

Abstract
Posttranslational modifications of core histones contribute to driving changes in chromatin conformation and compaction. Herein, we investigated the role of histone deacetylation on the mitotic process by inhibiting histone deacetylases shortly before mitosis in human primary fibroblasts. Cells entering mitosis with hyperacetylated histones displayed altered chromatin conformation associated with decreased reactivity to the anti-Ser 10 phospho H3 antibody, increased recruitment of protein phosphatase 1-δ on mitotic chromosomes, and depletion of heterochromatin protein 1 from the centromeric heterochromatin. Inhibition of histone deacetylation before mitosis produced defective chromosome condensation and impaired mitotic progression in living cells, suggesting that improper chromosome condensation may induce mitotic checkpoint activation. In situ hybridization analysis on anaphase cells demonstrated the presence of chromatin bridges, which were caused by persisting cohesion along sister chromatid arms after centromere separation. Thus, the presence of hyperacetylated chromatin during mitosis impairs proper chromosome condensation during the pre-anaphase stages, resulting in poor sister chromatid resolution. Lagging chromosomes consisting of single or paired sisters were also induced by the presence of hyperacetylated histones, indicating that the less constrained centromeric organization associated with heterochromatin protein 1 depletion may promote the attachment of kinetochores to microtubules coming from both poles.
INTRODUCTION
The successful segregation of chromosomes at mitosis relies on the coordinated execution of cytoskeletal and chromosomal events. The interphase microtubules reorganize to form a bipolar mitotic spindle and the relaxed interphase chromatin orderly compacts into highly condensed rod-like structures. Posttranslational modifications of nucleosome core histones contribute substantially to driving changes in chromatin conformation and compaction. Acetylation on lysine residues on the amino-terminal tails of histones contributes to the formation of a transcriptionally competent environment, by decreasing the affinity of the acetylated N termini for DNA and allowing access of general transcription factors to DNA. Conversely, deacetylated histones are mainly associated with compact chromatin states and transcriptional repression (for reviews, see Grunstein, 1997; Wade et al., 1997; Cheung et al., 2000). Underacetylated H3 and H4 histones abound in constitutive heterochromatin regions of human metaphase chromosomes and in the heterochromatic inactive X chromosome (Jeppesen et al., 1992; Belyaev et al., 1996). An overall reduction in acetylated H3 and H4 histones has also been observed in mitotic cells compared with interphase nuclei (Kruhlak et al., 2001).
Another histone posttranslational modification relevant to chromatin dynamics during mitosis is Ser 10 phosphorylation of histone H3. The presence of this modified histone has been correlated with mitotic chromosome condensation in several eukaryotic systems (Hendzel et al., 1997; Wei et al., 1998). H3 phosphorylation at Ser10 is governed by the activity of aurora/Ipl1 kinases (Speliotes et al., 2000; Adams et al., 2001; Giet and Glover 2001; Crosio et al., 2002) in competition with the phosphatase activity of the serine/threonine protein phosphatase 1 (Hsu et al., 2000; Murnion et al., 2001). It has been shown that Ser 10-modified histone colocalizes with the human condensin complex during G2/prophase (Schmiesing et al., 2000), suggesting that this modification may have a role in destabilizing local chromatin structure to allow access of condensation factors to DNA or in recruiting the condensation machinery on the modified histone amino-terminal tail (Wei et al., 1998; Schmiesing et al., 2000).
Although the importance of mitotic spindle alterations such as multipolarity or defective kinetochore–microtubule interactions in causing unbalanced chromosome segregation and aneuploidy is widely acknowledged, the role of defects in chromatin dynamics during mitosis in promoting this kind of genomic instability is still largely unexplored. Recently, a study with mutant Tetrahymena strains harboring unphosphorylatable H3 histone demonstrated that Ser-10 phosphorylation is essential for proper chromosome condensation and segregation (Wei et al., 1999). Moreover, in fission yeast a prolonged exposure to trichostatin A (TSA), a specific histone deacetylase inhibitor (Yoshida et al., 1995) was shown to lead to H3 and H4 hyperacetylation in centromeric heterochromatin, derepression of reporter genes in centromeric regions, and chromosome loss (Ekwall et al., 1997). Finally, treatment of mouse cells with TSA for several days induced a relocation of pericentromeric heterochromatic blocks to the nuclear periphery, defective association of heterochromatin protein 1 (HP1) to pericentromeric regions, centromere-positive micronuclei, and abnormal anaphases (Taddei et al., 2001).
