

Abstract
In this review, we summarize the present state of knowledge of the functional characteristics of the carotid body (CB) chemoreflex with respect to control of sympathetic nerve activity (SNA) in chronic heart failure (CHF). Evidence from both CHF patients and animal models of CHF has clearly established that the CB chemoreflex is enhanced in CHF and contributes to the tonic elevation in SNA. This adaptive change derives from altered function at the level of both the afferent and central nervous system (CNS) pathways of the reflex arc. At the level of the CB, an elevation in basal afferent discharge occurs under normoxic conditions in CHF rabbits, and the discharge responsiveness to hypoxia is enhanced. Outward voltage-gated K+ currents (IK) are suppressed in CB glomus cells from CHF rabbits, and their sensitivity to hypoxic inhibition is enhanced. These changes in IK derive partly from downregulation of nitric oxide synthase (NOS) / NO signaling and upregulation of angiotensin II (Ang II) / Ang II receptor (AT1R) signaling in glomus cells. At the level of the CNS, interactions of the enhanced input from CB chemoreceptors with altered input from baroreceptor and cardiac afferent pathways and from central Ang II further enhance sympathetic drive. In addition, impaired function of NO in the paraventricular nucleus of the hypothalamus participates in the increased SNA response to CB chemoreceptor activation. These results underscore the principle that multiple mechanisms involving Ang II and NO at the level of both the CB and CNS represent complementary and perhaps redundant adaptive mechanisms to enhance CB chemoreflex function in CHF.
1. Introduction
The chemoreflexes (central and peripheral) play a pivotal role in the control of alveolar ventilation to ensure that gas exchange rate in the lungs continually matches metabolic demand for O2 uptake and CO2 removal. Often overlooked, however, is the fact that these reflexes also exert important influences on cardiac and vascular control to regulate blood flow and thus gas exchange at the level of the tissues. An important component to chemoreflex activation is an increase in sympathetic outflow to the vascular beds. This response aids in the maintenance of arterial pressure in the face of the direct vasodilatory effects of hypoxemia or hypercapnia, and thus assists in the maintenance of a driving pressure for adequate blood flow and gas exchange in tissues. It should come as no surprise, therefore, that there are circumstances in which alterations in arterial chemoreflex function may contribute to the progression and severity of certain cardiovascular diseases that are influenced by sympathetic neural function, such as hypertension and heart failure.
An impediment to a better understanding of the significance of chemoreflexes in cardiovascular control has been the assumption that chemoreflexes normally are not activated at rest (normoxia/isocapnia) and thus should have little influence on the tonic control of sympathetic tone. This assumption however, has been shown to be incorrect. In normal humans at rest, hyperoxia, which inhibits peripheral chemoreceptor activity, decreases sympathetic nerve activity (Seals et al. 1991). In addition, animal models (Sun et al. 1999a, 1999b) and patients (Chua et al. 1996) with chronic heart failure (CHF) exhibit increased chemoreflex drive under normoxic conditions. Thus, it is not unreasonable to suggest that chemoreflexes contribute to sympathetic tone, even in the absence of hypoxia.
CHF is a major public health care problem. Acute coronary syndromes are the progenitor of CHF in many patients. The dramatic increase in survival rates after coronary infarcts due to more effective interventions and treatments, and the general aging of the global population are causing the incidence, prevalence, and economic burden of CHF to increase at a faster rate than any other cardiovascular disease. After age 60, more than 1 in 10 of all men and women develop CHF in the United States (Rosamond et al, 2007) and Europe (Tendera, 2005). Current prognosis is poor: 50% of all patients with CHF die within 5 years of diagnosis and less than 15% survive more than 10 years (Rosamond et al, 2007, Tendera, 2005). The need for a better understanding of the pathophysiological processes responsible for the progression of CHF is clear.
This review will focus specifically on a discussion of the alterations that occur in peripheral chemoreflex function in chronic heart failure (CHF), particularly that of the carotid body (CB) chemoreflex and its effects on sympathetic activity, the possible mechanisms that contribute to these alterations, and the role that these alterations may play in the pathophysiology of the disease.
2. Chemoreflex Control of Sympathetic Function in Heart Failure
Sympathohumoral activation is characteristic of all forms of chronic left ventricular failure and is a major influence on the rate of progression and ultimate mortality of CHF (Esler et al. 1997). To a limited extent, activation of the sympathetic nervous system is beneficial to the maintenance of arterial pressure as cardiac output declines with dysfunction of the left ventricle. However, the continually increasing sympathetic activation to the heart appears to engage a positive feedback cycle to hasten the progression of cardiac failure (Esler et al. 1997). It has been well documented for many years that the tonic restraint of sympathetic outflow by arterial and cardiopulmonary baroreflexes is depressed in CHF (Eckberg et al. 1971). This once popular notion that baroreceptor unloading is solely responsible for this sympathetic activation seems unlikely from evidence that has accumulated in the last 10 years. More recent studies indicate that maladaptive changes also occur in the central nervous system at integrative sites for autonomic control, such as the nucleus tractus solitarius (Hirooka, 2006), paraventricular nucleus of the hypothalamus (Patel, 2000), and rostral ventral lateral medulla (Zucker, 2006), which foster an enhanced sympathetic drive. Several lines of evidence also make it clear that augmented excitatory reflexes further boost activation of sympathetic outflow in CHF. These reflexes encompass sympathetic excitatory cardiac (Wang et al. 1999), somatic (Sinoway and Li, 2004), peripheral (Chua et al. 1996, Sun et al. 1999a) and central chemoreceptor reflexes (Narkiewicz et al. 1999).
The role of chemoreflex mechanisms in heart failure has received increased attention in the past several years (Schultz and Sun, 2000), but not without considerable controversy. An exaggerated ventilatory response to hypercapnia, indicative of an enhanced central chemoreflex, has been fairly consistently observed in humans with CHF (Kara et al. 2003). But the ventilatory response to isocapnic hypoxia, an index of peripheral chemoreflex function, in CHF patients seems less clear. Peripheral chemoreflex responses to hypoxia were not enhanced in patients with mild NYHA Class II-III CHF distinguished by dyspnea to mild (III) or moderate (II) exercise (Narkiewicz et al. 1999). Similarly, other clinical studies (Haque et al. 1996, van de Borne et al. 1996) reported that suppression of peripheral chemoreceptor activity by hyperoxia had no effect on muscle sympathetic nerve activity or arterial pressure. By contrast, other groups (Chua et al. 1996, 1997, Ponikowski & Banasiak, 2001, Ciarka et al. 2006) have found an enhanced ventilatory response to hypoxia in CHF patients, particularly those in more severe stages of heart failure. In addition, heightened peripheral chemoreflex function correlates significantly with the enhanced ventilatory response to exercise and dyspnea observed in these patients (Chua et al. 1997, Ciarka et al. 2006)).
The study of patient populations is complicated by variability in the etiology, duration, severity, and treatment of CHF. For this reason, we have chosen a more controlled analysis of chemoreflex function in a rabbit model of pacing-induced CHF (Sun et al. 1999a,1999b, Li et al. 2005, 2006) used extensively for the investigation of the regulation of sympathetic nerve activity in CHF (Zucker, 2006). Continuous rapid pacing of the rabbit heart (360-380 beats/min) over the course of 3-4 weeks results in a dilated cardiomyopathy with progressive deterioration of left ventricular function (Sun et al. 1999a). Characteristics of CHF in our paced rabbits used for study include a 40-50% reduction in ejection fraction and fractional shortening of the left ventricle without overt pulmonary edema, sympatho-humoral activation and altered autonomic reflexes as observed in patients with Class III CHF (Zucker, 2006, Sun et al. 1999b). Also, since the rabbit lacks functional aortic chemoreceptors (Verna et al. 1975), hypoxia-hyperoxia effects can be attributed to carotid body (CB) function.
CB chemoreflex control of sympathetic nerve activity and ventilation is clearly enhanced in CHF rabbits (Fig.1)(Sun et al. 1999a, Li, et al. 2005, Li et al. 2006). In fact, the augmented sympathetic drive occurs in the face of a potentiated ventilatory response that might be expected to attenuate the sympathetic response due to negative feedback from pulmonary afferents (Somers et al. 1989). In addition, central input from the CB provides a tonic excitatory influence on sympathetic outflow in rabbits with established CHF since hyperoxia reduces resting sympathetic nerve activity in CHF but not sham animals (Sun et al. 1999a), and selective CB denervation attenuates the elevated resting sympathetic nerve activity and plasma norepinephrine normally observed in CHF rabbits (Fig. 2). Enhanced CB chemoreflex sensitivity therefore represents a very potent contributory mechanism for sympathetic activation in heart failure.
Figure 1.
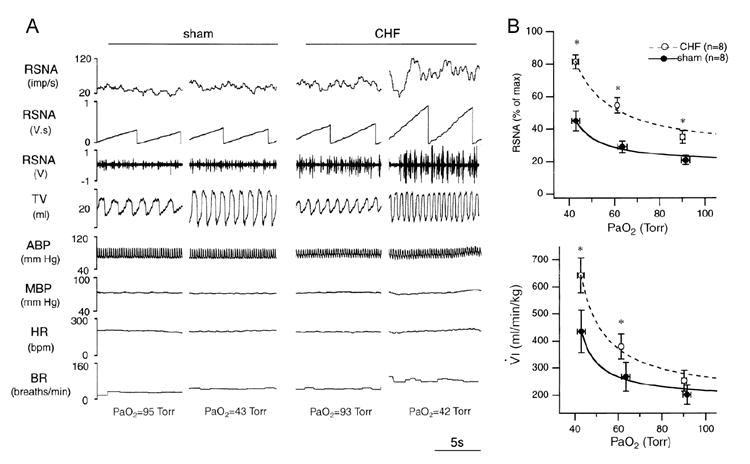
A. Chart recordings of renal sympathetic nerve activity (RSNA), tidal volume (TV), arterial blood pressure (ABP), mean arterial blood pressure (MBP), HR, and breathing rate (BR) under normoxic and hypoxic conditions from a sham and a chronic heart failure (CHF) rabbit. The CHF rabbit exhibited a higher level of baseline RSNA in the normoxic state and an enhanced RSNA and ventilatory response to hypoxia. PaO2, arterial PO2. B. RSNA-PaO2 (top) and minute ventilation (VÝI)-PaO2 relationships (bottom) in sham and CHF rabbits plotted by mean values of actual data (circles with error bars) and by fitted hyperbolic curves (lines). CHF rabbits exhibited a higher level of baseline RSNA in normoxic state (PaO2 at 90-95 Torr) and an enhanced response of RSNA andVÝI to hypoxia. * P < 0.05, CHF vs. sham. (Reprinted from Sun et al. 1999a, page 1268, with permission from the American Physiological Society.)
