

Abstract
In the yeast Saccharomyces cerevisiae, one of the two cytoplasmic lysine tRNAs, tRNACUULys, is partially associated with the mitochondrial matrix. Mitochondrial import of this tRNA requires binding to the precursor of the mitochondrial lysyl-tRNA synthetase, pre-MSK, and aminoacylation by the cytoplasmic lysyl-tRNA synthetase, KRS, appears to be a prerequisite for this binding. The second lysine isoacceptor tRNAmnmLys5s2UUU {where 5-[(methylamino)-methyl]-2-thiouridine is mnm5s2U} is exclusively localized in the cytoplasm. To study import determinants within the tRNACUULys molecule, we introduced a panel of replacements in the original sequences of the imported and nonimported lysine tRNAs that correspond to domains or individual residues that differ between these two isoacceptors. The mutant transcripts were tested for import, aminoacylation, and binding to pre-MSK. Import and aminoacylation efficiencies correlate well for the majority of mutant transcripts. However, some poorly aminoacylated transcripts were rather efficiently imported. Surprisingly, these transcripts retained binding capacity to pre-MSK. In fact, all imported transcripts retained pre-MSK binding capacity but nonimported versions did not, suggesting that this binding, rather than aminoacylation, is essential for import. Substitution of the anticodon arm of tRNACUULys with that of tRNAmnmLys5s2UUU abolished import without affecting aminoacylation. A version of tRNAmnmLys5s2UUU with an anticodon CUU was efficiently imported in vitro and was also found to be imported in vivo. This implies that the anticodon arm, especially position 34, is important for recognition by the import machinery. A nicked tRNACUULys transcript is still imported but its import requires reannealing of the two tRNA moieties, which implies that tRNACUULys is imported as a folded molecule.
Mitochondrial import of cytoplasmic tRNAs has been found in a wide range of organisms: plants, fungi, protozoa, and mammals (1–3). The yeast Saccharomyces cerevisiae imports a single nucleus-made tRNA, tRNACUULys (tRK1). This is one of two cytoplasmic lysine isoacceptors, the second one, tRNAmnmLys5s2UUU (tRK2) being localized exclusively in the cytoplasm (4, 5). A third yeast lysine tRNA, tRK3, is encoded by the mitochondrial DNA and its localization is restricted to the mitochondrial compartment. tRK1 is unequally distributed between the cytoplasm (95%) and the mitochondrial matrix (5%). Its mitochondrial import is energy- and protein-dependent and requires intact preprotein import machinery and the presence of the precursor form of the mitochondrial lysyl-tRNA synthetase (pre-MSK) (3, 6–8). Both the natural tRK1 and an in vitro-synthesized tRK1 transcript can be selectively imported into isolated mitochondria (9). The tRNA is probably imported in its aminoacylated form, and aminoacylation is a prerequisite for import of natural tRK1 and its in vitro transcript (3, 8, 9). Pre-MSK can form stable ribonucleoprotein complexes with tRK1, but only with its aminoacylated form (8). Therefore, aminoacylation of the tRNA might be necessary for interaction with its carrier protein, pre-MSK.
tRNA sequence requirements for mitochondrial import have recently been studied by in vivo experiments in Tetrahymena (10, 11) and trypanosomatids (12, 13). The anticodon in Tetrahymena tRNAGln and the D loop in Leishmania tRNAIle were found to determine the import. However, the lack of data on cytosolic factors directing tRNA import in these organisms and the difficulty to express some tRNA constructs in vivo delayed the understanding of molecular mechanisms of tRNA import. In yeast, the availability of an in vitro import system and the existence of a second nonimported cytoplasmic tRNALys offered an opportunity to use the in vitro approach for investigating possible import determinants within the tRK1 sequence.
Herein we present in vitro and in vivo import data for a collection of tRNALys variants that demonstrate that the anticodon arm of tRK1, especially position 34, and the acceptor stem contain determinants for its import selectivity. We also show that import requires binding of the mutant tRNAs to pre-MSK. In most cases, aminoacylation was found to be necessary for efficient pre-MSK recognition, but some mutations enable tRNALys variants to bind pre-MSK in their deacylated form. Finally, we present data implying that tRK1 is translocated across mitochondrial membranes as a folded molecule.
MATERIAL AND METHODS
Strains, Cloning, and Mutagenesis.
S. cerevisiae YPH499 (a,ρ+,ura3–52,lys2–801amber,ade2–101ochre,trp1-Δ63,his3-Δ200,leu2-Δ1) (14) was used for isolation of mitochondria and in vivo expression of tRK2CUU and tRK1UUU-7. Strain ΔMOM19irv (a,ρ+,mom19∷URA3,ade2–101ochre, his3-Δ200, leu2-Δ1,ura3–52,trp1-Δ63,lys2–801amber) (15) harbors plasmid pG11T6 containing the MSK1 gene (16). This strain, which is deficient in mitochondrial preprotein import and accumulates cytoplasmic precursors, was used for preparation of import-directing proteins and a pre-MSK-enriched fraction. Escherichia coli DH5αF′ and CJ236 were used for cloning and Kunkel mutagenesis, respectively (17, 18).
