Fig. S4, D). In vitro kinase assays showed that Aurora A directly phosphorylates GEF-H1 at Ser885 while Cdk1/Cyclin B (or Aurora B) surprisingly modified Ser959 instead (Fig S4, C,D). Together, these results indicate specific interactions between Aurora A and GEF-H1 during cell division and identify the C-terminus as the major phosphoacceptor region on GEF-H1.
Fig. 5. Phosphorylation of GEF-H1 by mitotic kinases Aurora A and Cdk1/Cyclin B.
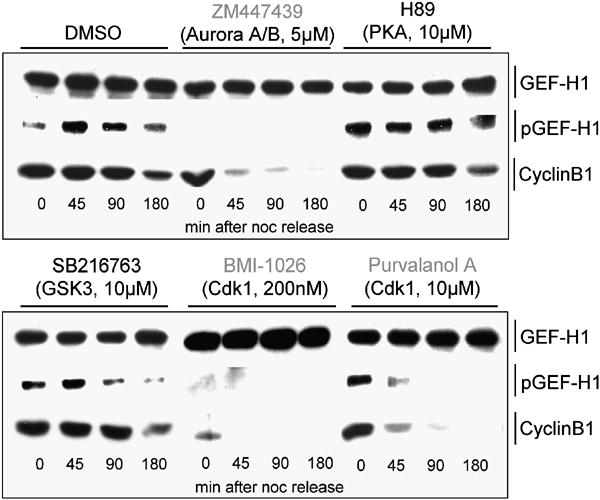
Inhibition of Aurora kinases and Cdk1/Cyclin B abolish GEF-H1 phosphorylation during mitosis. Before adherence, harvested synchronized HeLa cells were inhibitor treated for 15 min during release into fresh culture medium (w/o nocodazole) supplemented with the same inhibitors (at denoted concentrations and specificities). Inhibitors used here are specific for serine-threonine kinases that are either predicted by the Scansite algorithm (scansite.mit.edu) to phosphorylate GEF-H1 (PKA, GSK3β; data not shown) or are required for the control and timing of mitotic progression (Aurora A/B, Cdk1/Cyclin B) (Ferrari, 2006; Nigg, 2001). At the indicated times (after adherence), cells were lysed and analyzed by anti-pGEF-H1 and anti-GEF-H1 immunoblotting.
GEF-H1 is activated by dephosphorylation to promote GTP loading on RhoA
We examined whether phosphorylation could regulate the enzymatic activity of GEF-H1 in mitotic cells. Using calyculin A, a cell permeable inhibitor of protein phosphatases, we increased the levels of phosphorylated EGFP-GEF-H1 in transfected cells (Fig. 6, A). EGFP-GEF-H1 immunoprecipitates from these cells were then treated with calf intestinal phosphatase (CIP) ± phosphatase inhibitors, and used to catalyze the exchange of GDP for [35S]GTPγS on RhoA. While immunoprecipitates with high amounts of phospho-GEF-H1 did not catalyze nucleotide exchange on RhoA (Fig. 6, B; CIP+Inh), the dephosphorylation of GEF-H1 coincided with a dramatic increase in nucleotide exchange activity (Fig. 6, B; CIP), similar to the maximum exchange obtainable.
Fig. 6. Dephosphorylation stimulates GEF-H1 enzymatic activity.
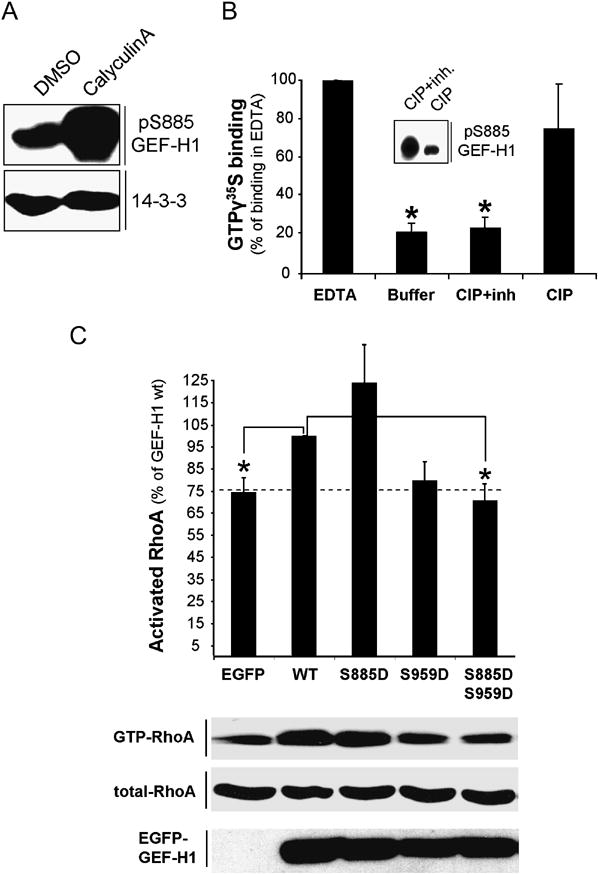
(A) Calyculin A induces phosphorylation of GEF-H1 in vivo. HeLa cells were treated with 100nM Calyculin A or DMSO for 45 min. The amount of phospho-GEF-H1 in the different lysates was analyzed by immunoblotting with anti-pGEF-H1 antibodies.
(B) Increase of GEF-H1 guanine nucleotide exchange activity in vitro after dephosphorylation. EGFP-GEF-H1 immunoprecipitates from Calyculin A-stimulated HeLa cells were treated with calf intestine alkaline phosphatase (CIP) in the presence (CIP+inh) or absence (CIP) of phosphatase inhibitors and subjected to in vitro GEF-assays as in Methods. The level of nucleotide binding in the presence of EDTA was set to 100%. pGEF-H1 levels in the immunoprecipitates after CIP treatment were analyzed by anti-pGEF-H1 immunoblotting (insert). Each value represents the mean (± SEM) of three independent experiments. Asterisk indicates values significantly different from EDTA controls (p≤ 0.0001).