Figure 2.
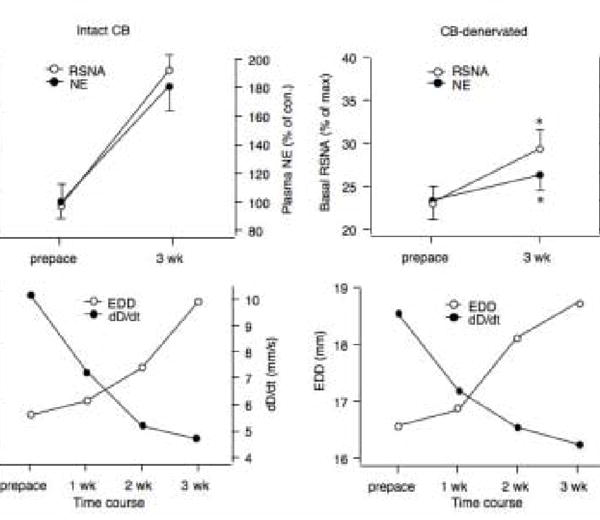
A. Resting renal sympathetic nerve activity (RSNA) and plasma norepinephrine concentration (NE) in rabbits paced for 3 weeks with (Intact CB) or without (bilateral CB-denervated) functional CBs. B. The decrease in left ventricular function was similar in both groups as evidenced by an increase in end diastolic diameter (EDD) and decrease in contractility (dD/dt). n = 4* P < 0.05, CB Denervated vs. Intact.
What are the mechanisms responsible for an enhanced peripheral chemoreflex in CHF? We will discuss evidence that the enhanced reflex sensitivity results from an alteration in the sensory characteristics of the CB chemoreceptors and from changes in the central integration of the chemoreceptor input.
3. Carotid Body Function in Heart Failure
There is an enhanced afferent input from CB chemoreceptors in the CHF rabbits (Sun et al. 1999b), which provides a primary contribution to the augmentation of reflex function. The baseline discharge in the normoxic state and the magnitude of the afferent response to corresponding levels of isocapnic hypoxia are greater in CHF rabbits than in sham rabbits (Fig. 3). These alterations were observed both in the intact (blood perfused) and isolated CB preparations (Sun et al. 1999b). This observation suggests that an intrinsic alteration within the CB, rather than a circulating factor, drives the altered afferent sensitivity in the CHF state.
Figure 3.
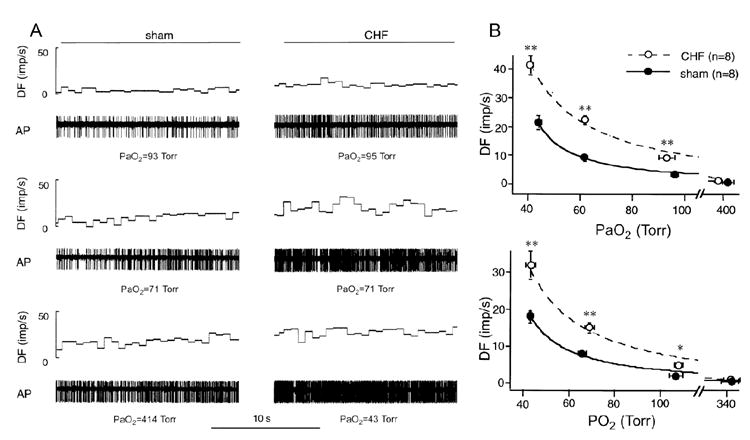
A. Representative recordings of action potentials from carotid body (CB) chemoreceptors during normoxia and 2 levels of isocapnic hypoxia in a sham (left) and a CHF rabbit (right). The CHF rabbit exhibited higher levels of discharge both in the normoxic state and at the equivalent levels of hypoxia, compared with the sham rabbit. B. PO2-discharge relationships of CB chemoreceptors in intact (A) and isolated (B) preparations in sham and CHF rabbits, showing an elevated baseline discharge in normoxic state and an enhanced response to hypoxia in both preparations from CHF rabbits. * P < 0.05; ** P < 0.01, CHF vs. sham. DF, discharge frequency [impulses (imp)/s]; AP, action potential; PaO2, arterial PO2.(Reprinted from Sun et al. 1999b, pages 1276 and 1277, with permission from the American Physiological Society.)
3.1 Role of Ang II on CB Function in CHF
There are numerous neurohumoral factors that are altered in CHF, such as angiotensin II (Ang II), nitric oxide (NO), catecholamines, endothelin, and atrial natriuretic peptide, among other factors that can affect CB chemoreceptor activity (Prabhakar, 1994). The role of many these factors in altered CB function in CHF remains largely unexplored, but there is good evidence that Ang II and NO play a major role.
Systemic and tissue Ang II levels are increased in CHF patients (Roig et al. 2000) and in CHF rabbits (Li et al. 2006). Ang II enhances CB chemoreceptor activity via the AT1 receptor (AT1R) in the CB (Allen, 1998). Furthermore, a locally generated Ang II system has been shown to be operational (Lam & Leung, 2002), and chronic hypoxia upregulates the expression of the AT1R in the rat CB (Fung et al. 2002).
Systemic administration of Ang II in conscious rabbits (to levels equivalent to the endogenous plasma Ang II level in CHF rabbits) enhances hypoxia-induced chemoreflex activation of sympathetic outflow (Li et al. 2006). This effect of Ang II in normal animals was similar to the enhanced CB chemoreflex function observed in CHF rabbits. Furthermore, blockade of AT1R by L-158,809 attenuated or normalized the exaggerated hypoxia-induced chemoreflex responses in CHF rabbits (Li et al., 2006). Thus, the ability of Ang II to enhance CB chemoreflex function is likely to contribute, along with its other known sympathetic excitatory effects, to the sympathetic activation observed in CHF.
This type of study, however, does not pinpoint the loci within the reflex arc where Ang II acts to enhance CB chemoreflex function. Circulating Ang II enhances sympathetic function at the level of the central nervous system (Reid, 1992) and studies have documented a role of central Ang II in enhancing sympathetic outflow in CHF (Zucker, 2006). Nevertheless, afferent recordings from the isolated CB confirm that a functional AT1R in the CB contributes to the elevated chemoreceptor afferent activity in CHF rabbits (Fig. 4) (Li et al. 2006). In these experiments, blockade of AT1R decreased CB chemoreceptor responses to hypoxia in the isolated CB (devoid of circulating Ang II) in CHF rabbits (Fig. 4) but not in sham rabbits. In addition, we found that the Ang II concentration and mRNA expression and protein levels of AT1R in the CB are increased in CHF rabbits (Fig. 4). These results suggest that locally produced Ang II in the CB is capable of altering CB chemoreceptor function and that elevation of tissue Ang II with upregulation of AT1R in the CB plays a major role in enhancing CB chemoreceptor sensitivity to hypoxia in CHF rabbits. Indeed, angiotensinogen and Ang converting enzyme are expressed in CB tissue, and the CB is capable of producing Ang II (Lam & Leung, 2002). And we have demonstrated that tissue Ang II and AT1R levels are elevated in the CB from CHF rabbits (Li et al. 2006). Further studies are needed to determine whether other components of the local Ang II system are upregulated in the CB in CHF.
Figure 4.
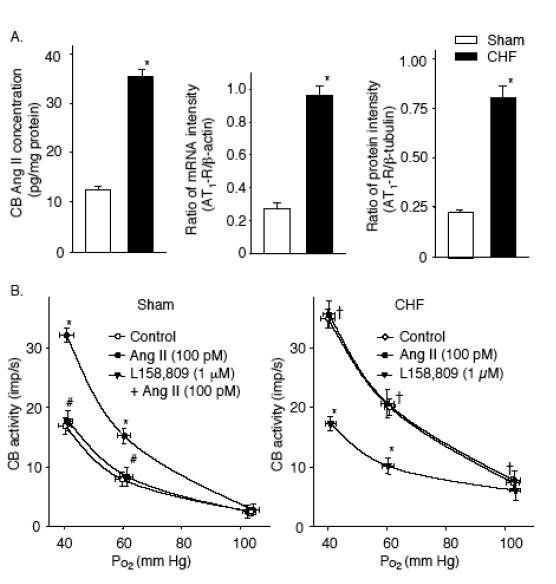
A. Angiotensin II (Ang II) concentration, relative expression of mRNA and protein of AT1 receptor in the CBs from sham and CHF rabbits. *p< 0.05 vs. sham rabbits. B. Effects of Ang II and the AT1R antagonist L-158,809 on the CB chemoreceptor activity in sham and CHF rabbits. Data are means ± SEM, n = 7 in each group. *P < 0.05 vs. control; #P < 0.05 vs. Ang II, †P < 0.05 vs. control in sham group. (Adapted and reprinted from Li et al. 2006, pages 136 and 137, with permission from the European Society of Cardiology.)
Results from this study (Li et al. 2006) also indicated that Ang II couldn’t explain all of the changes in CB chemoreceptor activities that occur in CHF rabbits. Our studies have consistently shown that CB chemoreceptor activity under normoxic (baseline) conditions is elevated in CHF rabbits (Sun et al. 1999b, Li et al. 2006). However, neither Ang II nor L158,809 had effects on baseline CB chemoreceptor activity in either sham or CHF rabbits (Li et al. 2006). On the other hand, perfusion of the CB with Ang II at levels much higher than we observed in CHF animals (> 1 nM) induces excitation of rat CB chemoreceptor activity under normoxic conditions (Allen, 1998). Although it is possible that very high levels of Ang II may contribute to an elevated basal CB chemoreceptor activity in the normoxic state in CHF rabbits, it is likely that other mechanisms are responsible for this effect, such as a decrease in CB nitric oxide (NO) production, as discussed below.