In our work, we have investigated the mechanisms promoting abnormal chromosome segregation when histones are maintained hyperacetylated during mitosis and the role of histone H3 phosphorylation in hyperacetylation-induced defective segregation in mammalian cells. To this aim, we inhibited histone deacetylation with TSA shortly before mitosis, rather than with long-term treatments, to avoid the known profound effects of histone deacetylase inhibition on gene expression and cell growth. Indeed, long-term treatments with histone deacetylase inhibitors have been shown to modulate gene expression (Van Lint et al., 1996), to influence acetylation of histones on promoters of target genes (Richon et al., 2000), and to promote growth arrest, differentiation, and apoptosis in different tumor cells (Kosugi et al., 1999; Saunders et al., 1999). To selectively investigate the role of histone modifications before mitosis on the fidelity of the mitotic process, the exposure regimen was built to influence histone deacetylation either during synthesis of late-replicating heterochromatic regions or during prophase mitotic condensation in exponentially growing human primary fibroblasts.
MATERIALS AND METHODS
Cell Cultures and Drug Treatments
Human lung diploid fibroblasts (MRC-5 cells), PtK1 cells, and HeLa cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained in minimal essential medium supplemented with fetal calf serum (MRC-5 cells: 15%; PtK1 and HeLa cells: 10%), antibiotics, and nonessential amino acids. Cells were grown at 37°C, in a humidified atmosphere, with 5% CO2. For experiments, MRC-5 and PtK1 cells were grown on sterilized coverslips inside 35-mm Petri dishes. For TSA (Calbiochem, San Diego, CA) treatment a 500-μg/ml stock solution was prepared in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO). MRC-5 cultures were treated with 200, 500, or 1000 ng/ml TSA for 1 h or 7 h or received the corresponding DMSO concentration as a control. Coverslips were then fixed and either Giemsa-stained for mitotic index evaluation, or processed for immunofluorescence or fluorescence in situ hybridization (FISH). For Giemsa staining, coverslips were fixed in a 3:1 methanol/acetic acid mixture and air dried before staining. The number of mitoses was recorded on 2000 cells/experimental point. Immunoblotting, immunofluorescence, and in vivo analysis of chromosome condensation were performed on cells treated for 1 or 7 h with 500 ng/ml TSA or receiving 0.1% DMSO.
Antibodies and Immunofluorescence
Anti-Ser 10 phospho H3, and anti-Lys 9 acetyl H3 (a gift from Dr. C. Nervi, University La Sapienza, Rome, Italy) rabbit antibodies were from Upstate Biotechnology (Lake Placid, NY). MPM-2 mouse antibody was from DAKO (Carpinteria, CA). Coverslips were washed in phosphate-buffered saline (PBS), fixed in absolute cold methanol, and then permeabilized in PBS + 0.5% Triton-X. After rinsing in PBS with 0.1% Tween 20, cells were blocked overnight in PBS/bovine serum albumin. Coverslips were incubated 2 h with the appropriate antibodies, rinsed in PBS with 0.1% Tween 20, and incubated for 45 min with the appropriate secondary antibodies (fluorescein isothiocyanate anti-rabbit and Texas Red anti-mouse antibodies from Vector Laboratories, Burlingame, CA). Cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and coverslips mounted in antifade solution (Vector Laboratories). Rabbit antibody against PP1-δ protein was kindly provided by Dr. E. Villa-Moruzzi (University of Pisa, Pisa, Italy), and PP1-δ immunostaining was performed according to Andreassen et al. (1998). For HP1 localization on mitotic chromosomes, MRC-5 or PtK1 cultures were fixed after 7- or 1-h treatment with TSA combined with a 2-h incubation in 0.5 μM colchicine before fixation to obtain mitotic spreads. Cells were then swollen in a hypotonic solution (37.5 mM KCl containing protease inhibitors and phenylmethylsulfonyl fluoride [PMSF]) and fixed in 4% (wt/vol) paraformaldehyde in PHEM buffer. Cells were then permeabilized with 0.5% Triton X-100 in PHEM buffer for 10 min and blocked for 1 h with 5% goat serum. They were then incubated overnight with CREST anti-kinetochore serum (Antibodies Incorporated, Davies, CA) and 2HP 1H5 anti-HP1α mouse antibody (a generous gift of Prof. P. Chambon, Centre National de la Recherche Scientifique, Strasbourg, France). Antibody detection and counterstaining were performed as described above except that anti-human and anti-mouse secondary antibodies (Vector Laboratories) were used.
FISH Analysis
MRC-5 cells treated for 1 or 7 h with 500 ng/ml TSA were fixed in a 3:1 methanol/acetic acid mixture. FISH staining was performed using 7 ng of biotin-labeled chromosome 16 alphoid probe (Oncor, Gaithersburg, MD) and 6 ng of digoxigenin-labeled chromosome 1 classical satellite DNA (pUC 1.77 probe; Cooke and Hindley, 1979) for each coverslip. FISH staining was performed as described previously (Cimini et al., 1999).