(C) Effects of GEF-H1 derivatives on accumulation of GTP-bound RhoA during mitosis. HeLa cells were transfected with the indicated EGFP-tagged expression vectors coding for different GEF-H1 mutants or EGFP alone for 24 h. Following a 15 h synchronization period with 100ng/ml noc, cells were released into fresh medium for 45min prior to lysis. Subsequently, the GTP-bound fraction of RhoA was extracted from the lysates by GST-RBD. The amount of RhoA bound to GST-RBD (GTP-RhoA) and the level of RhoA expression (total RhoA) in whole cell lysates was determined by immunoblotting using anti-RhoA antibodies. RhoA activation was expressed as percent activation relative to the EGFP-GEF-H1 wt-transfected control (set to 100%). Each datapoint represents the mean (± SEM) of at least three independent experiments. Asterisk indicates values significantly different from EGFP-GEF-H1wt controls (p ≤0.01). The dashed line marks the level of GTP-RhoA in EGFP-expressing cells. All values were normalized for expression efficiency of the individual constructs (anti-EGFP immunoblotting) and total RhoA levels by densitometric analysis as in (A). PonceauS staining was used to determine the amount of GST-RBD in the reaction.
To determine whether phosphorylation of GEF-H1 at Ser885and/or Ser959 regulated GEF-H1 activity in the context of mitosis, we expressed GEF-H1 phosphorylation mutants in metaphase-synchronized HeLa cells and measured GTP-bound RhoA. The increase in GTP-RhoA observed in mitotic lysates expressing wildtype GEF-H1 (compare with EGFP control) was used as a reference (Fig.6,C = 100%). Overexpression of the phosphomimetic S885D single mutant significantly increased GTP-RhoA as compared to cells expressing EGFP alone, while the S959D single mutant also increased GTP-RhoA, but was somewhat less active (Fig. 6, C). Phosphorylation-deficient GEF-H1 S885A and S959A mutants also induced RhoA activation similar to WT GEF-H1 (Fig.S5,C). In contrast, overexpression of the phosphomimetic double mutant SS885/959DD clearly did not significantly increase GTP-RhoA over control levels. These data suggest that both Aurora A and Cdk1/Cyclin B-mediated phosphorylation are necessary for full negative regulation of GEF-H1 activity during early stages of mitosis.
The phosphorylation of Ser885 in GEF-H1 is known to induce binding of 14-3-3 protein to GEF-H1 (Zenke et al., 2004), and we observed that 14-3-3 was associated with GEF-H1 in mitotic cell lysates (Fig.S5,B). Thus, part of the inhibitory effect of Aurora A-induced Ser885 phosphorylation in vivo may result from the stimulation of 14-3-3 binding to GEF-H1.
GEF-H1 depletion alters RhoA activation dynamics during cytokinesis in live cells
GEF-H1 is not phosphorylated and thereby most probably exists as an active form during telophase, a time-point when mitotic cells form the CF in a RhoA-dependent manner. To test whether GEF-H1 is involved in the activation of RhoA in telophase, we depleted HeLa cells of endogenous GEF-H1 and Ect2 using siRNA. Similar to cells in interphase (Fig. S6, B), GEF-H1 expression in mitotic cells was suppressed by approximately 79% (± 15, 8 %), while Ect2 siRNA-induced knock down was nearly complete, with 4% (± 3,4 %) protein expression remaining (Fig. 7, A; graph). Determination of GTP-RhoA accumulation in synchronized, siRNA-treated cells showed that control siRNA-treated cells exhibited increased GTP-bound RhoA during telophase (Fig. 7, A + B; CTRL siRNA). Depletion of Ect2 did not alter GTP-RhoA formation during telophase (Ect2 siRNA), which was surprising considering the known requirement for Ect2 to localize RhoA to the equatorial cortex prior to CF ingression. In contrast, GEF-H1 depletion led to a strong reduction in the amount of activated RhoA during telophase, while RhoA activation remained largely unaffected during pro- and metaphase (GEF-H1 siRNA). However, for both GEF-H1- and Ect2-ablated cells, high levels of RhoA activation were detected at 180 min, further suggesting hampered exit from mitosis. Taken together, these data indicate that GEF-H1 is involved in the activation of RhoA during cytokinesis.
Fig. 7. GEF-H1 modulates RhoA activation in mitotic cells.