3.2 Role of Ang II on K+ Currents in CB Glomus Cells in CHF
In recent studies, we explored the mechanism by which Ang II enhances CB chemoreceptor sensitivity to hypoxia (Li & Schultz, 2006). Although there is still some controversy about the detailed steps of the chemo-neurotransduction cascade in the CB, it is believed that the glomus or type I cells, which lie in synaptic apposition with afferent axons, are the initial sites of chemotransduction in the CB. The glomus cell expresses several types of membrane ion channels that influence its excitability. Of these channels, the hypoxia-inactivated outward K+ channels are proposed to play a key role for the initial depolarization, with subsequent activation of voltage-gated Ca2+ channels, release of neurotransmitter, and increases of sensory discharge in the carotid sinus nerve (Kemp, 2006).
The hypoxia-sensitive K+ channels in adult rabbit CB glomus cells are voltage-gated, Ca2+-insensitive K+ (Kv) channels, which encompass Kv4.1 and Kv4.3 subunits (Ganfornina & López-Barneo, 1992, Sanchez et al. 2002). Since Ang II enhances CB chemoreceptor sensitivity to hypoxia (Li et al. 2006), we reasoned that endogenous Ang II blunts the Kv currents (IK) and affects the sensitivity of Kv channels to hypoxia in the CB glomus cells in the CHF state.
Our studies confirm that hypoxia decreases IK and induces depolarization of the membrane potential in rabbit glomus cells (Fig. 5). Furthermore, the sensitivity of Kv channels to hypoxia is enhanced in isolated glomus cells from CHF rabbits, and blockade of AT1R alone is capable of reversing this enhanced hypoxic sensitivity (Fig. 5). In addition, exposing normal rabbit CB glomus cells to Ang II mimics this effect of CHF on Kv channel function. These observations demonstrate that Ang II is capable of enhancing the O2 sensitivity of Kv channels via an AT1R mediated pathway, and that this mechanism contributes to the enhanced sensitivity of Kv channels to hypoxia in glomus cells from CHF animals. Another deduction from these experiments is that this cellular Ang II/AT1R signaling mechanism is operational within isolated glomus cells.
Figure 5.
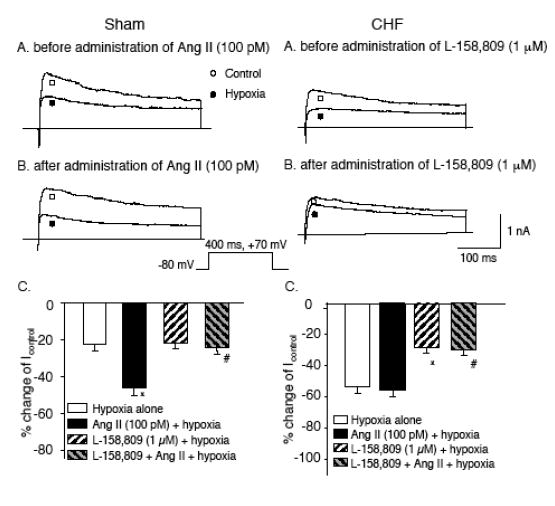
Panels A and B: IK from a sham (left) and CHF (right) glomus cell illustrating the effect of hypoxia on IK before (A) and after (B) administration of either 100 pM Ang II to the extracellular medium of the sham cell (left) or 1 μM L- 158,809 (AT1R antagonist) to the extracellular medium of the CHF cell (right). C. Percentage change of IK by hypoxia (Icontrol –Ihypoxia)/Icontrol) in sham glomus cells (left) and CHF glomus cells (right) before (hypoxia alone) and after exposure to either 100 pM Ang II, 1 μM L-158,809 or 100 pM Ang II + 1 μM L-158,809. Data are means ± S.E.M. n = 10 cells from 7 rabbits in each condition. *P < 0.05 versus hypoxia alone; #P < 0.05 versus Ang II (100 pM) + hypoxia).(Adapted and reprinted from Li & Schultz, 2006, pages 6 and 7, with permission from the Physiological Society.)
Ang II had no further effect on the O2 sensitivity of Kv channels in glomus cells from CHF rabbits. Conversely, AT1R blockade alone did not affect the O2 sensitivity of Kv channels in glomus cells from sham rabbits (Fig. 5). These results demonstrate that the ability of cellular Ang II/ AT1R mechanism to enhance the hypoxic sensitivity of Kv channels appears to be maximally expressed in glomus cells from CHF animals but is minimally functional (or at least not demonstrable in our isolated experimental conditions) in the glomus cells of normal rabbits.
Although our results suggest that elevation of local tissue Ang II in the CB plays an important role on the hypersensitivity of IK to hypoxia in CHF, it is not clear how Ang II within an isolated glomus cell interacts with AT1R to affect the sensitivity of Kv channels. Recent studies have shown that intracellular administration of Ang II increases the peak inward calcium current density and decreases the junctional conductance via intracellular AT1R in cardiac myocytes (De Mello & Monterrubio, 2004), and intracellular application of an AT1R antagonist abolishes the effect of intracellular Ang II. It seems likely that the enhanced sensitivity of IK to hypoxia in CB glomus cells in CHF may be due to elevation of intracellular Ang II binding to intracellular AT1R. This notion is evidenced by our observation that intracellular administration of L158,809 (added to the recording pipette solution) blunted the sensitivity of IK to hypoxia in the CB glomus cells from CHF rabbits (Li & Schultz, 2006).
The downstream mechanism by which Ang II/AT1R activation enhances the sensitivity of IK to hypoxia in CB glomus cells is not known. One possible candidate is protein kinase C (PKC) signaling pathway. AT1R activation results in a rapid, phospholipase C-dependent sustained release of diacylglycerol, which leads to activation of PKC (Ushio-Fukaiet al. 1998). PKC was shown to cause inhibition of KCa currents in CB glomus cells of rats, but PKC activation could not to account for inhibition of these channels by acute hypoxia (Peers & Carpenter 1998). The effect of PKC on the O2 sensitivity of Kv channels in rabbit CB glomus cells has not been addressed yet.
Alternatively, Ang II recently has been implicated in activation of ROS, specifically superoxide anion, in CHF (Zucker, 2006). AT1R activation promotes activation of NADPH oxidase with superoxide anion production (Griendling et al. 1994). Our preliminary experiments have shown that mRNA and protein expressions of NADPH oxidase subunits are enhanced in CBs from CHF rabbits, and a NADPH oxidase inhibitor (apocynin) and superoxide dismutase mimetic (tempol) inhibit the effect of Ang II on the sensitivity of IK to acute hypoxia (Li et al. 2004a). These results suggest that the NADPH oxidase-superoxide anion pathway may mediate the effects of Ang II in CB glomus cells.
The hypoxia-sensitive Kv channels in adult rabbit CB encompass those with Kv4.1, and Kv4.3 subunits (Ganfornina & López-Barneo, 1992, Sanchez et al. 2002). But the rabbit glomus cells also express the Kv3.4 channel, which appears to be insensitive to hypoxia (Sanchezet al. 2002). Thus the extent to which hypoxia influences the total IK in rabbit glomus cells may be altered by changes in the relative expression of these channel subtypes. Indeed, Kääb, et al. (2005) showed that chronic exposure of rabbit CB glomus cells to hypoxia induces downregulation of Kv3.4 channels, but not the hypoxia-sensitive Kv4.3 channels in rabbit CB glomus cells. This shift in balance of Kv channels resulted in a more predominant role of the hypoxia-sensitive Kv4.3 to the total outward K+ current, with a subsequent relative increase in the magnitude of acute hypoxia-induced inhibition of IK. We found (Li & Schultz, 2006) that in CHF, there is a similar decrease the expression of Kv3.4 but not Kv4.3 in rabbit CB glomus cells (Fig. 6). It is possible that the increased proportion of Kv4.3 channels to the total outward K+ current contributes somewhat to the enhanced sensitivity of IK to hypoxia in glomus cells in CHF. This observation, however, does not downplay the significance of the marked effect of AT1R blockade to reverse the exaggerated hypoxic sensitivity of IK observed in glomus cells in CHF rabbits, and points to an important regulatory role for Ang II on post-translational Kv activity in the glomus cell in the CHF state.
Figure 6.
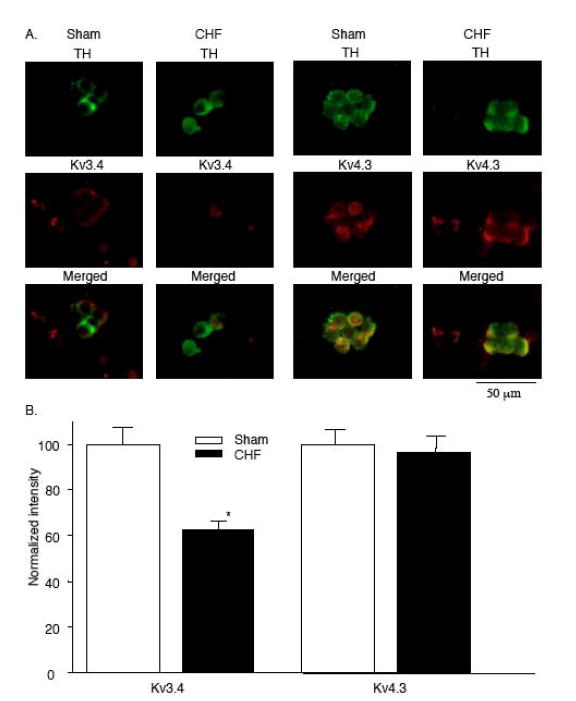
A. Co-localization of tyrosine hydroxylase (TH) and Kv3.4 (left) and Kv4.3 (right) in CB glomus cells from sham and CHF rabbits. Top row: Green immunofluorescent image for TH. Middle row: red immunofluorescent image for Kv3.4 (left) or Kv4.3 (right). Bottom row: Merged image for overlap of TH and Kv3 or Kv4.3. B. Normalized fluorescence intensity for Kv3.4 and Kv4.3 in CB glomus cells from sham and CHF rabbits Data are means ± S.E.M. n = 5 rabbits for each group. *P < 0.05 versus sham.(Adapted and reprinted from Li & Schultz, 2006, page 9, with permission from the Physiological Society.)