Cloning, isolation, analysis, and sequencing of recombinant DNA were performed by standard methods (19). The tRK1 gene from plasmid pY109 (20) provided by H. Feldmann, was subcloned by PCR into the BamHI site of M13mp19 by using two oligonucleotides. The 5′ oligonucleotide contained a T7 RNA polymerase promoter for in vitro transcription and the 3′ oligonucleotide contained a BstNI site to recreate the 3′ terminal CCA sequence upon in vitro transcription. The tRK2 gene from plasmid pFD17 (21), provided by G. Fink, was subcloned in a similar way but the first residue of the tRNA coding sequence T1 was changed into G to allow efficient in vitro transcription and, accordingly, residue A72 was changed into C72 to restore base pairing at the end of the acceptor stem. In addition, the 23-bp intron of the tRK2 gene was removed by PCR deletion mutagenesis. For in vivo expression of tRK2CUU, the entire coding sequence with its intron and 50 bp of 5′ and 3′ flanking sequences was PCR-cloned into the BamHI site of plasmid pRS416 (22) and mutagenized to yield pTRK2-CUU. Oligonucleotide-directed mutagenesis was performed by the method of Kunkel (18) on single-stranded DNAs. For in vivo expression of the tRK1UUU-7 variant, a 300-bp fragment containing the tRK1 gene and flanking sequences was PCR-cloned from the pY109 plasmid (20) into the BamHI site of pRS416 and oligonucleotide-mutagenized to yield pTRK1–7.
tRNAs, in Vitro Transcription and Aminoacylation.
Total yeast tRNA was prepared by phenol extraction. tRK1 and tRK2 were purified by reverse-phase chromatography followed by preparative gel electrophoresis and verified by partial sequencing and oligonucleotide hybridization. For isolation of mitochondrial tRNA, partially purified organelles were treated with a mixture of RNases (5) followed by sucrose gradient centrifugation, and mitochondrial RNA was extracted by a hot phenol treatment in the presence of 1% SDS and 0.05% diethyl pyrocarbonate. tRNAsLys were detected in cytoplasmic and mitochondrial tRNA preparations by either dot hybridization (23) or a modified Northern blot procedure (24) using the following 5′-end 32P-labeled oligonucleotides probes: 5′-CCTAACCTTATGATTAAG-3′ (tRK1), 5′-ACCTTTCGGTTAAAAGCCGAA-3′ (tRK1UUU-7), 5′-CCTGACATTTCGGTTAAA (tRK2), 5′-GACATTTCGGTTAAGAGC-3′ (tRK2CUU), and 5′-AACCATGGGTTGGTTAAA-3′ (tRK3).
For in vitro transcription, 5–10 μg of BstNI-linearized phage replicative form DNAs (or BstNI-treated PCR products) were transcribed with T7 RNA polymerase (Promega) by a standard procedure (25). Transcripts were gel-purified, dephosphorylated with calf intestine phosphatase (Boheringer Mannheim) in the presence of RNAsine (Promega), and labeled with [γ-32P]ATP to a specific activity of 106 cpm/μg by using T4 polynucleotide kinase. Gel-repurified transcripts were treated by a heat denaturation–renaturation cycle in the presence of 0.5 mM MgCl2. Aminoacylation was done with either purified yeast cytoplasmic lysyl-tRNA synthetase (KRS) or a mixture of yeast aminoacylating enzymes as described (26–29).
Import-Directing Proteins, Aminoacyl-tRNA Synthetases, and Antibodies.
Import-directing proteins were prepared from ΔMOM19irv (pG11T6) cells disrupted with glass beads in breakage buffer [10 mM Hepes, pH 6.8/50 mM KCl/5 mM DTT/1 mM EDTA/protease inhibitor mixture (Boheringer Mannheim)/10% glycerol]. After removal of cell debris, nucleic acids were precipitated by polyethylenimine treatment (26), and proteins were collected from the supernatant by (NH4)2SO4 precipitation (70% of saturation) and dialyzed against breakage buffer with 50% glycerol. Import-directing protein can be stored at −20°C for several months.
For preparation of a pre-MSK-enriched cytosolic protein fraction, ΔMOM19irv cells overexpressing the MSK1 gene from plasmid pG11T6 were sonicated for 30 s in 10 mM Tris⋅HCl, pH 7.0/50 mM KCl/5 mM DTT/1 mM EDTA/10% glycerol in the presence of a protease inhibitor mixture. After removal of cell debris, the supernatant was adjusted to 150 mM KCl, loaded onto a DE-52 column equilibrated in 10 mM Tris⋅HCl, pH 7.0/150 mM KCl/1 mM EDTA/10% glycerol, and the flow-through fraction was collected. Proteins were desalted and adjusted to 10 mM Hepes, pH 6.8/20 mM KCl/2 mM DTT/50% glycerol by Sephadex G-75 gel-filtration (30) and stored at −20°C. The obtained cytosolic fraction was enriched in pre-MSK and contained only minor amounts of mitochondrial proteins (as verified by Western blot analysis).
Purified KRS was a gift of M. Mirande (Centre National de La Recherche Scientifique, Gif-sur-Yvette). The mixture of yeast aminoacyl-tRNA synthetases was prepared as described (28, 29). Rabbit anti-MSK polyclonal antibodies were provided by A. Tzagoloff (Columbia University).
tRNA Import and Pre-MSK Binding.
Mitochondria isolation and tRNA import assays were as described (6–9). In all cases in vitro-synthesized transcripts were incubated with KRS and lysine in a standard aminoacylation mixture (26) before the import assay. Quantitation of tRNA import was done by scanning autoradiographs on a PhosphorImager (Fuji). Import efficiency of the preaminoacylated tRK1 transcript was about 0.5 pmol of tRNA per mg of mitochondrial protein. For pre-MSK binding, aliquots of 5′-32P-labeled transcripts (or tRNAs) were mixed with 5 μg of a pre-MSK-enriched fraction in 50 μl of the standard import mixture (without mitochondria) and incubated for 10 min at 30°C. The mixture was centrifuged to remove insoluble aggregates and 50 μl of lysis buffer (0.1% Nonidet P-40/0.1 M Tris⋅HCl, pH 7.5), and 1 μl of an appropriate dilution of anti-MSK antibodies was added to the supernatant. After incubation for 1 h on ice, 10% (vol/vol) protein A-Sepharose (Pharmacia) was added and the mixture was shaken for 1 h at 4°C. Sepharose beads were collected by brief centrifugation and washed with lysis buffer, and the radioactivity determined by measuring Cerenkov radiation. As a control, the same assay was done with preimmune antibodies. On average, values of nonspecific binding observed with preimmune antibodies were less than 5% of the value of pre-MSK binding to aminoacylated tRK1 detected with anti-MSK antibodies.