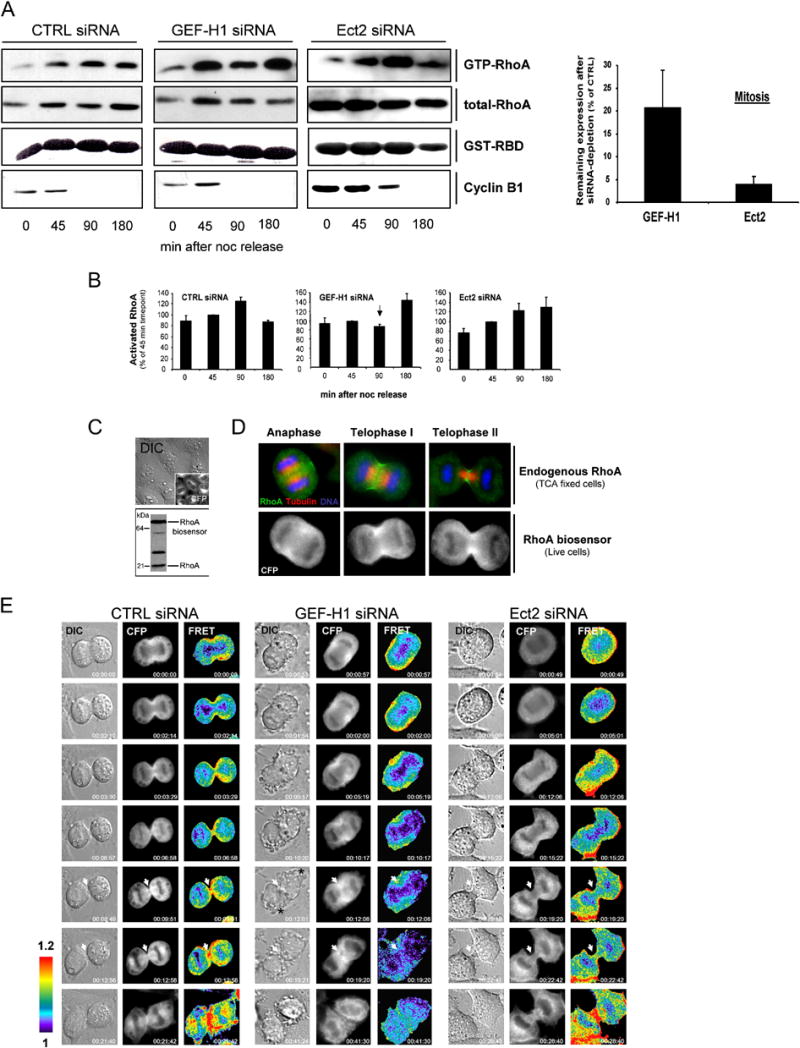
(A + B) Cell cycle-associated changes in the level of GTP-RhoA after GEF-H1 depletion. Synchronized HeLa cells were subjected to GST-RBD pull down assays to detect GTP-RhoA during different stages of mitosis in lysates depleted of GEF-H1, Ect2 or control-depleted lysates. A representative experiment is shown in (A). At least four independent assays for each condition were used to quantify the activation profile of RhoA as inFig. 6D (B). The amount of GTP-RhoA at 45 min after noc release was set to 100%. All values were normalized for the total amount of RhoA. Error bars indicate ± SEM. (A; graph) The remaining expression of GEF-H1 and Ect2 in the mitotic lysates was quantitated densitometrically after immunoblotting and normalized to CTRL-siRNA treated cells. Values represent the mean of four independent determinations ±SEM. Note that Ect2 ablation was more efficient than depletion of GEF-H1.
(C) HeLa cells stably transfected with a FRET-based RhoA biosensor were analyzed for expression of the latter by live microscopy (upper panels; CFP channel) and immunoblotting of respective lysates using anti-RhoA antibodies (lower panel). Biosensor expression was ~2-fold increased over endogenous RhoA levels as determined by densitometric analysis.
(D) Localization of the RhoA biosensor in live cells during cytokinesis. The subcellular distribution of the RhoA biosensor in live cells (CFP panel) was compared to that of endogenous RhoA in TCA-fixed cells during different stages of mitosis. Fixed cells were stained with anti-RhoA (green) and anti-tubulin (red) antibodies and DAPI (blue) for DNA.
(E) RhoA activation patterns during cytokinesis. HeLa cells stably expressing the RhoA biosensor were transfected with the indicated siRNAs for 30 h before progression through cytokinesis was analyzed by DIC microscopy and FRET/CFP ratio imaging to represent FRET efficiency (FRET). The localization of the biosensor is shown in the CFP channel. FRET pictures denote activation patterns of the biosensor. All images are processed and scaled identically so that regions of intense RhoA activity are shown in red. The elapsed time is denoted at the bottom right corners of each picture. A representative image of at least 6 similar images acquired for each condition is shown. White arrows indicate equatorial regions with high RhoA activation in CTRL-depleted cells and reduced RhoA activation in GEF-H1 and Ect2 siRNA-treated cells during late stages of cytokinesis. Asterisks indicate regions of aberrant membrane activities in cells treated with GEF-H1 siRNA.
To test the hypothesis that GEF-H1-induced mitotic aberrations involve perturbed RhoA activation, we generated cells stably expressing a single chain FRET-based biosensor that responds to GTP-loading on RhoA (Pertz et al., 2006) (Fig. 7, C). Analogous to endogenous RhoA in TCA-fixed cells (Yonemura et al., 2004), CFP fluorescence of the biosensor concentrates at the equatorial cortex at the onset of furrowing (Fig. 7, D; CFP panel, anaphase) and during membrane ingression (telophase I + II), indicating that it likely reflects regulation of endogenous RhoA. We examined spatio-temporal changes in the activity of HeLa cell RhoA during cytokinesis (Figs. 7, E and S10). Images were taken from biosensor-expressing cells that entered anaphase, as evidenced by initiation of chromosome separation. The emission ratio FRET/CFP was used to represent RhoA activation, as described (Pertz et al., 2006). In control cells, activated RhoA was found to accumulate at the equatorial cortex during anaphase (Fig. 7, E; CTRL siRNA, FRET panel, time point 0). As furrowing progressed, RhoA activity increased in the CF, reaching a peak level in the late phase of cytokinesis when the midbody matrix was formed and daughter cells started to separate (13 out of 15 cells analyzed) (here, time point 21.42). In cells treated with GEF-H1 siRNA, early cortical activation of RhoA was not affected (Fig. 7, E; GEF-H1 siRNA, FRET panel, time points 0-10.17). However, as constriction proceeded, RhoA activation in the forming CF decreased. Of particular interest, in 62% of the cells analyzed (8 out of 13), localized RhoA activation was strongly diminished at late stages of cytokinesis shortly before midbody formation, although RhoA protein was still concentrated at sites of cleavage as in control cells (CFP + FRET panels, time points 12.06-19.20). The loss of localized RhoA activation in the CF was correlated with dramatic cortical blebbing and aberrant membrane activities, as described (Fig. 3). (See also Fig.S10).