Despite the potent effects of Ang II-AT1R on the hypoxic sensitivity of IK, neither Ang II nor AT1R blockade has effects on IK atnormoxia in either sham or CHF glomus cells (Li & Schultz, 2006). These results are consistent with our study described above (Li et al. 2006) where exposure of the isolated CB to a similar level of Ang II or to L-158,809 had no effect on the activity of chemoreceptor discharge at normoxia in either sham or CHF rabbits. Thus endogenous Ang II cannot account for the suppression of Kv channel activity in glomus cells (Li & Schultz, 2006) or the enhanced chemoreceptor activity observed in CHF rabbits (Sun et al. 1999b, Li et al. 2005, Li et al. 2006) in normoxic (baseline) conditions. We have other evidence however, that suggests that a NO mechanism is likely to participate in the enhanced baseline activity in the CB under resting normoxic conditions in CHF (Sun et al. 1999b, Li et al. 2005).
3.3 Role of NO on CB Function in CHF
In addition to Ang II, we have explored the possibility that nitric oxide (NO) also plays a role in altered CB function in CHF. NO is of importance for several reasons. The NO synthesizing enzyme, nitric oxide synthase (NOS), is localized to nerve fibers and vascular endothelium in the CB, and NO is inhibitory to the carotid body sensory activity (Prabhakar, 1999). Because O2 is essential for biosynthesis of NO, during normoxia in normal conditions, basal production of NO acts as an amplifier of O2 to keep CB chemoreceptor discharge suppressed (Prabhakar, 1999).
It is well documented that either NOS activity or NO production is decreased in various tissues in the CHF state (Zucker et al. 2004). These data imply that a possible reduction of NO release in the CB in the CHF state would lead to a disinhibition of CB chemoreceptors and elevate activity during normoxia, and even contribute to the enhanced afferent sensitivity to hypoxia. Indeed, we found that basal NO production from the carotid body (Sun et al. 1999b, Li et al. 2005) and the density of NADPH-diaphorase positive cells (a marker of NOS) within the carotid body (Sun et al. 1999b) were less in CHF than in sham rabbits. These results suggest an attenuated NOS activity and/or a lower content of NOS within the CB in CHF rabbits. Moreover, we found that NOS inhibition (N-nitro-L-arginine, L-NNA) in the CB increased the baseline discharge of CB chemoreceptors in the normoxic state and their response to isocapnic hypoxia in sham rabbits, but had very little effect in either condition in CHF rabbits (Sun et al. 1999b). Conversely, the NO donor, S-nitroso-N-acetylpenicillamine (SNAP), inhibited CB chemoreceptor activity to a much greater extent in CHF than in sham rabbits. These data indicate that a tonic inhibitory effect of NO on the activity of the CB chemoreceptors is near maximally expressed in sham rabbits but virtually absent in CHF rabbits. These data also strongly support the idea that an attenuated NOS activity in the carotid body of CHF rabbits, rather than an inability to respond to NO, contributes to the enhanced activity of CB chemoreceptors in this condition.
The two constitutive isomers of NOS, neuronal NOS (nNOS) and endothelial NOS (eNOS), are present in the CB (Prabhakar, 1999). The density of NADPH-diaphorase positive fibers is decreased in the CB of CHF rabbits (Sun et al. 1999b). We have confirmed a decreased presence of nNOS in nerve fibers innervating the CB using immunohistochemistry, and a decreased total nNOS protein expression in the carotid body from CHF rabbits (Li et al. 2005). Furthermore, gene transfer of nNOS using an adenoviral vector (Ad.nNOS) to the CB in CHF rabbits enhanced protein expression and NO production in the CB and reversed the enhanced CB chemoreceptor activity seen in the CHF state (Fig. 7)(Li et al. 2005). The specific nNOS inhibitor,S-methyl-L-thiocitrulline (SMTC) abolished the effect of Ad.nNOS on CB chemoreceptor activity (Fig. 7). Equally important, SMTC alone enhanced CB chemoreceptor activity in sham rabbits, indicating that, in this species, nNOS provides a tonic inhibitory influence on CB chemoreceptor activity under normal conditions. By contrast, SMTC failed to increase CB chemoreceptor activity in CHF rabbits without nNOS gene transfer, indicating a loss of this tonic inhibitory influence in the CHF state (Fig. 7). These results, taken together, demonstrate that a marked down regulation of endogenous nNOS in the CB is involved in the enhanced CB chemoreceptor activity in CHF rabbits.
Figure 7.
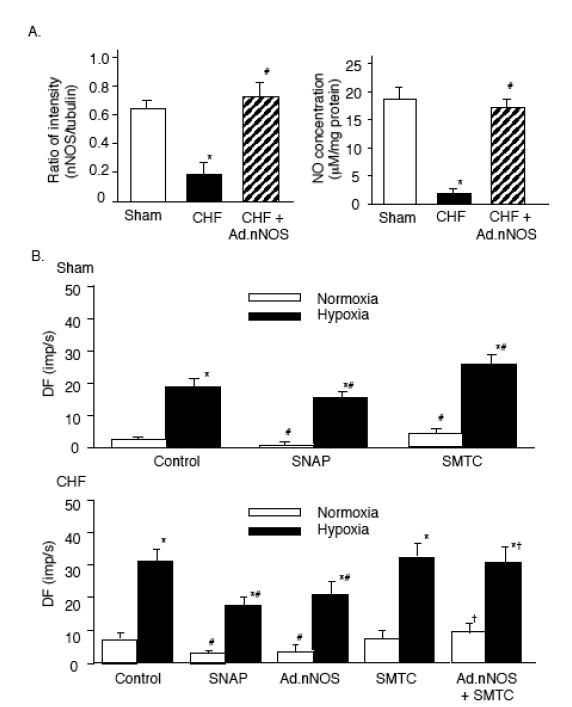
A. Relative nNOS protein expression and NO concentration in sham, CHF, and CHF+Ad.nNOS treated CBs. n=6 in each group. Data are mean±SEM, *P<0.05 vs sham; #P<0.05 vs CHF rabbits. B. Effect of SNAP (100 μM) or SMTC (1 μM) on the activity of CB chemoreceptors in sham, CHF, and CHF+Ad.nNOS treated CBs. Hypoxia: PaO2=40±2.4 mm Hg. Data are mean±SEM, n=8 in each group. *P<0.05 vs normoxia; #P<0.05 vs control, †P<0.05 vs CHF Control or CHF+Ad.EGFP control.(Adapted and reprinted from Li et al. 2005, pages 264 and 265, with permission from the American Heart Association.)
Ad.nNOS to the CB proved efficacious in elevating nNOS protein expression and NO production in treated CBs of CHF rabbits to levels found in sham rabbits (Fig. 7). Yet, even though ad.nNOS treatment reduced CB chemoreceptor activity and chemoreflex function in CHF rabbits toward that seen in sham rabbits (Li et al. 2005), the gene transfer did not completely normalize CB function (Fig. 7). It is possible that the inability of the technique to target specific cell types within the CB influenced the efficacy of the gene transfer on chemoreceptor function. More likely, the inability of nNOS upregulation to fully normalize CB function in CHF rabbits points to important complementary or synergistic role of Ang II on CB chemoreceptor activity in CHF.
Our observation that NADPH oxidase-derived superoxide anion mediates the Ang II-enhanced CB chemoreceptor activity in CHF rabbits (Li et al. 2004a) suggests an important interaction between these two signaling pathways. Ang II may contribute to depressed bioavailable NO in the CB by suppressing nNOS gene expression (Kihara et al. 1997) and/or increased scavenging of NO through superoxide anion production. Conversely, the downregulation of NO production in the CB in CHF may act to enhance the effects of Ang II by reduced scavenging of superoxide anion by NO. The relationship among NO, Ang II and superoxide anion on CB chemoreceptor function is not yet clear and deserves further study. But it is clear that upregulation of Ang II/superoxide anion pathways and downregulation of NOS/NO pathways in the CB in CHF have complementary effects on CB chemoreceptor activity and glomus cells K+ channel function (see below).
NNOS gene transfer to both CBs also significantly blunted but did not completely normalize the elevated resting sympathetic nerve activity seen in conscious CHF rabbits (Li et al. 2005). These results demonstrate the important contribution of enhanced CB chemoreceptor input to elevated sympathetic outflow in CHF, and the contribution of nNOS down regulation in the CB to this effect. The fact that enhanced gene expression of CB nNOS did not completely normalize resting sympathetic nerve activity in CHF rabbits is not unexpected, since it did not completely normalize CB chemoreflex function as described above. In addition, it is well known that a number of other cardiovascular reflex and central neural alterations contribute to elevated sympathetic activity in CHF (Zucker, 2006). These results underscore the significance of a multiplicity of factors contributing to sympathetic hyperactivity in CHF.
A participation of eNOS in the alteration of the characteristics of the CB chemoreceptors in CHF rabbits cannot be confirmed or excluded at this time. Existing evidence of the role of eNOS vs nNOS in the CB is somewhat controversial. Studies have shown that a nonspecific NOS inhibitor,NG-nitro-L-arginine methyl ester (L-NAME), significantly enhances the CB ventilatory response in rats (Haxhiu et al. 1995) and cats (Valdés et al. 2003); but specific nNOS inhibitors were ineffective. On the other hand, using mutant mice deficient in nNOS and eNOS isoforms, (Kline et al. 1998) found that mice lacking nNOS showed greater ventilatory responses to hypoxia than wild-type controls; whereas responses to hypoxia were blunted in mutant mice lacking eNOS compared with the wild-type. We have observed a downregulation eNOS in the CB of CHF rabbits (Li and Schultz, unpublished), but have not yet investigated the functional consequence of this change.
3.4 Role of NO on K+ Currents in CB Glomus Cells in CHF
In other studies, we have addressed the mechanism by which NO suppresses carotid body chemoreceptor afferent activity. As summarized above, IK is attenuated and the resting membrane potential is depolarized in CB glomus cells from CHF rabbits as compared with those in sham rabbits (Li & Schultz 2006 ). This depression in IK from glomus cells is consistent with the elevated CB chemoreceptor activity observed in CHF rabbits at rest. While our studies described above suggest that Ang II does not affect baseline CB chemoreceptor activity (Li et al. 2006) or IK during normoxia (Li & Schultz, 2006), NO donors inhibited baseline discharge of CB chemoreceptor afferents (Sun et al. 1999b, Li et al. 2005)
As predicted, NO donors enhanced the IK in glomus cells in sham and CHF rabbits (Fig. 8) (Li et al. 2004b). Conversely, L-NNA blunted IK in sham rabbits but not in CHF rabbits (Fig. 8). Based upon these results, it is reasonable to assume that the blunted IK in glomus cells under normoxic onditions is a consequence of NO depletion in the CB and that this effect contributes to the enhancement of CB hemoreceptor function.