RESULTS
In Vitro Import of Mutated tRNALys Transcripts.
We have previously shown that an in vitro transcript of tRK1, once aminoacylated, is a good substrate for mitochondrial import in vitro (9). The transcript (which is not expected to carry modified nucleosides) is an even better import substrate than the corresponding fully modified natural tRNA. To identify import determinants within the tRK1 sequence, we have used oligonucleotide mutagenesis coupled with in vitro transcription to generate a set of mutant transcripts and studied their in vitro import efficiencies after aminoacylation with KRS. The sequences of tRK1 and tRK2 differ in 21 positions located in the acceptor arm (including the “discriminator” base at position 73), D arm, anticodon arm, and variable loop (Fig. 1). Because tRK1 is imported but tRK2 is not (4, 5, 7–9), we reasoned that determinants for import selectivity should lie within regions of sequence divergence between the two isoacceptors. We have therefore introduced a panel of replacements in the original tRK1 and tRK2 sequences that correspond to domains (or individual residues) differing between the two isoacceptors.
Figure 1.
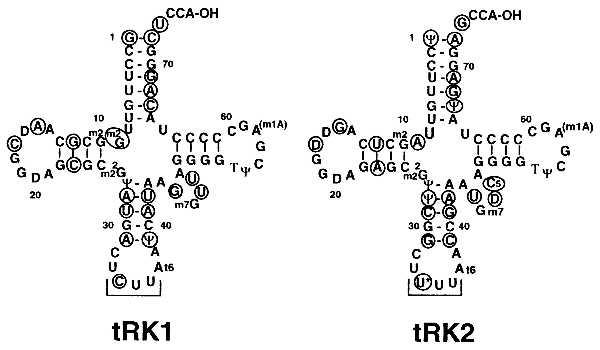
Cloverleaf structures of tRK1 and tRK2 (based on refs. 31 and 32). Residues that differ between the two isoacceptors are circled; the anticodons are underlined.
Import properties of the mutant tRK1 and tRK2 transcripts are summarized in Table 1. For tRK2, it was not possible to obtain a transcript with the wild-type sequence by in vitro transcription with T7 RNA polymerase because the original coding sequence begins with a thymidine. Therefore, the pair Ψ1-A72 of wild-type tRK2 was replaced by G1-C72. The resulting tRK2G1C72 transcript was found to be slightly imported in the in vitro import assay, whereas the natural tRK2 was not (see Fig. 2 and Table 1, transcript 30). Import of tRK2G1C72 may be due to either the replacement of the first base pair in the acceptor stem or the absence of modified nucleosides in the transcript that may have a negative effect onto import of the natural tRK2. A change of the tRK2G1C72 anticodon from UUU to CUU (transcript 2) resulted in a significant increase of the import efficiency (Fig. 2 and Table 1). In fact, transcript 2 was even a better import substrate than the tRK1 transcript. This suggests that residue C34 is at least one of the determinants for import selectivity of tRK1. However, the sequence context around this position appears also to be important because a version of tRK1 with the anticodon UUU (transcript 8) was still imported, though with a lesser efficiency than tRK1, but replacement of the entire anticodon arm of tRK1 with that of tRK2 (transcript 7) resulted in a total loss of import capacity (Fig. 2 and Table 1).
Table 1.
Mutant tRK1 and tRK2 transcripts and their import efficiencies
| Transcript | tRNA | Original bases | New bases | Import efficiency, % |
|---|---|---|---|---|
| 1 | tRK1 | No change | 100 | |
| 40 | tRK1 | C67, A68, G69 | U67, G68, A69 | <5 |
| 4 | tRK1 | C67, A68, G69, C72, U73 | U67, G68, A69, A72, G73 | 50–75 |
| 29 | tRK1 | U73 | G73 | 75–100 |
| 25 | tRK1 | m2G9 | A9 | 20–30 |
| 26 | tRK1 | m2G9, G12, A15, C17, C23 | A9, U12, G15, U17, A23 | 50–75 |
| 8 | tRK1 | C34 | U34 | 50–75 |
| 6 | tRK1 | A28, U29, A31, Ψ39, A41, U42 | U28, C29, G31, C39, G41, A42 | 75–100 |
| 7 | tRK1 | A28, U29, A31, C34, Ψ39, A41, U42 | U28, C29, G31, U34, C39, G41, A42 | <5 |
| 5 | tRK1 | G45, U48 | U45, C48 | 75–100 |
| 30 | tRK2 | Ψ1, A72 | G1, C72 | 5–10 |
| 2 | tRK2 | Ψ1, U34, A72 | G1, C34, C72 | 150–200 |
| 93 | tRK2 | Ψ1, A72, G73 | G1, C72, U73 | 75–100 |
| 33 | tRK2 | Ψ1, U34, A72, G73 | G1, C34, U72, U73 | <5 |
Import of transcript 1 was taken as 100%
Figure 2.
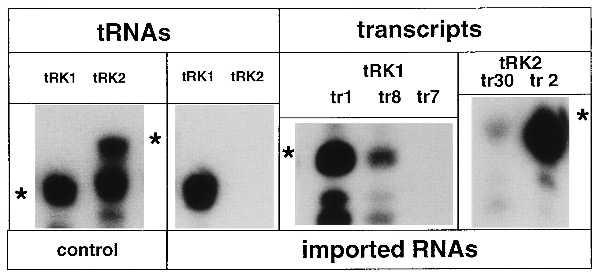
Import of mutant tRNALys transcripts into isolated mitochondria. Autoradiographic detection of 5′-32P-labeled tRK1, tRK2, and in vitro transcripts after separation on a denaturing polyacrylamide gel. In vitro import assays, preparation of mitoplasts, and extraction of imported RNAs were as described (6) with modifications (9). Three pmoles of 32P-labeled RNA (tRNAs or transcripts) were used in each assay. Lanes: control, labeled tRK1 and tRK2, used as markers; imported RNAs, detection of RNase-protected RNAs extracted from mitoplasts after the import reaction; tr1, tr2, tr7, tr8, tr30, in vitro transcripts numbers (see Table 1). Asterisks indicate the positions of the nondegraded RNAs.