In contrast, in Ect2-depleted cells CFP fluorescence of the RhoA biosensor was no longer accumulated at equatorial sites of CF formation (Ect2 siRNA, CFP panels). Instead, the biosensor was randomly distributed along the cell periphery, correlated with strong but unlocalized cortical activation of RhoA throughout all stages of mitosis analyzed (Ect2 siRNA, FRET panels) (5 out of 6 cells analyzed). RhoA activation in the CF was only marginal (FRET panel, time points 19.20-22.24). Concomitantly, CF formation and constriction was either markedly slowed (Fig. 7, E; Ect2 siRNA, DIC panels) or incomplete and often resulted in regression of the CF and binucleated cells (See Fig. S10; Ect2 siRNA, DIC panels. See also Fig. S3, b). In combination with the pull-down activity assay data presented, these results demonstrate distinct roles for GEF-H1 and Ect2 in RhoA activation during cytokinesis. While Ect2 appears to initially direct the localization of RhoA protein to equatorial sites prior to CF formation, GEF-H1 participates in GTP-loading of RhoA after furrowing has been induced.
DISCUSSION
GEF-H1 localization may link cortical activation of RhoA to MTs
MT-derived signals are important for RhoA translocation to and activation at the equatorial cortex to allow positioned assembly and contraction of the acto-myosin CF (Bement et al., 2005). We have shown in this study that GEF-H1 meets important requirements for a regulator of RhoA activation at the cell cortex. First, GEF-H1 localizes to the tips of cortical MTs of the mitotic spindle, thereby placing its catalytic activity in close proximity to the cell cortex (Fig. 1). Second, as the mitotic spindle becomes compressed by the ingressing cortex, short GEF-H1 immunoreactive segments in the equatorial plane adopt a condensed circular structure that is aligned with the inner face of the acto-myosin contractile ring. A similar structure has been described in Drosophila that is made up of GAP50C-PAV, linking the MT network with the acto-myosin contractile ring through interaction with the Ect2 homolog Pbl (Somers and Saint, 2003). GEF-H1 could therefore represent another structural element to link MTs with the cell cortex in mammalian cells.
GEF-H1 is a target of Aurora A and Cdk1/Cyclin B kinases during early mitosis
In the present work we established the mitotic kinases Aurora A/B and Cdk1/Cyclin B as important regulators of GEF-H1 activity during cell division. We identified Ser885 of GEF-H1 as the principle phospho-acceptor site for Aurora A (Fig. S4, B) and Ser959 as the primary Cdk1/Cyclin B GEF-H1 phosphorylation site. It is possible that upon mitotic entry phosphorylation by Cdk1/Cyclin B induces a conformational change in the GEF-H1 C-terminal regulatory domain allowing Aurora A to access and phosphorylate Ser885, which would be otherwise masked by structural constraints. Similar regulation has been described for Ect2 in which pre-phosphorylation by Cdk1/Cyclin B stimulated the association of Ect2 with Polo-like kinase 1, likely by releasing Ect2 from intramolecular auto-inhibitory interactions (Hara et al., 2006).
Reducing GEF-H1 phosphorylation by phosphatase treatment resulted in increased guanine nucleotide exchange activity of wt GEF-H1 (Fig. 6, B). Consistently, phosphomimetic Ser885D/Ser959D double mutants of GEF-H1 significantly exhibited less RhoA activation in mitotic lysates (Fig. 6, C). We conclude that phosphorylation of GEF-H1 during cell division negatively regulates its enzymatic activity. Our results do not address whether phosphorylation might inhibit GEF-H1 activity by affecting MT binding. However, the reduction in exchange activity may involve 14-3-3 protein binding induced by Ser885 phosphorylation (Zenke et al., 2004), as we have observed association of 14-3-3 with GEF-H1 in mitotic cell immunoprecipitates (Fig.S5, B). Since MT depolymerization rapidly activates GEF-H1 to enhance cell contractility (Krendel et al., 2002), GEF-H1-induced contractions would likely interfere with cortical stabilization required for mitotic cell rounding. Given that the interphase-mitosis transition is accompanied by a rapid breakdown of the cytoplasmic MT network (Zhai et al., 1996), immediate inactivation of GEF-H1 (by phosphorylation) may be necessary for mitotic rounding.
GEF-H1 and Ect2 mediate different aspects of RhoA activation during cytokinesis
Ect2 is essential for RhoA localization at the equatorial cortex, a prerequisite for CF formation and ingression (Nishimura and Yonemura, 2006; Yuce et al., 2005). Inhibition of Ect2 often relies on overexpression of the inhibitory N-terminus (Saito et al., 2004), and mitotic cells expressing Ect2-N show reduced RhoA activation during cytokinesis (Kimura et al., 2000). It is not clear however, if the reduced GTP-loading on RhoA is due to interference with Ect2 exchange activity, as opposed to the activation and/or localization of other GEFs (e.g. GEF-H1) that might be triggered by full-length Ect2. Accordingly, N-terminal mutants of Ect2 that retained full GEF activity in vitro failed to rescue the cytokinesis defect in cells depleted of endogenous Ect2 , suggesting that recruitment of additional regulators apart from Ect2 exchange activity itself is crucial for RhoA activation. We observed that siRNA-mediated depletion of Ect2, while quite effective (see Fig.S6), did not interfere with bulk RhoA activation during telophase (Fig. 7, A and B), also indicating that Ect2 does not directly activate RhoA. In clear contrast, while inhibition of GEF-H1 did not affect recruitment of RhoA and other regulators of cytokinesis to cortical regions (Fig. 7, E and Fig. S7), ablation of GEF-H1 caused a strong decrease in overall RhoA activation during telophase (Fig. 7, A and E). Furthermore, we observe that the activation profile of GEF-H1 during mitosis is inversely correlated to that of Ect2. While GEF-H1 is inhibited during early stages of mitosis and is activated by dephosphorylation during cytokinesis (this study), phosphorylation increases the catalytic activity of Ect2 in vitro (Tatsumoto et al., 1999) and in M-phase cells (Hara et al., 2006;Niiya et al., 2006). This indicates distinct roles for GEF-H1 and Ect2 during cell division.