Figure 8.
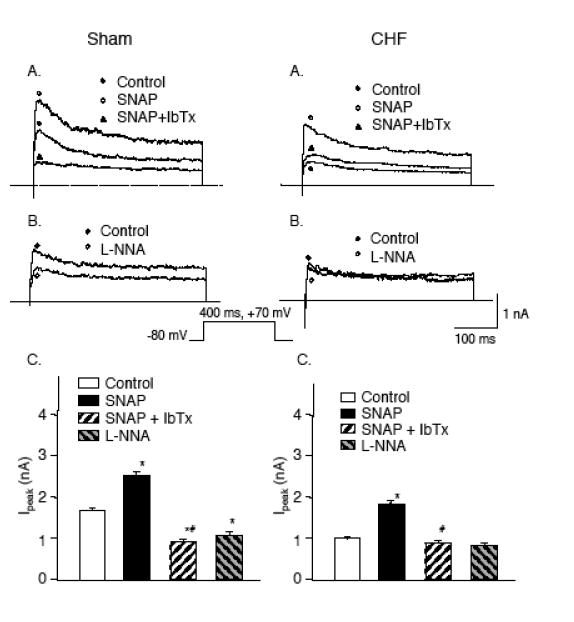
Panels A and B: IK elicited in a glomus cell from a sham (left) and CHF (right) rabbit (A) before (control) and after administration of 100 μM SNAP alone and 100 μM SNAP plus 100 nM IbTx to the extracellular medium, (B) before (control) and after administration of 10 μM L-NNA to the extracellular medium. C. Peak Ik (-80 to +70 mV pulse) obtained from glomus cells in control condition with 100 μM SNAP alone with 100 μM SNAP plus 100 nM IbTx, and with 10 μM L-NNA added to the extracellular medium. Data are mean±SEM n= 8 cells from 6 rabbits in each group. *P < 0.05 versus control; #P < 0.05 versus SNAP.(Adapted and reprinted from Li et al. 2004a, pages 225 and 226, with permission from the Physiological Society.)
It remains an unresolved question how NO production and NOS inhibitors can influence IK in isolated CB glomus cells, when existing evidence can not confirm significant immunostaining of NOS in CB glomus cells (Prabhakar, 1999, Li et al. 2005). However, in recent experiments using single cell PCR, we successfully detected expression of nNOS mRNA in rabbit glomus cells (Li and Schultz, unpublished). Thus it appears that NOS is present in rabbit CB glomus cells at levels that are difficult to detect with immunofluorescence but sufficient to influence K+ channel activity, or NOS is sequestered, as in mitochondria, and inaccessible to immunofluorescence antibodies. An important remaining object will be to assess where is nNOS is located within CB glomus cells, and whether there is a change in expression of this nNOS in glomus cells from CHF rabbits.
The glomus cell membrane in rabbits possesses calcium dependent, voltage–gated K (KCa or maxi K) channels in addition to Kv channels described above that contributes to the total outward IK (López-López et al. 1993, 1997 ). Our study (Li et al. 2004b) showed that iberiotoxin (IbTx), a specific blocker of KCa channels, reduced IK in sham rabbits but had no effect in CHF rabbits. These results indicated that KCa channel activity is markedly suppressed in glomus cells from CHF rabbits, and the attenuated IK in glomus cells of CHF rabbits is largely due to decreased activity of these KCa channels. This is not to say, however, that the suppressed KCa channel activity exclusively mediates the attenuated IK in CB glomus cells in CHF rabbits. Kv channel blockers, 4-AP and TEA, confirmed that other Kv channels, probably Kv3.4 as discussed above, are also involved, but to a lesser extent.
Our results also indicate that the attenuated KCa activity contributes to the relative depolarization of resting membrane potential observed in the CHF glomus cells (Li et al. 2004b). SNAP lowered the resting potential to normal levels in CHF cells, and IbTx reversed this effect. However, a HERG-like K+ channel has been reported in rabbit glomus cells which could also influence the resting membrane potential (Overholtet al.2000). The contribution of HERG channels to the depolarized membrane potential in the CHF glomus cells remains to be determined.
The effect of NO to enhance Ik in rabbit glomus cells appears to be mediated by these KCa channels (Li et al. 2004b). Our results confirmed that IbTx completely abolished the ability of SNAP to enhance IK in glomus cells from sham and CHF rabbits (Fig. 8). From these data, we propose that decreased NO production contributes to the blunted IK in glomus cells from CHF rabbits by preventing the activation of KCa channels.
NO activates soluble guanylate cyclase, and many of its effects are mediated by stimulation of cGMP production (Murad, 2006). Our results also indicate that the effect of NO on the KCa channel activity in CB glomus cells is cGMP-dependent because the guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3,-a]quinoxalin-1-one (ODQ) inhibited the effects of SNAP on the IK and the cGMP analog 8-bromo-cGMP could mimic the effect of SNAP on the IK in both sham and CHF rabbits (Li et al. 2004b). In rat CB glomus cells, NO enhances KCa channel activity through cGMP-dependent protein kinase G (Silva & Lewis, 2002). It seems likely, but has not been shown, that the cGMP effect on KCa channel rabbit CB glomus cells is also mediated by protein kinase G.
3.5 Role of Other Channels in Glomus Cells in Heart Failure
Our studies reveal that steady state K+ currents (IKss), as well as the peak transient outward currents, are similarly influenced in CHF (Li et al. 2004b, Li & Schultz, 2006). The Kv3.4, Kv4.3 and KCa channels that we described in our studies are known to be principle conduits of the peak transient outward K+ current in rabbit glomus cells (Ganfornina & López-Barneo, 1992, Sanchezet al. 2002). The family of K+ channels that comprise the IKss in the CB glomus cells from rabbit are not well described, nor extensively studied. HERG-like K+ channel (IKr), one component of delayed rectifier K+ channels, has been identified in the CB glomus cells of the rabbit (Overholtet al. 2000) but its sensitivity to the conditions we describe in this study have yet to be studied. Other components of delayed rectifier K+ channels (such as rapidly activating, slowly inactivating delayed rectifier K+ channel and slowly activated delayed rectifier K+ channels) are not reported in rabbit CB glomus cells. Background K+ channels (such as TASK channels) that are O2 sensitive in rat CB glomus cells (Buckleret al. 2000), also have not been described in rabbit CB glomus cells. It also is possible that changes in inactivation kinetics of Kv channels could influence the magnitude of the IKss. Therefore, further study is needed to explore the underlying mechanisms of alterations of IKss induced by CHF, hypoxia, and Ang II in rabbit glomus cells.
Voltage-gated Ca2+ channels also play an important role in the function of CB glomus cells (Overholt & Prabhakar, 1997). To date however, very little is known of the function of these channels in the CB in the setting of CHF. Indirectly, it is known that NO inhibits L-type Ca2+ channels in rabbit glomus cells via a cGMP independent mechanism (Summers et al. 1999). The role of Ang II on L-type Ca2+ channel activity in the CB has not been addressed. However, Ang II is known to activate L-type Ca2+ channels in vascular smooth muscle (Fuller et al. 2005). It would seem likely, therefore, that L-type Ca2+ channel activity would be enhanced in CB glomus cells from CHF animals as a result of downregulation of NO production and/or increased production of Ang II.
3.6 Role of Reduced Blood Flow on CB Function in CHF
The process that occurs in the course of the development of CHF that causes an upregulation of local Ang II/AT1R and downregulation of NOS/NO in CB glomus cells is not evident from our present studies. The changes in Kv channel function we describe in glomus cells in CHF rabbits are similar in most respects to those described in rabbit glomus cells exposed to chronic hypoxia (Kääb, et al. 2005). This observation raises the question whether the chronically decreased cardiac output of CHF may be sufficient to produce a decrease in O2 delivery to the carotid body (akin to chronic hypoxia) that leads to the functional changes that occur in glomus cells. This issue warrants further study.
The question also arises whether enhanced CB Ang II/AT1R and/or reduced NOS/NO signaling in the CB, at least in part, enhances CB chemoreceptor sensitivity by reducing blood flow to the CB. We cannot discount the possibility that a reduction in CB blood flow may contribute to the enhanced afferent chemoreceptor activity of the CB in CHF. Nevertheless, we were able to observe changes in K+ channel function in isolated CB glomus cells from the CHF state that were directly influenced by pharmacological manipulation of both signaling pathways in the isolated cells. Thus there is evidence to support an effect of Ang II and NO on glomus cell function independent of the vasculature.
Although the CB is known to acutely autoregulate its blood flow such that moderate changes in systemic hemodynamics have little effect on CB afferent function (Marshall, 1994), the effects of chronic reductions in cardiac output on flow stability of the CB and CB afferent function are not clear. An important objective of future studies should address the role of chronic a reduction in blood supply to the carotid body, similar to that observed in CHF, on CB chemoreceptor activity and glomus cell ion channel function.
4. Central integration of CB Input in CHF
4.1 Central Interaction with other Reflexes and Brainstem Mechanisms
Considerable interaction is known to occur between baroreceptor and CB chemoreceptor control of sympathetic nerve activity (Trzebski et al. 1975, Somers et al.1991). The two afferent inputs have a mutual inhibitory interaction on sympathetic outflow: baroreceptor input suppresses chemoreflex activation of sympathetic outflow and chemoreceptor input suppresses baroreflex inhibition of sympathetic outflow (Wennergren et al. 1976).
The impaired baroreflex sensitivity (Zucker et al. 2004) and enhanced CB chemoreflex sensitivity, both characteristic of patients and experimental animals with CHF, are likely then to act synergistically to enhance sympathetic activity. There is a close correlation between enhanced peripheral chemoreflex sensitivity and impaired baroreflex sensitivity in CHF patients (Ponikowski & Banasiak, 2001). Inhibition of peripheral chemoreceptors by breathing 100% O2 improved baroreflex function in CHF patients. In addition, we have found that selective denervation of the carotid bodies improves baroreflex sensitivity in heart failure rabbits (Xia et al. 2000). These studies suggest a causative role of the chemoreflex on impaired baroreflex function in heart failure. Nevertheless, the extent of establishing cause and effect between alterations in baroreflex and chemoreflex function as a result of their central interaction in heart failure remains to be resolved.