Another region of tRK1 that might also be relevant for its import efficiency is the acceptor stem. Indeed, replacement of bases 67–69 of tRK1 by the corresponding residues of tRK2 (transcript 40), though not disturbing helix formation, significantly inhibits the import (Table 1). However, the same replacement completed by a change of the discriminator base U73 to G and unpairing of the first acceptor helix pair (C72 to A) restored import (Table 1, transcript 4). The importance of the first base pair of the acceptor stem seems to depend on the sequence context, because replacement of the strong G⋅C pair in the well-imported transcript 2 by a weak G⋅U pair and a change of the discriminator base (G73 to U) dramatically reduces the import efficiency (Table 1, transcript 33). It is unlikely that this effect is due to the change of the discriminator base, because such a change alone conferred efficient import capacity to a poorly imported variant of tRK2 (Table 1, compare transcript 93 versus transcript 30).
Transcript 25, in which there is only a single m2G9 (of tRK1) → A replacement, is still imported but with a 2–3 times lower efficiency than the nonmutated transcript. However, a m2G9 → A change and replacement of the complete D arm of tRK1 with that of tRK2 (transcript 26) had little or no effect onto import efficiency. These results suggest that the D arm of tRK1 does not contain determinants for its import selectivity. The same may be said about the variable loop whose replacement (in transcript 5) does not affect import.
In Vivo Import of Mutant tRNAsLys.
The above results showed that a U → C change in the first position of the anticodon of a tRK2 transcript conferred efficient import in our in vitro system. To test whether this is also the case in vivo, we constructed a version of the tRK2 gene with the anticodon CUU (tRK2CUU) and expressed it from a centromeric plasmid, pTRK2-CUU. The construct was made on the basis of one of the seven chromosomal tRK2 gene copies, cloned in plasmid pFD17 (21). The tRK2 gene copies contain all an identical intron of 23 bases (Fig. 3a). The U34 → C change should not interfere with correct folding of the pre-tRNA but replaces a weak U⋅G base pair by a strong C⋅G pair (Fig. 3b). The mutated gene was found to be expressed in vivo and to give rise to a tRNA molecule with a slightly lower electrophoretic mobility than the natural tRK2 (Fig. 3c). The altered mobility might be due to a difference in base modification (absence of mnm5s2U in position 34) but not to the absence of intron splicing, because the unspliced precursor should have a significantly lower gel mobility and would not have been detected with the oligonucleotide probe used (which corresponds to the anticodon arm and variable loop of tRK2CUU). Cell fractionation and subsequent hybridization with a tRK2CUU-specific oligonucleotide probe revealed that a small fraction of the mutated tRNA is associated with the mitochondrial matrix (Fig. 3 c and d). A tRK2UUU-specific probe, used as a negative control, did not detect the import. The level of expression of tRK2CUU was comparable to that of tRK2 and approximately 2 times lower than that of tRK1 (Fig. 3d). The amount of imported tRK2CUU is about 10 times lower than that of imported tRK1, which suggests that tRK2CUU has an approximately 5 times lower import efficiency. The fact that a single base change in the anticodon of a normally nonimported tRNALys results in its import in vivo is in agreement with our in vitro results and demonstrates that residue C34 is important for import selectivity. However, the weakness of the import signal of tRK2CUU indicates that the first position of the anticodon is not the only position affecting import efficiency.
Figure 3.
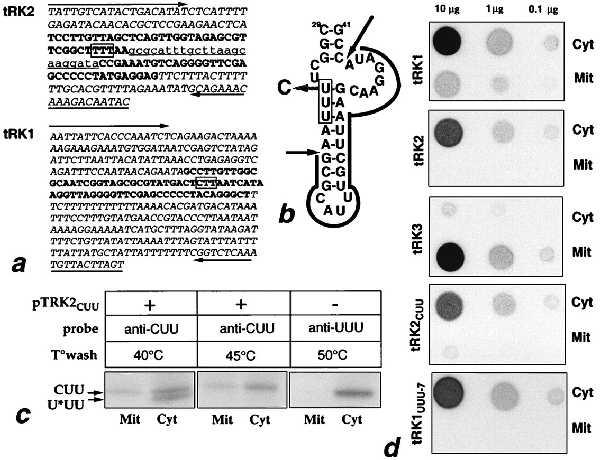
In vivo import of tRK2cuu. (a) Sequences of the tRK2 (GenBank accession no. U40828; chromosome XVI) and tRK1 (20) genes; arrows indicate sequences corresponding to oligonucleotides used for the subcloning of the two genes; 5′ and 3′ flanking regions are in italic type; underlined lowercase type shows intron sequences of the tRK2 gene; boldface type shows the “coding” sequence; anticodons are boxed. (b) Predicted secondary structure of the intron containing part of the tRK2 gene; intron sequences are outlined with plain arrows indicating sites of cleavage; the shadowed arrow indicates the U34 to C replacement in tRK2CUU; the anticodon is boxed. (c) Northern blot hybridization of total (Cyt) and mitochondrial (Mit) tRNAs from wild-type and pTRK2-CUU transformed cells, using oligonucleotide probes specific for tRK2CUU (anti-CUU) and tRK2UUU (anti-UUU). Temperatures of washes are indicated above the autoradiographs. At 40°C, the anti-CUU probe recognizes both mutant and wild-type tRK2, whereas at 45°C, only tRK2CUU is detected. Arrows indicate the tRK2CUU (CUU) and tRK2mnmLys5s2UUU (U*UU) specific bands; 5 μg of total and 15 μg of mitochondrial tRNA were loaded in each slot. (d) Dot hybridization of total (Cyt) and mitochondrial (Mit) tRNA with tRK1, tRK2, tRK3, tRK1UUU-7, and tRK2CUU-specific probes; the amount of dotted RNA is indicated above the autoradiographs.