Inhibition of GEF-H1 causes late defects in cytokinesis that correlate with decreased RhoA activation at the CF
Using time-lapse video microscopy, we identified aberrations during CF formation in cells treated with GEF-H1-specific RNAi duplexes. A majority of the GEF-H1-depleted cells demonstrated loss of membrane stability in the CF during late stages of mitosis after normal furrow induction (Fig. 3; panel c and d). These perturbations did not necessarily coincide with cytokinesis failure, as membranous aberrations often subsided, and a compact midbody bridge formed (Fig. 3, panel c). The membrane phenotype observed here is reminiscent of the depletion of either the actin bundling protein, anillin, or the RhoA effector, citron kinase (D’Avino et al., 2004; Echard et al., 2004), whose down-regulation led to reduced cortical stability of the cleavage plane and, later, the intercellular midbody bridge (Echard et al., 2004). This resulted in the loss of membrane attachment to this structure, causing a blebbing phenotype. As GEF-H1 might be important for both linking cortical components (e.g. anillin-bundled actin filaments of the contractile ring) to the mitotic spindle, as well as regulating the activity of RhoA effector proteins in the context of contractile ring formation and ingression, our findings are consistent with GEF-H1 RNAi-induced aberrations of the CF.
Using a FRET-based biosensor, we detected high RhoA activity at sites of constriction and in the CF during all stages of cytokinesis (Fig. 7, E and S10). In contrast, Yoshizaki et al. were not able to detect high levels of RhoA activation in the CF of HeLa cells using their FRET-based Raichu-RhoA probes (Yoshizaki et al., 2003). These conflicting observations may be explained by the inherent insensitivity of Raichu probes to RhoGDI regulation, as well their constitutive localization to membranes (Nakamura et al., 2005; Yoshizaki et al., 2003; Yoshizaki et al., 2004). The localization of the RhoA biosensor (Pertz et al., 2006) used in this study coincides with that of endogenous RhoA, perhaps due to its normal binding to RhoGDI, which controls reversible RhoA membrane targeting (DerMardirossian and Bokoch, 2005). In fact, RhoGDI activity appears to be important for RhoA function during cytokinesis, since cells lacking RhoGDI are multinucleated (Rivero et al., 2002). We conclude that the patterns of RhoA activity we detect reflect the endogenous RhoA activation profile and, hence, increase in the CF during cytokinesis.
GEF-H1 ablation in HeLa cells causes a decrease in RhoA activation during late stages of cytokinesis as demonstrated in this paper by FRET- and RBD-affinity-based assays (Fig. 7, A and E). Early cortical RhoA activation was, however, not perturbed by reducing GEF-H1 protein levels. Therefore, GEF-H1 may act as a modulator of late mitosis to ensure that midbody formation and daughter cell abscission occur correctly. From this perspective, GEF-H1 might be involved in sustaining high RhoA activity in the CF to ensure contractility and cortical stiffness (O’Connell et al., 1999). By providing cortical tension in the CF, this would also guarantee that polarized daughter cell spreading is initiated from the poles and migration occurs in opposite directions. Loss of tension in the CF would consequently coincide with plasma membrane instabilities, especially as new membrane material might still be added to the CF to adjust the normally high furrow cortical tensions. Reduced RhoA activation in the CF caused by GEF-H1 depletion is therefore consistent with the observed membrane perturbations during cell cleavage. In contrast to our results, Bakal et al (Bakal et al., 2005) observed that Lfc, the mouse homolog of GEF-H1, had an early influence on mitotic spindle assembly in Rat-2 cells. We clearly established here that early mitotic effects, including mitotic spindle formation, are not observed upon disruption of GEF-H1 function in HeLa cells (Fig. S8). We note that Bakal et.al. did not directly assess the regulation of RhoA activity in Rat-2 cells by Lfc, precluding comparisons of how RhoA activation might influence the phenotypic differences observed. However, such differences in GEF-H1/Lfc function may be accounted for by a number of variables, including differences in the adherent properties of the cells used, in regulatory mechanisms modulating GEF activity, or by intrinsic differences in mitotic mechanisms that have been noted between cells (Burgess and Chang, 2005).
Our data also establish that Ect2 and GEF-H1 mediate different aspects of RhoA activation at the cell cortex in HeLa cells. Bulk RhoA activation is not affected by Ect2 depletion, and RhoA activation appears to be mislocalized, rather than diminished (Fig.7,E and S10). This suggests that Ect2 does not directly catalyze GTP-loading on RhoA during cytokinesis. We propose that Ect2 serves as a core signaling platform essential for the recruitment of important modules of the cleavage machinery such as RhoA and its regulators to equatorial sites (see Fig. S9). Interactions of Ect2 with several components implicated in the regulation of acto-myosin-based contractility of the CF are known (Chalamalasetty et al., 2006; Kamijo et al., 2006; Yuce et al., 2005). Consequently, loss of Ect2 would cause delocalization of cleavage factors from the equator, resulting in dispersed RhoA activation around the cortex. In good agreement, as a result of Ect2 siRNA treatment RhoA activation is no longer concentrated at the CF and becomes scattered around the cell cortex, as indicated by our FRET experiments. We suggest that once a RhoA-containing signaling complex is assembled and activated at the cell cortex in a MT- and Ect2-dependent manner, RhoA cycling is intensified by localized activation of GTPase regulators such as GEF-H1. Accordingly, perturbation of GEF-H1 was accompanied by late cytokinetic defects but did not affect localization of RhoA and other regulators of cytokinesis (Fig. S7).