There are numerous other factors that may affect central integration of peripheral chemoreceptor control of sympathetic outflow in CHF. Central Ang II is known to be an important component to the central mechanisms of elevated sympathetic function in CHF animals (Zucker, 2006). Our lab has found that intraceberoventricular (ICV) administration of losartan reduced peripheral chemoreflex activation of sympathetic outflow to hypoxia in CHF rabbits but not in sham rabbits (Sun et al. 1999c). By contrast, ICV Ang II augmented the hypoxia-induced sympathetic response in sham rabbits but not in CCHF rabbits. Our results support the idea that central Ang II as well as CB Ang II is involved in the alteration of CB chemoreflex function in CHF.
The central locus of this Ang II effect remains to be revealed. However, recent evidence suggests an important component of Ang II effects on CB chemoreflex function reside at the level of the nucleus tractus solitarius (NTS) in the medulla. In rats, CB chemoreceptor activation of sympathetic nerve activity was enhanced when cardiac sympathetic afferent input was concomitantly activated by either electrical stimulation of central end of the left cardiac sympathetic nerve or epicardial application of capsaicin to the left ventricle (Gao et al. 2007). Microinjection of an AT1R antagonist into the NTS completely abolished this facilitatory effect of cardiac sympathetic afferent stimulation to enhance CB chemoreflex activation of sympathetic outflow. These results suggest that cardiac sympathetic afferents augment the CB chemoreceptor reflex and that central Ang II, specifically located in the NTS, plays a major role in these reflex interactions. Since the cardiac sympathetic afferent reflex also is enhanced in CHF (Wang et al. 1999), it is likely that this central interaction plays an important role in the enhanced chemoreceptor reflex in the CHF state.
4.2 CB Chemoreflex at the Level of the Paraventricular Nucleus of the Hypothalamus (PVN)
Given the significance of the CB chemoreceptors in cardiorespiratory reflex regulation in CHF, it is important to understand the central neural anatomical pathways and neurotransmitters involved in processing and modulation of the cardiovascular and respiratory responses to chemoreceptor activation. Unfortunately, these central anatomical substrates are not yet well understood. Several studies have identified central neural structures involved in the sympathoexcitatory component of chemoreflexes; however, most have focused on the pons and the medulla (Guyenet 2000), which are the primary projection areas of chemosensitive afferents and the primary sites for respiratory and sympathetic integration. Apart from these areas of the brain, other studies suggest an involvement of the hypothalamus in modulating the respiratory and cardiovascular responses to hypoxia and hypercapnia (Horn & Waldrop 1994, Kubo et al. 1997, Olivan et al. 2001). In particular, hypoxia and hypercapnia stimulate neurons in various hypothalamic nuclei, including the PVN (Berquin et al. 2000).
Because the PVN is known to influence sympathetic outflow (Zhang et al. 1997), we speculated that the PVN plays an important role in the modulation of sympathoexcitation induced by the activation of the CB chemoreflex. To further support this notion, Kubo et al. (1997) reported that the excitatory amino acid antagonist kynurenate injected into the PVN inhibited the chemoreceptor reflex-induced pressor response in anesthetized rats. Later, Olivan et al. (2001) demonstrated bilateral electrolytic lesion of the PVN in conscious rats produced a significant reduction in the magnitude and duration of the pressor response induced by chemoreflex activation, suggesting that the PVN is involved in the regulation of cardiovascular responses to CB chemoreflex activation.
Reddy et al. (2005) investigated the involvement of the PVN in the modulation of sympathoexcitatory reflex activated by peripheral and central chemoreceptors in rats. Selective blockade of neurotransmission in the PVN with microinjection of lidocaine had no effect on baseline sympathetic and cardiorespiratory variables; however, the sympathetic and phrenic nerve responses evoked by CB chemoreceptor stimulation were significantly attenuated. On the other hand, blocking neurotransmission within the PVN had no effect on the hypercapnia-induced central chemoreflex responses in carotid body denervated animals. Furthermore, bilateral microinjection of the gamma aminobutyric acid receptor type A (GABAA) antagonist bicuculline into the PVN augmented the sympathetic and phrenic responses to CB chemoreceptor stimulation. Conversely, the GABAA agonist muscimol injected into the PVN attenuated these reflex responses. These results suggest a selective role of the PVN in processing the sympathoexcitatory and ventilatory component of the CB chemoreflex, but not the central chemoreflex, and that this CB chemoreflex pathway is negatively modulated by GABAA receptors in the PVN.
Previous studies have demonstrated that NO within the PVN influences cardiovascular function and sympathetic out flow (Zhang & Patel, 1998). Furthermore, these investigators documented that the endogenous NO-mediated effect within the PVN of CHF rats is less potent in suppressing sympathetic outflow compared with control rats (Zhang et al. 2001). Reddy et al. (2007) examined the role of NO in the PVN on CB chemoreflex responses in CHF rats. Bilateral microinjection of the NOS inhibitorNG-monomethyl-L-arginine (L-NMMA) into the PVN augmented the sympathetic and phrenic nerve responses to CB chemoreceptor stimulation in normal rats, but was less effective in enhancing in chemoreflex responses in CHF rats, in which sympathetic and phrenic nerve responses were already exaggerated. These results are consistent with previous observations which demonstrated a decrease message for nNOS and protein levels of nNOS in the PVN of CHF rats (Zheng et al. 2005). Conversely, after microinjection of the NO donor sodium nitroprusside SNP into the PVN, there was a significant decrease in CB chemoreflex mediated activation of sympathetic activity in both sham and CHF rats. However, the magnitude of the SNP mediated attenuation in sympathetic activation was significantly smaller in CHF rats compared to the sham rats.
Based on these results, it appears that NO within the PVN is involved in regulating the sympathetic response to CB chemoreflex activation by providing a tonic restraining influence on the reflex. In the CHF state, this restraining influence on CB chemoreflex function is lost due to the impairment of NO mechanisms in the PVN. However, the exact neural pathways through which the NO synthesized within the PVN influences the reflex responses of CB chemoreceptors remain to be determined. Previous studies have proposed that the effect of NO within the PVN on the modulation of sympathetic activity may be mediated by the release of the inhibitory neurotransmitter, GABA (Zhang & Patel, 1998). It seems reasonable to speculate that the excitatory input of CB chemoreceptors via projections from the NTS activates excitatory receptors within the PVN, possibly NMDA receptors, on efferent neurons that send projections to the presympathetic neurons in the RVLM or spinal cord, and that GABA mechanisms within the PVN that normally act to restrain this pathway are impaired in CHF due to the down regulation of NOS and NO production in the PVN.
Our evidence that central Ang II also is involved in the enhancement of the CB chemoreflex in CHF (Sun et al. 1999c) raises the question whether this effect is also mediated at the level of the PVN. Ang II in the PVN is known to exert an excitatory influence on sympathetic activity and antagonistically interact with NO effects (Li, Y.F. et al. 2006). Whether the excitatory effects of central Ang II on CB chemoreflex function in CHF are mediated, at least in part, by an effect on these pathways at the level of the PVN remains to be determined.
4.3 CB Chemoreflex and Central Control of Breathing in CHF
It is important to mention briefly that the ramifications of enhanced central input from CB chemoreceptors extend not simply to central integration of sympathetic function, but to control of breathing as well. Sleep apnea and periodic or Cheyne-Stokes breathing is a common complication in CHF patients. The theoretic underpinnings and experimental evidence for the relationship between enhanced chemoreflex function with breathing instability in CHF extend well beyond the limits of our review, but are well documented in other recent reviews (Ponikowski & Banasiak, 2001, Cherniack et al. 2005, Caples et al. 2005).
5. Summary
In this review, we have summarized the present state of knowledge on the functional characteristics of the carotid body chemoreflex with respect to control of sympathetic function in heart failure. Evidence in both CHF patients and animal models of CHF has clearly established that the CB chemoreflex is enhanced in CHF and contributes to the tonic elevation in sympathetic function. This derangement derives from altered function at the level of both the afferent and central pathways of the reflex arc (Fig. 9).
Figure 9.
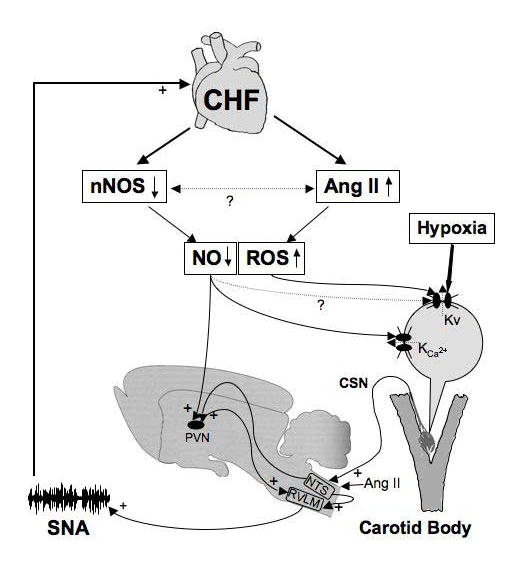
A schematic overview of known mechanisms contributing to altered carotid body (CB) chemoreflex function in chronic heart failure (CHF). Impairment of cardiac output leads to downregulation of nNOS/NO and upregulation of Ang II/AT1 receptor/ROS signaling pathways to inhibit voltage-gated, calcium dependant (KCa) and O2 sensitive (Kv) K+ channels in CB glomus cells, respectively. The resultant enhanced excitability of glomus cells increases afferent discharge of the carotid sinus nerve (CSN) to activate a reflex increase in sympathetic nerve activity (SNA). In addition, effects of increased Ang II at the nucleus tractus solitarius (NTS) in the medulla and decreased NO in the paraventricular nucleus of the hypothalamus (PVN) further enhance CB chemoreflex activation of SNA in CHF. The scheme illustrates the feed-forward effect of CB chemoreflex activation of SNA to further advance deterioration of cardiac function, which then further engages CB chemoreflex activation of SNA via escalation and reinforcement of the afferent and central mechanisms described.