To verify if changes in the tRK1 anticodon arm that block the import in vitro have the same effect in vivo, we constructed a tRK1 gene variant, referred to as tRK1UUU-7, containing the tRK2 anticodon arm in the tRK1 backbone. Although well expressed in vivo, tRK1UUU-7 was not found to be imported into mitochondria (Fig. 3d). This result is in perfect agreement with our in vitro import results (see Table 1, transcript 7) and confirms that the anticodon arm of tRK1 contains residues critical for import selectivity.
Aminoacylation and Pre-MSK Binding Properties of Mutant Transcripts.
We have previously shown that aminoacylation of tRK1 by the cytoplasmic lysyl-tRNA synthetase (KRS) is a prerequisite for its mitochondrial import (8, 9). To see whether the same holds true for in vitro-synthesized mutant transcripts, we have determined their aminoacylation parameters by using purified KRS. Table 2 shows that among the 13 mutant transcripts studied, transcripts 2, 5, 6, 7, 8, and 29 showed aminoacylation efficiencies comparable to the nonmutated tRK1 transcript, whereas 6 other transcripts, transcripts 4, 40, 33, 93, 25, and 26, showed efficiencies decreased by 30–50 times. All the transcripts with low aminoacylation levels contained mutations in either the acceptor stem (transcripts 4, 40, 33, and 93) or the D-arm region (transcripts 25 and 26) and in all cases the Km remained almost unchanged and the Vmax was strongly decreased (Table 2).
Table 2.
Aminoacylation and pre-MSK-binding capacities of tRK1/tRK2 mutant transcripts
| RNA | Aminoacylation with [3H]lysine
|
Pre-MSK binding, %
|
|||
|---|---|---|---|---|---|
| Km, μM | Vmax, pmol/min | Vmax/Km | + KRS | − KRS | |
| 1 | 2.2 | 250 | 1.00 | 85 | 20 |
| 40 | 1.8 | 5 | 0.03 | 10 | 5 |
| 4 | 6.3 | 15 | 0.02 | 5 | 80 |
| 29 | 6.7 | 290 | 0.40 | 75 | 45 |
| 25 | 4.8 | 20 | 0.03 | 15 | 20 |
| 26 | 1.4 | 5 | 0.04 | 20 | 70 |
| 8 | 2.3 | 115 | 0.44 | 70 | 85 |
| 6 | 2.1 | 90 | 0.35 | 85 | 20 |
| 7 | 2.2 | 85 | 0.33 | 10 | <5 |
| 5 | 3.0 | 75 | 0.20 | 100 | 35 |
| 30 | 1.2 | 15 | 0.10 | 20 | 20 |
| 2 | 1.5 | 450 | 2.60 | 100 | 40 |
| 93 | 5.9 | 10 | 0.02 | 45 | 100 |
| 33 | 4.2 | 10 | 0.02 | 30 | <5 |
| tRK1 | 1.4 | 2,600 | 16.20 | 100 | 5 |
| tRK2 | 1.5 | 2,500 | 14.50 | <5 | 15 |
Pre-MSK binding of aminoacylated tRK1 was taken as 100%; values of nonspecific binding detected with preimmune antibodies have been subtracted. + KRS, in preaminoacylated form (preincubation with KRS and lysine); − KRS, without preincubation with KRS.
In the majority of cases, import efficiencies of the mutant transcripts can be correlated to their respective aminoacylation efficiencies. For example, transcripts 2, 5, 6, 8, and 29 show both efficient import and high aminoacylation levels. On the other hand, transcripts 33 and 40, which are poorly aminoacylated, are not imported, and transcript 30, which has an intermediate aminoacylation rate, also has an intermediate import efficiency. Surprisingly, among the poorly aminoacylated transcripts at least four (transcripts 4, 25, 26, and 93) are imported rather efficiently. It is therefore possible that these transcripts can be imported without being aminoacylated by KRS.
Our previous results indicated that aminoacylation of tRK1 might be necessary to allow interaction with pre-MSK, which is likely to act as a carrier for mitochondrial translocation of the tRNA (8). We therefore tested all our in vitro constructs for their abilities to bind pre-MSK in an immunoassay (Table 2). A clear correlation was found between pre-MSK binding properties and import efficiencies; i.e., all transcripts that efficiently bind to pre-MSK are imported, whereas those that do not bind are not imported, irrespective of their aminoacylation abilities by KRS. For example, the capacities of transcripts 4, 26, and 93 to bind pre-MSK in their nonacylated form (i.e., in absence of KRS) could explain their ability to be imported. This implies that, contrary to the natural tRK1, some in vitro-synthesized mutant transcripts can be recognized by pre-MSK and imported without previous aminoacylation by KRS. Conversely, the poor binding to pre-MSK of transcript 7 (in which the anticodon arm of tRK1 was replaced by that of tRK2) in both absence or presence of KRS explains its lack of import. This implies that some residues in the anticodon region of tRK1 (including position 34) are directly involved in interaction with pre-MSK.
In Vitro Import of a Nicked tRK1 Transcript.