Since CF induction was not affected by GEF-H1 depletion, this also suggests the existence of additional RhoA activators. Recently, a new exchange factor, MyoGEF, (Wu et al., 2006) was identified that associates with non-muscle myosin II and whose depletion also causes multinucleation. For the future, it will be interesting to analyze how different RhoA GEFs are coordinated through MTs, mitotic kinases, and associated signaling factors to modulate and fine tune different aspects of mammalian cell contractile ring formation and constriction.
MATERIALS AND METHODS
Expression constructs
EGFP-GEF-H1 full length constructs in the mammalian expression vector pCMV5 and GST-tagged individual domain constructs of GEF-H1 in the bacterial expression vector pGEX-KG have been described (Krendel et al., 2002; Zenke et al., 2004). An EGFP-GEF-H1 C53R/Y393A construct was generated by inserting a N-terminal KpnI/BstBI fragment including the C53R mutation into KpnI/BstBI cut pCMV5-EGFP-GEF-H1 DHmut vector which contained the Y393A mutation (Krendel et al., 2002). Phospho-mimetic (S885D, S959D, SS885/959DD) and non phosphorylatable GEF-H1 mutants (S885A, S959A, SS885/959AA) were generated by site-directed mutagenesis and confirmed by sequencing.
Antibodies
Generation and affinity purification of the polyclonal anti-GEF-H1 antibody used in this study has been described (Zenke et al., 2004). The polyclonal anti-pS885 GEF-H1 antibody was from Cell Signaling Technology. Both anti-GEF-H1 antibodies were used at 1:1000 in immunoblots and 1:100 in IF experiments. Additional antibodies used in this work are detailed in the Supp. Methods.
RNAi interference
GEF-H1- and Ect2-directed pools of four individual siRNA duplex oligonucleotides (SMARTpool® reagent) were purchased from Dharmacon RNA Technologies (Lafayette, CO) and selected according to the SMARTselection algorithm (see Supp. Methods). siRNA pools were transfected using LipofectAmine 2000 (Invitrogen), as indicated in the Supp. Methods.
Cell culture, DNA transfections, synchronization and drug treatments
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium containing 8% fetal bovine serum, 10 mM glutamine, 10 mM Hepes and antibiotics (100 U/ml penicillin G, and 100 μg/ml streptomycin). For biochemical analysis, cells were grown in 10 cm dishes and transfected with 5-10μg EGFP-tagged GEF-H1 constructs using LipofectAmine 2000 per the manufacturer. For immunostaining, HeLa cells were grown on glass in 24-well plates and transfected with 0.1-0.5μg of plasmid DNA using LipofectAmine Plus reagent (Invitrogen), per manufacturer’s recommendations.
Cell synchronization in prometaphase was achieved by the thymidine/nocadazole method and mitotic cells processed as previously described, with minor modifications (Gohla et al., 2005) – see Supp. Methods.
Aurora A kinase activity was inhibited with 5 μM ZM477439 (kindly provided by Astra Zeneca), Aurora B with 0.5 μM Hesperadin (a gift of Boehringer Ingelheim), Cdk1/Cyclin B with either 10 μM Purvalanol A (Calbiochem) or 200 nM BMI-1026 (kindly provided by Kyung Lee, NCI), protein kinase A was blocked with 10 μM H89 (Calbiochem), and GSK3 with 10 μM SB216763 (Tocris). Solvent (DMSO) controls were performed in parallel.
Immunofluorescence and microscopy
For microscopic observations, HeLa cells were grown on glass coverslips in 24-well plates in DMEM containing 8% fetal calf serum and transfected as described above. For localization of endogenous GEF-H1, cells were fixed in ice-cold MeOH or MeOH/acetone (50:50) for 20 min. To enhance visualization of GEF-H1 at MTs, soluble cellular components were extracted for 30 sec in extraction buffer (80 mM Pipes, pH 6.8, 1 mM MgCl2, 4 mM EGTA, 10 μM Taxol, 0.5% (w/v) Triton X-100) prior to fixation. To stain for endogenous RhoA, cells were fixed in 10% TCA at 4°C (Yonemura et al., 2004). Cells were processed for immunofluorescence staining according to (Gohla et al., 2005) using Alexa-488 and Alexa-568 secondary antibody conjugates. DNA was visualized with DAPI (1:2000; Sigma). Quadruple stainings were obtained using biotinylated-phalloidin together with Alexa-750-labelled streptavidin in accordance with the manufacturer’s suggestions (Molecular Probes).
Dominant negative (d/n) GEF-H1 mutants were transfected as above and fixed in ice-cold MeOH. Only those cells that expressed the EGFP-fusion proteins of interest at comparable levels (similar to cells expressing EGFP alone) were scored. For this study, multinucleated cells were defined as cells with more than one nucleus, including lobulated-, macro- and micronuclei (Gohla et al., 2005).
Live cell imaging
Live cell imaging of dividing cells was performed in a sealed chamber at 37°C on a Nikon TE2000-U microscope with a 40x/1.3 oil objective and DIC optics. Multi dimensional acquisition was controlled by MetaMorph software. For RNAi experiments, cells were transfected with siRNA duplexes for 24 h and then synchronized with 100ng/ml noc for 6-9 h. After a 45 min release period in fresh medium, mitotic cells were imaged every 20 sec for a period of 7 h.