The mechanisms responsible for elevated afferent activity from the CB in CHF are not yet fully understood. Recent studies have revealed that an upregulation of the local Ang II/ AT1R signaling pathway in CB glomus cells enhances the sensitivity of Kv channels to hypoxic inhibition and may be mediated by ADPH oxidase derived superoxide anion. In addition, downregulation of NOS/NO signaling pathway in glomus cells in CHF contributes to suppress of KCa channel function. It is also clear that the effects of Ang II and NO extend beyond the CB and influence integration of CB afferent input at various sites within the CNS to further enhance chemoreflex control of sympathetic function in CHF.
Because the chronic sympathetic activation driven from the CB and other autonomic pathways leads to further deterioration of cardiac function, a positive feedback cycle is likely to be engaged. As CHF ensues, the increasing severity of cardiac dysfunction leads to progressive escalation of these alterations in CB chemoreflex function to further elevate sympathetic activity and cardiac deterioration (Fig. 9). The trigger or causative factors that occur in the development of CHF that sets this cascade of events in motion and the time course over which they occur remain obscure. Ultimately, however, causative factors must surely be tied to cardiac pump failure and reduced cardiac output. Within the carotid body, a progressive and chronic reduction in blood flow may be the key to initiating the maladaptive changes that occur in CB chemoreflex function in CHF.
Acknowledgments
Supported by: National Heart, Lung, Blood Institute PPO-1 HL062222
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimersthat apply to the journal pertain.
References
- Allen AM. Angiotensin AT1 receptor-mediated excitation of rat carotid body chemoreceptor afferent activity. J Physiol (Lond) 1998;510:773–781. doi: 10.1111/j.1469-7793.1998.773bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berquin P, Bodineau L, Gros F, Larnicol N. Brainstem and hypothalamic areas involved in respiratory chemoreflexes: a Fos study in adult rats. Brain Res. 2000;857:30–40. doi: 10.1016/s0006-8993(99)02304-5. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid-, and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caples SM, Wolk R, Somers VK. Influence of cardiac function and failure on sleep-disordered breathing: evidence for a causative role. J Appl Physiol. 2005;99:2433–2439. doi: 10.1152/japplphysiol.00676.2005. [DOI] [PubMed] [Google Scholar]
- Cherniack NS, Longobardo G, Evangelista CJ. Causes of Cheyne-Stokes respiration. Neurocrit Care. 2005;3:271–279. doi: 10.1385/NCC:3:3:271. [DOI] [PubMed] [Google Scholar]
- Chua T, Clark AL, Amadi AA, Coats AJ. Relation between chemosensitivity and the ventilatory response to exercise in chronic heart failure. J Am Coll Cardiol. 1996;27:650–657. doi: 10.1016/0735-1097(95)00523-4. [DOI] [PubMed] [Google Scholar]
- Chua TP, Ponikowski P, Webb-Peploe K, Harrington D, Anker S, Piepoli M, Coats AJ. Clinical characteristics of patients with an augmented peripheral chemoreflex in chronic heart failure. Eur Heart J. 1997;18:480–487. doi: 10.1093/oxfordjournals.eurheartj.a015269. [DOI] [PubMed] [Google Scholar]
- Chugh SS, Chua TP, Coats AJ. Peripheral chemoreflex in chronic heart failure: Friend and foe. Am Heart J. 1996;132:900–904. doi: 10.1016/s0002-8703(96)90333-6. [DOI] [PubMed] [Google Scholar]
- Ciarka A, Cuylits N, Vachiery JL, Lamotte M, Degaute JP, Naeije R, van de Borne P. Increased peripheral chemoreceptors sensitivity and exercise ventilation in heart transplant recipients. Circulation. 2006;113:252–257. doi: 10.1161/CIRCULATIONAHA.105.560649. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Monterrubio J. Intracellular and extracellular angiotensin II enhance the L-type calcium current in the failing heart. Hypertension. 2004;44:360–364. doi: 10.1161/01.HYP.0000139914.52686.74. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Drabinsky M, Braunwald E. Defective cardiac parasympathetic control in patients with heart disease. New Eng J Med. 1971;285:877–883. doi: 10.1056/NEJM197110142851602. [DOI] [PubMed] [Google Scholar]
- Esler M, Kaye D, Lambert G, Esler D, Jennings G. Adrenergic nervous system in heart failure. Am J Cardiol. 1997;80:7L–14L. doi: 10.1016/s0002-9149(97)00844-8. [DOI] [PubMed] [Google Scholar]
- Fuller AJ, Hauschild BC, Gonzalez-Villalobos R, Awayda MS, Imig JD, Inscho EW, Navar LG. Calcium and chloride channel activation by angiotensin II-AT1 receptors in preglomerular vascular smooth muscle cells. Am J Physiol Renal Physiol. 2005;289: F760–F767. doi: 10.1152/ajprenal.00422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung ML, Lam SY, Chen Y, Dong X, Leung PS. Functional expression of angiotensin II receptors in type-1 cells of the rat carotid body. Pflugers Arch. 2001;441:474–480. doi: 10.1007/s004240000445. [DOI] [PubMed] [Google Scholar]
- Fung ML, Lam SY, Dong X, Chen Y, Leung PS. Postnatal hypoxemia increases angiotensin II sensitivity and up-regulates AT1a angiotensin receptors in rat carotid body chemoreceptors. J Endocrinol. 2002;173:305–313. doi: 10.1677/joe.0.1730305. [DOI] [PubMed] [Google Scholar]
- Ganfornina MD, López-Barneo J. Potassium channel types in arterial chemoreeptor cells and their selective modulation by oxygen. J Gen Physiol. 1992;100:401–426. doi: 10.1085/jgp.100.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Pan YX, Wang W, Li YL, Schulz HD, Zucker IH, Wang W. Cardiac Sympathetic Afferent Stimulation Augments the Arterial Chemoreceptor Reflex in Anesthetized Rats. J Appl Physiol. 2007;102:37–43. doi: 10.1152/japplphysiol.00681.2006. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol. 2000;121:147–162. doi: 10.1016/s0034-5687(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Haque WA, Boehmer J, Clemson BS, Leuenberger UA, Silber DH, Sinoway LI. Hemodynamic effects of supplemental oxygen administration in congestive heart failure. J Am Coll Cardiol. 1996;27:353–357. doi: 10.1016/0735-1097(95)00474-2. [DOI] [PubMed] [Google Scholar]
- Hirooka Y. Localized gene transfer and its application for the study of central cardiovascular control. Auton Neurosci . 2006:126–127. 120–129. doi: 10.1016/j.autneu.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Horn EM, Waldrop TG. Modulation of the respiratory responses to hypoxia and hypercapnia by synaptic input onto caudal hypothalamic neurons. Brain Res. 1994;664:25–33. doi: 10.1016/0006-8993(94)91949-6. [DOI] [PubMed] [Google Scholar]
- Kääb S, Miguel-Velado E, López-López JR, Pérez-García MT. Downregulaion of Kv3.4 channels by chronic hypoxia increases acute oxygen sensitivity in rabbit carotid body. J Physiol. 2005;566:395–408. doi: 10.1113/jphysiol.2005.085837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes--physiology and clinical implications. Acta Physiol Scand. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- Kemp PJ. Detecting acute changes in oxygen: will the real sensor please stand up? . Exp Physiol. 2006;91:829–834. doi: 10.1113/expphysiol.2006.034587. [DOI] [PubMed] [Google Scholar]
- Kihara M, Umemura S, Kadota T, Yabana M, Tamura K, Nyuui N, Ogawa N, Murakami K, Fukamizu A, Ishii M. The neuronal isoform of constitutive nitric oxide synthase is up-regulated in the macula densa of angiotensinogen gene-knockout mice. Lab Invest. 1997;76:285–294. [PubMed] [Google Scholar]
- Kline D, Yang T, Huang P, Prabhakar NR. Altered response to hypoxia in mutant mice deficient in neuronal nitric oxide synthase. J Physiol. 1998;511:273–287. doi: 10.1111/j.1469-7793.1998.273bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Yanagihara Y, Yamaguchi H, Fukumori R. Excitatory amino acid receptors in the paraventricular hypothalamic nucleus mediate pressor response induced by carotid body chemoreceptor stimulation in rats. Clin Exp Hypertens. 1997;19:1117–1134. doi: 10.3109/10641969709083208. [DOI] [PubMed] [Google Scholar]
- Lam SY, Leung PS. A locally generated angiotensin system in rat carotid body. Regul Pept. 2002;107:97–103. doi: 10.1016/s0167-0115(02)00068-x. [DOI] [PubMed] [Google Scholar]
- Li YF, Wang W, Mayhan WG, Patel KP. Angiotensin-mediated increase in renal sympathetic nerve discharge within the PVN: role of nitric oxide. Am J Physiol Regul Integr Comp Physiol. 2006 ;290:R1035–R1043. doi: 10.1152/ajpregu.00338.2004. [DOI] [PubMed] [Google Scholar]
- Li YL, Gao L, Wang W, Zucker IH, Schultz HD. NADPH oxidase derived superoxide anion mediates the angiotensin II-enhanced peripheral chemoreceptor sensitivity in heart failure rabbits. Circulation. 2004a;110(III):265. doi: 10.1016/j.cardiores.2007.04.006. [Abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Sun SY, Overholt JL, Prabhakar NR, Rozanski GJ, Zucker IH, Schultz HD. Attenuated outward potassium currents in carotid body glomus cells of heart failure rabbit: involvement of nitric oxide. J Physiol. 2004b;555:219–229. doi: 10.1113/jphysiol.2003.057422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Li YF, Liu D, Cornish KG, Patel KP, Zucker IH, Channon KM, Schultz HD. Gene transfer of neuronal nitric oxide synthase to carotid body reverses enhanced chemoreceptor function in heart failure rabbits. Circ Res. 2005;97:260–267. doi: 10.1161/01.RES.0000175722.21555.55. [DOI] [PubMed] [Google Scholar]
- Li YL, Schultz HD. Enhanced sensitivity of Kv channels to hypoxia in the rabbit carotid body in heart failure: role of angiotensin II. J Physiol. 2006;575:215–227. doi: 10.1113/jphysiol.2006.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W, Schultz HD. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res. 2006;71:129–138. doi: 10.1016/j.cardiores.2006.03.017. [DOI] [PubMed] [Google Scholar]