The mechanism of translocation of a tRNA across mitochondrial membranes, though shown in yeast to depend on the mitochondrial preprotein translocation machinery and on cytoplasmic import-directing factors (7, 8), remains unclear. For example, it is not known whether the tRNA molecule is unfolded during translocation across mitochondrial membranes or is imported as a folded molecule. To test these possibilities, we constructed two truncated forms of tRK1 (Fig. 4a). Fragment A represents the first 52 bases of the tRNA and fragment B represents its last 24 residues including the 3′ terminal CCA. When annealed to one another by a cycle of heat denaturation and renaturation in the presence of MgCl2, the A and B moieties form a duplex with a mobility on a nondenaturing gel comparable to that of the noninterrupted tRK1 transcript (Fig. 4b).
Figure 4.
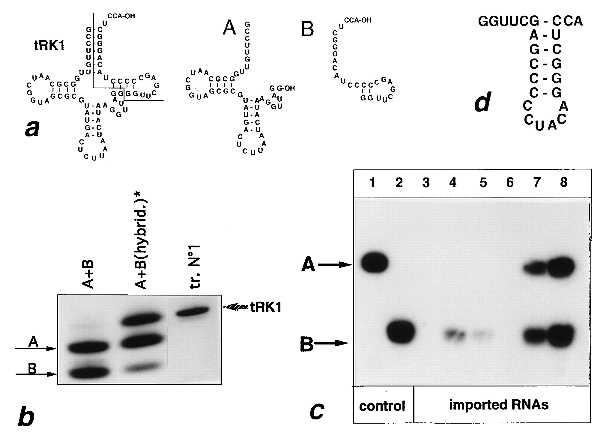
In vitro import of a nicked tRK1 transcript. (a) Secondary structures of tRK1 and of the truncated A and B fragments. (b) Nondenaturing gel electrophoresis of fragments A and B: simple mixture of the two fragments (lane A+B), reannealed A+B fragments [lane A+B(hybrid.)], renatured tRK1 transcript (lane tr.N°1). (c) Autoradiographic detection on a denaturing polyacrylamide gel of imported A and B fragments (lanes 3–6) and of the nicked tRK1 transcript (lanes 7 and 8). Before the import reaction, the truncated fragments were aminoacylated with a mixture of yeast synthetases and a mixture of amino acids and the reannealed fragments with KRS and lysine. The amount of labeled A and B fragment used per assay was 6 pmol of fragment A (lanes 1–8) and either 12 pmol (lanes 1–6 and 8) or 3 pmol (lane 7) of fragment B. Control, labeled fragments A (lane 1) and B (lane 2) used as markers; imported RNAs, in vitro import assays of fragment A (lane 3), fragment B (lane 4), fragment B in the presence of either 400 pmol (lane 5) or 800 pmol (lane 6) of unlabeled tRK1 and of the hybridized A+B fragments (lanes 7 and 8). (d) Possible secondary structure of the B fragment.
In vitro import of fragments A and B into isolated mitochondria were performed after preaminoacylation. The 5′ fragment alone was not detectable within mitochondria but, surprisingly, the 3′ fragment was slightly imported (Fig. 4c, lanes 3 and 4). Import of fragment B strongly decreased in the presence of a 33-fold molar excess of unlabeled tRK1 and was no longer detectable in the presence of a 66-fold excess (Fig. 4c, lanes 5 and 6). We have previously shown that a similar excess of unlabeled tRK1 totally blocks the import of labeled tRK1 (9). This suggests that import of the B fragment and of tRK1 proceed by a similar mechanism. Furthermore, aminoacylated fragment B showed low but detectable binding to pre-MSK, but fragment A lacked pre-MSK binding capacity (Table 3). This suggests that, as for tRK1, the import of the B fragment is mediated by pre-MSK. Transcript B could not be aminoacylated by KRS and lysine but was charged by a mixture of labeled amino acids in the presence of a mixture of synthetases (Table 3), suggesting that it is imported in a misacylated form. Misacylation of fragment B may be due to formation of a potentially stable stem–loop structure with a 3′ terminal CCA (Fig. 4d). This structure partially resembles the valine minihelix that can be aminoacylated by the yeast ValRS (33). It is possible that the B fragment can better mimic a substrate for pre-MSK binding than the A fragment, which although it contains the anticodon arm, cannot adopt a tRNA-like structure.
Table 3.
Import, aminoacylation, and pre-MSK binding properties of truncated and nicked tRK1 transcripts
| RNA | Import, % | Aminoacylation, %
|
Pre-MSK binding, % | |
|---|---|---|---|---|
| With aa mix | With lysine | |||
| tRK1 transcript | 100 | 100 | 100 | 100 |
| A fragment | <5 | <1 | <1 | 6 |
| B fragment | 24 | 15 | <1 | 18 |
| A + B | 28* | 17 | <1 | ND |
| A + B (hybrid) | 75 | 35 | 25 | ND |
Data for import are percent of transcript 1 import efficiency (average of three experiments). Data for aminoacylation are percent transcript 1 maximal aminoacylation. The pre-MSK binding of the aminoacylated tRK1 transcript was taken as 100% (values of nonspecific binding detected with preimmune antibodies have been subtracted); the tRK1 transcript was aminoacylated with lysine and fragments A and B with a mixture of amino acids (aa mix). ND, not determined.
Only fragment B is imported if fragments A and B are mixed without annealing.
After annealing of fragments A and B, the duplex can be aminoacylated by KRS and lysine (Table 3) and becomes imported with a significantly higher efficiency than the B fragment alone (Fig. 4c and Table 3). The amount of imported A fragment is dependent upon the amount of added B fragment and the two fragments are imported in an equimolar ratio, implying that they are coimported as a duplex molecule. This result demonstrates that a nicked tRK1 transcript can be imported in vitro, which suggests that in vivo tRK1 is likely to be translocated across mitochondrial membranes as a folded molecule.