RhoA activation in dividing cells
A HeLa cell line stably expressing a RhoA-biosensor (Pertz et al., 2006) was generated by co-transfecting 4μg RhoA-biosensor plasmid DNA together with 1μg of pPURO vector (Clontech). Stable clonal populations were isolated based on puromycin resistance and YFP fluorescence, then further purified by cell sorting (FACS). FRET imaging was performed as described (Pertz et al., 2006) – see also Supp. Methods.
Wildtype Hela cells or HeLa cells transfected for 24 h with EGFP-GEF-H1 constructs or siRNA were synchronized with thymidine/noc or noc alone, respectively (see text) and released for the indicated times in fresh medium prior to performing Rhotekin RBD pull-down assays to quantify GTP-RhoA (Ren and Schwartz, 2000).
Kinase assays
Active His-tagged Aurora A/B and Cdk1/Cyclin B kinases were purchased from Upstate Cell Signaling Solutions. In vitro phosphorylation assays were carried out as described (Knaus et al., 1995) - See Supp. Methods for details.
Phosphatase treatment, (co)-immunoprecipitation and in vitro exchange assays
Phosphatase treatments, chemical cross-linking with DSP, coimmunoprecipitation and in vitro exchange assays are detailed in the Supp. Methods or as described elsewhere (Downward, 1995; Krendel et al., 2002; Zenke et al., 2004).
Supplementary Material
Acknowledgments
We thank B. Anliker and C. Roberson for critical reading and comments on the manuscript. We are grateful to K. Matter (London, UK), C. Der (U. N. Carolina, Chapel Hill), Claire Crafter (Astra Zeneca), Kyung S. Lee (NCI), and Boehringer Ingelheim for kindly providing reagents, and to R. Rottapel (Toronto, Canada) for sharing unpublished data. This work was supported by NIH grant GM39434 (to G.M.B.) and post-doctoral fellowships by the Deutsche Forschungsgemeinschaft (DFG) [Bi814/1-1] and the American Heart Association [0425020Y] (to J.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alsop GB, Zhang D. Microtubules continuously dictate distribution of actin filaments and positioning of cell cleavage in grasshopper spermatocytes. J Cell Sci. 2004;117:1591–1602. doi: 10.1242/jcs.01007. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Bakal CJ, Finan D, LaRose J, Wells CD, Gish G, Kulkarni S, DeSepulveda P, Wilde A, Rottapel R. The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis. Proc Natl Acad Sci U S A. 2005;102:9529–9534. doi: 10.1073/pnas.0504190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Benink HA, von Dassow G. A microtubule-dependent zone of active RhoA during cleavage plane specification. J Cell Biol. 2005;170:91–101. doi: 10.1083/jcb.200501131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann H. Cytokinesis and the spindle midzone. Cell Cycle. 2005;4:1709–1712. doi: 10.4161/cc.4.12.2215. [DOI] [PubMed] [Google Scholar]
- Burgess DR, Chang F. Site selection for the cleavage furrow at cytokinesis. Trends Cell Biol. 2005;15:156–162. doi: 10.1016/j.tcb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Canman JC, Cameron LA, Maddox PS, Straight A, Tirnauer JS, Mitchison TJ, Fang G, Kapoor TM, Salmon ED. Determining the position of the cell division plane. Nature. 2003;424:1074–1078. doi: 10.1038/nature01860. [DOI] [PubMed] [Google Scholar]
- Chalamalasetty RB, Hummer S, Nigg EA, Sillje HH. Influence of human Ect2 depletion and overexpression on cleavage furrow formation and abscission. J Cell Sci. 2006;119:3008–3019. doi: 10.1242/jcs.03032. [DOI] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- D’Avino PP, Savoian MS, Glover DM. Mutations in sticky lead to defective organization of the contractile ring during cytokinesis and are enhanced by Rho and suppressed by Rac. J Cell Biol. 2004;166:61–71. doi: 10.1083/jcb.200402157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avino PP, Savoian MS, Glover DM. Cleavage furrow formation and ingression during animal cytokinesis: a microtubule legacy. J Cell Sci. 2005;118:1549–1558. doi: 10.1242/jcs.02335. [DOI] [PubMed] [Google Scholar]
- Dean SO, Rogers SL, Stuurman N, Vale RD, Spudich JA. Distinct pathways control recruitment and maintenance of myosin II at the cleavage furrow during cytokinesis. Proc Natl Acad Sci U S A. 2005;102:13473–13478. doi: 10.1073/pnas.0506810102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant R, Glotzer M. Centrosome separation and central spindle assembly act in redundant pathways that regulate microtubule density and trigger cleavage furrow formation. Dev Cell. 2003;4:333–344. doi: 10.1016/s1534-5807(03)00057-1. [DOI] [PubMed] [Google Scholar]
- DerMardirossian C, Bokoch GM. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Downward J. Measurement of nucleotide exchange and hydrolysis activities in immunoprecipitates. Methods Enzymol. 1995;255:110–117. doi: 10.1016/s0076-6879(95)55013-5. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Hall A, Glotzer M. A requirement for Rho and Cdc42 during cytokinesis in Xenopus embryos. Curr Biol. 1997;7:12–23. doi: 10.1016/s0960-9822(06)00023-6. [DOI] [PubMed] [Google Scholar]
- Echard A, Hickson GR, Foley E, O’Farrell PH. Terminal cytokinesis events uncovered after an RNAi screen. Curr Biol. 2004;14:1685–1693. doi: 10.1016/j.cub.2004.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S. Protein kinases controlling the onset of mitosis. Cell Mol Life Sci. 2006;63:781–795. doi: 10.1007/s00018-005-5515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M. Cleavage furrow positioning. J Cell Biol. 2004;164:347–351. doi: 10.1083/jcb.200310112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. NatCell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- Hamilton BT, Snyder JA. Acceleration of cytokinesis in PtK1 cells treated with microtubule inhibitors. Exp Cell Res. 1983;144:345–351. doi: 10.1016/0014-4827(83)90413-5. [DOI] [PubMed] [Google Scholar]