- López-López JR, DeLuis DA, González C. Properties of a transient K+ current in chemoreceptor cells of rabbit carotid body. J Physiol. 1993;460:15–32. doi: 10.1113/jphysiol.1993.sp019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-López JR, González C, Pérez-García MT. Properties of ionic currents from isolated adult rat carotid body chemoreceptor cells: effect of hypoxia. J Physiol. 1997;499:429–441. doi: 10.1113/jphysiol.1997.sp021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol. 1994;74:543–594. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- Murad F. Shattuck Lecture: Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, van de Borne PJ, Kato M, Somers VK. Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation. 1999;100:262–267. doi: 10.1161/01.cir.100.3.262. [DOI] [PubMed] [Google Scholar]
- Olivan MV, Bonagamba LG, Machado BH. Involvement of the paraventricular nucleus of the hypothalamus in the pressor response to chemoreflex activation in awake rats. Brain Res. 2001;895:167–172. doi: 10.1016/s0006-8993(01)02067-4. [DOI] [PubMed] [Google Scholar]
- Overholt JL, Prabhakar NR. Ca2+ current in rabbit carotid body glomus cells is conducted by multiple types of high-voltage-activated Ca2+ channels. J Neurophysiol. 1997;78:2467–2474. doi: 10.1152/jn.1997.78.5.2467. [DOI] [PubMed] [Google Scholar]
- Overholt JL, Ficker E, Yang T, Shams H, Bright GR, Prabhakar NR. HERG-like potassium current regulates the resting membrane potential in glomus cells of the rabbit carotid body. J Neurophysiol. 2000;83:1150–1157. doi: 10.1152/jn.2000.83.3.1150. [DOI] [PubMed] [Google Scholar]
- Patel KP. Role of paraventricular nucleus in mediating sympathetic outflow in heart failure. Heart Fail Rev. 2000;5:73–86. doi: 10.1023/A:1009850224802. [DOI] [PubMed] [Google Scholar]
- Peers C, Carpenter E. Inhibition of Ca2+-dependent K+ channels in rat carotid body type I cells by protein kinase C. J Physiol. 1998;512:743–750. doi: 10.1111/j.1469-7793.1998.743bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponikowski P, Banasiak W. Chemosensitivity in chronic heart failure. Heart Fail Monit. 2001;1:126–131. [PubMed] [Google Scholar]
- Prabhakar NR. Neurotransmitters in the carotid body. In: O’Regan, et al., editors. Arterial Chemoreceptors: Cell to System. Plenum Press; New York: 1994. pp. 57–69. [Google Scholar]
- Prabhakar NR. NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol. 1999;115:161–168. doi: 10.1016/s0034-5687(99)00019-5. [DOI] [PubMed] [Google Scholar]
- Reddy MK, Patel KP, Schultz HD. Differential role of the paraventricular nucleus of the hypothalamus in modulating the sympathoexcitatory component of peripheral and central chemoreflexes. Am J Physiol Regul Integr Comp Physiol. 2005;289: R789–R797. doi: 10.1152/ajpregu.00222.2005. [DOI] [PubMed] [Google Scholar]
- Reddy MK, Schultz HD, Zheng H, Patel KP. Altered nitric oxide mechanism within the paraventricular nucleus contributes to the augmented carotid body chemoreflex in heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H149–H157. doi: 10.1152/ajpheart.00117.2006. [DOI] [PubMed] [Google Scholar]
- Reid IA. Interactions between ANGII, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am J Physiol Endocrinol Metab. 1992;262:E763–E778. doi: 10.1152/ajpendo.1992.262.6.E763. [DOI] [PubMed] [Google Scholar]
- Roig E, Perez-Villa F, Morales M, Jiménez W, Orús J, Heras M, Sanz G. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. 2000;21:53–57. doi: 10.1053/euhj.1999.1740. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart Disease and Stroke Statistics--2007 Update: A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115 doi: 10.1161/CIRCULATIONAHA.106.179918. in press. [DOI] [PubMed] [Google Scholar]
- Sanchez D, López-López JR, Pérez-García MT, Sanz-Alfayate G, Obeso A, Ganfornina MD, González C. Molecular identification of Kvα subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J Physiol. 2002;542:369–382. doi: 10.1113/jphysiol.2002.018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz HD, Sun SY. Chemoreflex function in heart failure. Heart Fail Rev. 2000;5:45–56. doi: 10.1023/A:1009846123893. [DOI] [PubMed] [Google Scholar]
- Seals DR, Johnson DG, Fregosi RF. Hyperoxia lowers sympathetic nerve activity at rest but not during exercise in humans. Am J Physiol Regul Integr Comp Physiol. 1991;260:R873–R878. doi: 10.1152/ajpregu.1991.260.5.R873. [DOI] [PubMed] [Google Scholar]
- Silva JM, Lewis DL. Nitric oxide enhances Ca2+-dependent K+ channel activity in rat carotid body cells. Pflugers Arch. 2002;443:671–675. doi: 10.1007/s00424-001-0745-1. [DOI] [PubMed] [Google Scholar]
- Sinoway LI, Li J. A perspective on the muscle reflex: implications for congestive heart failure. J Appl Physiol. 2005;99:5–22. doi: 10.1152/japplphysiol.01405.2004. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Abboud FM. Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest. 1991;87:1953–1957. doi: 10.1172/JCI115221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Influence of ventilation and hypocapnia on sympathetic nerve responses to hypoxia in normal humans. J Appl Physiol. 1989;67:2095–2100. doi: 10.1152/jappl.1989.67.5.2095. [DOI] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR. Nitric oxide inhibits L-type Ca2+ current in glomus cells of the rabbit carotid body via a cGMP-independent mechanism. J Neurophysiol. 1999;81:1449–1457. doi: 10.1152/jn.1999.81.4.1449. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced peripheral chemoreflex function in conscious rabbits with pacing-induced heart failure. J Appl Physiol. 1999a;86:1264–1272. doi: 10.1152/jappl.1999.86.4.1264. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol. 1999b;86:1273–82. doi: 10.1152/jappl.1999.86.4.1273. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Alteration of peripheral chemoreflex and arterial baroreflex in heart failure: Role of central angiotensin II. The FASEB J. 1999c;13 [Abstract] [Google Scholar]
- Tendera M. Epidemiology, treatment, and guidelines for the treatment of heart failure in Europe. Eur Heart J Suppl. 2005;7:J5–J9. [Google Scholar]
- Trzebski A, Lipski J, Majcherczyk S, Szulczyk P, Chruscielewski L. Central organization and interaction of the carotid body baroreceptor and chemoreceptor sympathetic reflex. Brain Res. 1975;87:227–237. doi: 10.1016/0006-8993(75)90420-5. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Griendling KK, Akers M, Lyons PR, Alexander RW. Temporal dispersion of activation of phospholipase C-beta1 and -gamma isoforms by angiotensin II in vascular smooth muscle cells. Role of alphaq/11, alpha 12, and beta gamma G protein subunits. J Biol Chem. 1998;273:19772–19777. doi: 10.1074/jbc.273.31.19772. [DOI] [PubMed] [Google Scholar]
- Valdés V, Mosqueira M, Rey S, Rio RD, Iturriaga R. Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms. Am J Physiol Lung Cell Mol Physiol. 2003;284:L57–L68. doi: 10.1152/ajplung.00494.2001. [DOI] [PubMed] [Google Scholar]
- van de Borne P, Oren R, Anderson EA, Mark AL, Somers VK. Tonic chemoreflex activation does not contribute to elevated muscle sympathetic nerve activity in heart failure. Circulation. 1996;94:1325–1328. doi: 10.1161/01.cir.94.6.1325. [DOI] [PubMed] [Google Scholar]
- Verna A, Roumy M, Leitner LM. Loss of chemoreceptive properties of the rabbit carotid body after destruction of the glomus cells. Brain Res. 1975;100:13–23. doi: 10.1016/0006-8993(75)90239-5. [DOI] [PubMed] [Google Scholar]
- Xia XH, Sun SY, Cornish KG, Zeng YC, Wang W, Schultz HD. Effect of carotid body denerveation on Cheyne-Stokes respiration and baroreflex function in heart failure. Circulation. 2000;102( Suppl S ):700–701. [Abstract] [Google Scholar]
- Wang W, Schultz HD, Ma R. Cardiac sympathetic afferent activity is enhanced in heart failure. Am J Physiol Heart Circ Physiol. 1999;277:H812–H817. doi: 10.1152/ajpheart.1999.277.2.H812. [DOI] [PubMed] [Google Scholar]
- Wennergren G, Little R, Oberg B. Studies on the central integration of excitatory chemoreceptor influences and inhibitory baroreceptor and cardiac receptor influences. Acta Physiol Scand. 1976;96:1–18. doi: 10.1111/j.1748-1716.1976.tb10166.x. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li YF, Patel KP. Blunted nitric oxide-mediated inhibition of renal nerve discharge within PVN of rats with heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H995–H1004. doi: 10.1152/ajpheart.2001.281.3.H995. [DOI] [PubMed] [Google Scholar]
- Zhang K, Patel KP. Effect of nitric oxide within the paraventricular nucleus on renal sympathetic nerve discharge: role of GABA. Am J Physiol Regul Integr Comp Physiol. 1998;275:R728–R734. doi: 10.1152/ajpregu.1998.275.3.R728. [DOI] [PubMed] [Google Scholar]
- Zheng H, Li YF, Cornish KG, Zucker IH, Patel KP. Exercise training improves endogenous nitric oxide mechanisms within the paraventricular nucleus in rats with heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2332–H2341. doi: 10.1152/ajpheart.00473.2004. [DOI] [PubMed] [Google Scholar]
- Zucker IH. Novel mechanisms of sympathetic regulation in chronic heart failure. Hypertension. 2006;48:1005–1011. doi: 10.1161/01.HYP.0000246614.47231.25. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Li YF, Wang Y, Wang W, Patel KP. The origin of sympathetic outflow in heart failure: the roles of angiotensin II and nitric oxide. Prog Biophys Mol Biol. 2004;84:217–232. doi: 10.1016/j.pbiomolbio.2003.11.010. [DOI] [PubMed] [Google Scholar]