DISCUSSION
We have studied, by in vitro and in vivo experiments, structural features of tRK1 that are relevant to its mitochondrial import. The data presented indicate that the anticodon region of tRK1 contains determinants for its import selectivity. In fact, replacement of the entire anticodon arm of tRK1 by the one of tRK2 totally blocked the import, both in vitro and in vivo. The most important position in this region appears to be the wobble position of the anticodon (C34), because introduction of C34 into a tRK2 transcript directs its efficient import in vitro. This conclusion is strengthened by the fact that this single-base change in the tRK2 anticodon conferred import in vivo to this normally nonimported tRNA. However, in vivo import of the tRK2CUU version is low when compared with tRK1. In fact, in vitro experiments revealed that position 34 is not only important by itself but also in its context, because no significant loss of import efficiency was found with a tRK1UUU transcript (transcript 8). We suggest that in tRK2, the uridine modification {5-[(methylamino)-methyl]-2-thiouridine, mnm5s2U} in position 34 may act as an antideterminant for import, because a tRK2 transcript is weakly imported but natural (fully modified) tRK2 is not. The mitochondrial import apparatus seems to be able to discriminate between mnm5s2U and cytidine in position 34 of the tRNA molecule but not (or, at least, with a much lesser efficiency) between unmodified uridine and cytidine in this position.
A second domain of the tRK1 molecule which might be important for its import selectivity is the acceptor stem, because replacement of residues 67–69 of tRK1 with those of tRK2 (transcript 40) blocks the import. However, this effect can be reversed by additional changes in positions 72 and 73 (transcript 4), which restore the import. Such a differential effect of mutations in the acceptor stem onto import can be explained by differences in pre-MSK-binding properties of the two transcripts, due to conformational changes in the acceptor helix (see below).
Our previous results indicated that pre-MSK (which is essential for tRK1 import) can form a stable complex with tRK1 but only with its aminoacylated form. We therefore hypothesized that aminoacylation is essential for binding to pre-MSK, which in turn would serve as a carrier for mitochondrial transport of the tRNA (3, 8, 9). We show herein that pre-MSK binding is indeed of critical importance for the import of mutant tRNA transcripts, because all transcripts with high pre-MSK binding capacities are imported well (i.e., transcripts 1, 2, 4, 5, 6, 8, 26, 29, and 93) but those with low binding are poorly imported (i.e., transcripts 7, 30, 33, and 40). However, comparison of aminoacylation capacities of mutant tRNAs showed that aminoacylation is needed for pre-MSK binding and mitochondrial import of only a portion of the mutant tRNAs. The well-imported transcripts 1, 2, 5, 6, and 29 show higher pre-MSK binding in their aminoacylated form. The poorly imported transcripts 30, 33, and 40 (which are also poorly aminoacylated) show low pre-MSK binding capacities. In contrast, transcripts 4, 26, and 93 (which are poorly aminoacylated) retain high pre-MSK binding capacities in their deacylated form and are well imported.
We suggest that the role of aminoacylation in import is to induce a conformational change in tRK1 to facilitate its binding to pre-MSK. As a matter of fact, the role of aminoacylation as a factor of conformational change in tRNA was recently described for the tRNA-like domain of several viral RNAs (34). It was shown that the role of aminoacylation is to induce productive interaction of the tRNA-like domain with replication factors, although the nature of the amino acid at the 3′ terminus of the viral RNA is not crucial. If we accept the idea that aminoacylation of tRK1 facilitates its binding to pre-MSK, how then can some tRNALys variants (i.e., transcripts 4, 26, and 93) bind to pre-MSK in their deacylated form? It is worth noting that these transcripts either contain mutations in the acceptor stem or have an altered tertiary interaction. Transcript 26, for example, has a disrupted A15-U48 tertiary interaction replaced by a potential G15-U48 pair, which is not found in any known tRNA (35). On the other hand, transcripts 4 and 40, which differ by residues 72 and 73 in the acceptor stem, have low aminoacylation rates but differ greatly in their pre-MSK binding and import capacities. It is therefore possible that some particular alterations in the core of the tRK1 L shape structure or in the acceptor helix might induce conformational changes that can mimic the effect of aminoacylation on tRK1 and allow pre-MSK binding.
We have used artificial import substrates to study the mechanism of translocation of tRK1 across mitochondrial membranes in vitro. It has been shown previously that polynucleotides cross-linked to a signal peptide for preprotein targeting into mitochondria can be imported into isolated mitochondria (36, 37). Likewise, if we assume that tRK1 import involves formation of a covalent link with its carrier protein (presumably pre-MSK), then folding of the tRNA should not be required for its mitochondrial translocation. However, our results with a nicked tRK1 transcript are clearly in favor of the importance of the tRNA cloverleaf structure, because the imported 3′ moiety of tRK1 can direct mitochondrial transport of the 5′ portion of the tRNA, which alone is not imported. This suggests translocation of the tRNA as a folded molecule.
Acknowledgments
We thank H. Kazakova for obtaining mutant 93; A. Tzagoloff, N. Pfanner, G. Fink, H. Feldmann, and M. Mirande for providing strains, plasmids, enzymes, and antibodies; and L. Brubacher for technical assistance. This work was supported by the Centre National de la Recherche Scientifique, the Université Louis Pasteur, the Association Française contre les Myopathies, the Moscow State University, and the Russian Foundation for Basic Research (Grant 96.04.48027). N.S.E. was supported by an European Molecular Biology Organization fellowship for Eastern molecular biologists and by the Université Louis Pasteur.