- Hara T, Abe M, Inoue H, Yu LR, Veenstra TD, Kang YH, Lee KS, Miki T. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr-341 in G2/M phase. Oncogene. 2006;25:566–578. doi: 10.1038/sj.onc.1209078. [DOI] [PubMed] [Google Scholar]
- Kamijo K, Ohara N, Abe M, Uchimura T, Hosoya H, Lee JS, Miki T. Dissecting the role of Rho-mediated signaling in contractile ring formation. Mol Biol Cell. 2006;17:43–55. doi: 10.1091/mbc.E05-06-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Billadeau DD, Chen J. The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem. 2005;280:5733–5739. doi: 10.1074/jbc.M409298200. [DOI] [PubMed] [Google Scholar]
- Kimura K, Tsuji T, Takada Y, Miki T, Narumiya S. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J Biol Chem. 2000;275:17233–17236. doi: 10.1074/jbc.C000212200. [DOI] [PubMed] [Google Scholar]
- Kishi K, Sasaki T, Kuroda S, Itoh T, Takai Y. Regulation of cytoplasmic division of Xenopus embryo by rho p21 and its inhibitory GDP/GTP exchange protein (rho GDI) J Cell Biol. 1993;120:1187–1195. doi: 10.1083/jcb.120.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. Regulation of human leukocyte p21-activated kinases through G protein--coupled receptors. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- Mandato CA, Benink HA, Bement WM. Microtubule-actomyosin interactions in cortical flow and cytokinesis. Cell Motil Cytoskeleton. 2000;45:87–92. doi: 10.1002/(SICI)1097-0169(200002)45:2<87::AID-CM1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Minoshima Y, Kawashima T, Hirose K, Tonozuka Y, Kawajiri A, Bao Y C, Deng X, Tatsuka M, Narumiya S, May WS, Jr, et al. Phosphorylation by aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev Cell. 2003;4:549–560. doi: 10.1016/s1534-5807(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Aoki K, Matsuda M. Monitoring spatio-temporal regulation of Ras and Rho GTPase with GFP-based FRET probes. Methods. 2005;37:146–153. doi: 10.1016/j.ymeth.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays. 1995;17:471–480. doi: 10.1002/bies.950170603. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- Niiya F, Tatsumoto T, Lee KS, Miki T. Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene. 2006;25:827–837. doi: 10.1038/sj.onc.1209124. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Yonemura S. Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci. 2006;119:104–114. doi: 10.1242/jcs.02737. [DOI] [PubMed] [Google Scholar]
- O’Connell CB, Wheatley SP, Ahmed S, Wang YL. The small GTP-binding protein rho regulates cortical activities in cultured cells during division. J Cell Biol. 1999;144:305–313. doi: 10.1083/jcb.144.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Piekny A, Werner M, Glotzer M. Cytokinesis: welcome to the Rho zone. Trends Cell Biol. 2005;15:651–658. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Rappaport R. Cleavage furrow establishment by the moving mitotic apparatus. Dev Growth Differ. 1997;39:221–226. doi: 10.1046/j.1440-169x.1997.t01-1-00010.x. [DOI] [PubMed] [Google Scholar]
- Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Ren Y, Li R, Zheng Y, Busch H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem. 1998;273:34954–34960. doi: 10.1074/jbc.273.52.34954. [DOI] [PubMed] [Google Scholar]
- Rivero F, Illenberger D, Somesh BP, Dislich H, Adam N, Meyer AK. Defects in cytokinesis, actin reorganization and the contractile vacuole in cells deficient in RhoGDI. Embo J. 2002;21:4539–4549. doi: 10.1093/emboj/cdf449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Liu XF, Kamijo K, Raziuddin R, Tatsumoto T, Okamoto I, Chen X, Lee CC, Lorenzi MV, Ohara N, Miki T. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J Biol Chem. 2004;279:7169–7179. doi: 10.1074/jbc.M306725200. [DOI] [PubMed] [Google Scholar]
- Somers WG, Saint R. A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis. Dev Cell. 2003;4:29–39. doi: 10.1016/s1534-5807(02)00402-1. [DOI] [PubMed] [Google Scholar]
- Strickland LI, Donnelly EJ, Burgess DR. Induction of cytokinesis is independent of precisely regulated microtubule dynamics. Mol Biol Cell. 2005;16:4485–4494. doi: 10.1091/mbc.E05-04-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147:921–928. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Asiedu M, Adelstein RS, Wei Q. A novel guanine nucleotide exchange factor MyoGEF is required for cytokinesis. Cell Cycle. 2006;5:1234–1239. doi: 10.4161/cc.5.11.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localization in cells and tissues. Exp Cell Res. 2004;295:300–314. doi: 10.1016/j.yexcr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223–232. doi: 10.1083/jcb.200212049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Parrini MC, Dulyaninova NG, Bresnick AR, Mochizuki N, Matsuda M. Cell type-specific regulation of RhoA activity during cytokinesis. J Biol Chem. 2004;279:44756–44762. doi: 10.1074/jbc.M402292200. [DOI] [PubMed] [Google Scholar]
- Yuce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol. 2005;170:571–582. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke FT, Krendel M, DerMardirossian C, King CC, Bohl BP, Bokoch GM. p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J Biol Chem. 2004;279:18392–18400. doi: 10.1074/jbc.M400084200. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Kronebusch PJ, Simon PM, Borisy GG. Microtubule dynamics at the G2/M transition: abrupt breakdown of cytoplasmic microtubules at nuclear envelope breakdown and implications for spindle morphogenesis. J Cell Biol. 1996;135:201–214. doi: 10.1083/jcb.135.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.