ABBREVIATIONS
- MSK
mitochondrial lysyl-tRNA synthetase
- KRS
cytoplasmic lysyl-tRNA synthetase
References
- 1.Schneider A. Trends Cell Biol. 1994;4:282–286. doi: 10.1016/0962-8924(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 2.Dietrich A, Small I, Cosset A, Weil J-H, Maréchal-Drouard L. Biochimie. 1996;78:518–529. doi: 10.1016/0300-9084(96)84758-4. [DOI] [PubMed] [Google Scholar]
- 3.Tarassov I A, Martin R P. Biochimie. 1996;78:502–510. doi: 10.1016/0300-9084(96)84756-0. [DOI] [PubMed] [Google Scholar]
- 4.Martin R P, Schneller J M, Stahl A J C, Dirheimer G. Nucleic Acids Res. 1977;4:3497–3510. doi: 10.1093/nar/4.10.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin R P, Schneller J M, Stahl A J C, Dirheimer G. Biochemistry. 1979;18:4600–4605. doi: 10.1021/bi00588a021. [DOI] [PubMed] [Google Scholar]
- 6.Tarassov I A, Entelis N S. Nucleic Acids Res. 1992;20:1277–1281. doi: 10.1093/nar/20.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarassov I A, Entelis N S, Martin R P. J Mol Biol. 1995;245:315–323. doi: 10.1006/jmbi.1994.0026. [DOI] [PubMed] [Google Scholar]
- 8.Tarassov I A, Entelis N S, Martin R P. EMBO J. 1995;14:3461–3471. doi: 10.1002/j.1460-2075.1995.tb07352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Entelis N S, Krasheninnikov I A, Martin R P, Tarassov I A. FEBS Lett. 1996;384:38–42. doi: 10.1016/0014-5793(96)00259-1. [DOI] [PubMed] [Google Scholar]
- 10.Rusconi C P, Cech T R. EMBO J. 1996;15:3286–3295. [PMC free article] [PubMed] [Google Scholar]
- 11.Rusconi C P, Cech T R. Genes Dev. 1996;10:2870–2880. doi: 10.1101/gad.10.22.2870. [DOI] [PubMed] [Google Scholar]
- 12.Lima B D, Simpson L. RNA. 1996;2:429–440. [PMC free article] [PubMed] [Google Scholar]
- 13.Hauser R, Schneider A. EMBO J. 1995;14:4212–4220. doi: 10.1002/j.1460-2075.1995.tb00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sikorski R S, Hieter P. Genetics. 1989;122:7–18. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moczko M, Ehrmann B, Gaärtner F, Hönlinger A, Schäfer E, Pfanner N. J Biol Chem. 1994;269:9045–9051. [PubMed] [Google Scholar]
- 16.Gatti D L, Tzagoloff A. J Mol Biol. 1991;218:557–568. doi: 10.1016/0022-2836(91)90701-7. [DOI] [PubMed] [Google Scholar]
- 17.Woodcock D M, Crowther P J, Doherty J, Jefferson S, DeCruz E, Noyez-Weidnez M, Smith S S, Michael M Z, Graham M W. Nucleic Acids Res. 1989;17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunkel T A. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 20.Nelbök P, Feldmann H. Biol Chem Hoppe-Seyler. 1985;366:1041–1051. doi: 10.1515/bchm3.1985.366.2.1041. [DOI] [PubMed] [Google Scholar]
- 21.Del Rey F J, Donahue T F, Fink G. Proc Natl Acad Sci USA. 1982;79:4138–4142. doi: 10.1073/pnas.79.13.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heitzler J, Maréchal-Drouard L, Dirheimer G, Keith G. Biochim Biophys Acta. 1992;1129:273–277. doi: 10.1016/0167-4781(92)90503-r. [DOI] [PubMed] [Google Scholar]
- 24.Wilhelm M L, Baranowski W, Keith G, Wilhelm F X. Nucleic Acids Res. 1992;20:4106. doi: 10.1093/nar/20.15.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampson J R, Uhlenbeck O C. Proc Natl Acad Sci USA. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cirakoglu B, Waller J-P. Eur J Biochem. 1985;149:353–361. doi: 10.1111/j.1432-1033.1985.tb08933.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinez R, Mirande M. Eur J Biochem. 1992;207:1–11. doi: 10.1111/j.1432-1033.1992.tb17012.x. [DOI] [PubMed] [Google Scholar]
- 28.Von der Haar F. Methods Enzymol. 1979;49:257–267. doi: 10.1016/0076-6879(79)59088-0. [DOI] [PubMed] [Google Scholar]
- 29.Ebel J P, Giegé R, Bonnet J, Kern D, Befort N, Bollack C, Fasiolo F, Gangloff J, Dirheimer G. Biochimie. 1973;55:547–564. doi: 10.1016/s0300-9084(73)80415-8. [DOI] [PubMed] [Google Scholar]
- 30.Maréchal-Drouard L, Small I, Weil J-H, Dietrich A. Methods Enzymol. 1995;260:310–327. doi: 10.1016/0076-6879(95)60148-1. [DOI] [PubMed] [Google Scholar]
- 31.Smith C J, Teh H S, Ley A N, D’Obrenan P. J Biol Chem. 1972;248:4475–4485. [PubMed] [Google Scholar]
- 32.Madison J T, Boguslawski S J. Biochemistry. 1974;13:524–527. doi: 10.1021/bi00700a019. [DOI] [PubMed] [Google Scholar]
- 33.Frugier M, Florentz C, Giegé R. Proc Natl Acad Sci USA. 1992;89:3990–2994. doi: 10.1073/pnas.89.9.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giegé R. Proc Natl Acad Sci USA. 1996;93:12078–12081. doi: 10.1073/pnas.93.22.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giegé R, Puglisi J D, Florentz C. Prog Nucleic Acids Res Mol Biol. 1993;45:129–206. doi: 10.1016/s0079-6603(08)60869-7. [DOI] [PubMed] [Google Scholar]
- 36.Vestweber D, Schatz G. Nature (London) 1989;338:170–172. doi: 10.1038/338170a0. [DOI] [PubMed] [Google Scholar]
- 37.Seibel P, Trappe J, Villani G, Klopstock T, Papa G, Richmann H. Nucleic Acids Res. 1995;23:10–17. doi: 10.1093/nar/23.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]