Microscopy
All preparations were examined under an Olympus Vanox microscope equipped with a 100× (1.35 numerical aperture) oil immersion objective and a SPOT charge-couple device camera (Diagnostic Instruments, Sterling Heights, MI). Color encoded images were acquired using ISO 2000 software (DeltaSistemi, Rome, Italy) and processed with Adobe Photoshop software. For measuring HP1 fluorescence at centromeres, all digital images were obtained with the same camera settings. The best in-focus image of a kinetochore was determined visually and, based on the CREST signal, a region corresponding to the kinetochore was generated using Photoshop software. The mean fluorescence intensity in the region was then recorded together with the mean fluorescence intensity of a close area outside the kinetochore (background fluorescence) and the fluorescence intensity of the background was subtracted from the kinetochore fluorescence. The average value of HP1 fluorescence was calculated for at least 51 kinetochores per each experimental condition, and results were compared using a statistical t test.
Analysis of Mitotic Progression and Chromosome Dynamics in Live Cells
A plasmid carrying the full-length coding sequence for H2B histone subcloned into the pEGFP-N1 mammalian expression vector (a generous gift from Dr. P. Magalhaes, University of Padua, Padua, Italy) was used to obtain a PtK1 cell population enriched in H2B-green fluorescent protein (GFP)–expressing cells as described in Cimini et al. (2002). H2B-GFP–expressing PtK1 cultures were incubated in 500 ng/ml TSA for 30 min or 5 h and then observed by fluorescence and phase contrast microscopy under a Nikon Eclipse 300 inverted microscope equipped with a 37°C heated stage, 60× (0.7 numerical aperture) objective. Prophase cells were localized and mitotic progression was observed. Time intervals from nuclear envelope breakdown to alignment of all chromosomes to the metaphase plate (prometaphase), from chromosome alignment to anaphase onset (metaphase), from anaphase onset to cleavage furrow appearance (anaphase), and from cleavage furrow appearance to completion of cytokinesis (telophase/cytokinesis) were recorded.
Immunoblotting
Cell pellets were resuspended in 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM Na3VO4, 20 mM NaF, 1 mM PMSF, 1 mM dithiothreitol, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 0.5% NP-40. Cells were passed through a 26-gauge needle 10 times and then briefly centrifuged at 13,000 rpm. Nuclear pellets were resuspended in high salt buffer (50 mM HEPES, 500 mM NaCl, 1 mM PMSF, 1 mM Na3VO4, 20 mM NaF, 10 μg/ml aprotinin, 1% NP-40) and sonicated on ice for 10 s at 14-μm amplitude in a Soniprep 150 apparatus equipped with a microtip. Thereafter, pellets were extracted for 30 min on ice in the high salt solution and supernatant from a 13,000 rpm centrifugation was kept at –80°C. Protein content was measured using the Bradford reagent (Sigma-Aldrich). Equal amounts of nuclear proteins from TSA-treated and untreated cells were electrophoresed through SDS-PAGE and then electrotransferred onto nitrocellulose membranes in a Trans-Blot apparatus. Membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 5% low fat dry milk for 1 h and then incubated with anti-acetylated H3 or anti-phospho H3 antibody. After multiple washes in Tris-buffered saline containing 0.1% Tween 20 and incubation with anti-rabbit horseradish peroxidase-linked antibody, immunocomplexes were revealed by a chemiluminescence kit (Amersham Biosciences, Piscataway, NJ).
RESULTS
Cells Entering Mitosis with Hyperaceylated Histones Display Distorted Chromatin Conformation
To investigate the role of histone deacetylase inhibition before mitosis on the accuracy of cell division, we first studied whether entry into mitosis was affected by deacetylase inhibition by analyzing the mitotic index in TSA-treated human primary MRC-5 fibroblasts. The frequency of mitotic cells was reduced by 30% already at 200 ng/ml TSA (Figure 1A), when cells were kept in the presence of the drug during prophase or during synthesis of late-replicating heterochromatic regions (1 or 7 h before fixation, respectively), in accordance with available data showing a G2 checkpoint induction by deacetylase inhibitors in normal cells (Qiu et al., 2000). For 1-h TSA treatment, a further 30% decrease of the mitotic index was observed in cells treated with 500 ng/ml TSA and no further reduction was observed at 1000 ng/ml, indicating that inhibition of deacetylation and biological effects were fully accomplished with 500 ng/ml TSA. Based on these results, we decided to perform our subsequent studies on chromosome dynamics during mitosis at the dose of 500 ng/ml TSA. Then, we directly tested whether histone deacetylase activity was efficiently inhibited in our experimental conditions. We performed immunofluorescent detection of acetylated H3 histone in TSA-treated cells by using an anti-Lys 9 acetyl H3 antibody. The acetylated form of histone H3 was undetectable on control interphase and mitotic MRC-5 cells after immunostaining (Figure 1B, –TSA), whereas an intense staining was observed both on nuclei and mitotic cells in cultures treated for 1 h with TSA (Figure 1B, +TSA). In TSA-treated cultures the strong antibody reactivity of mitotic cells (arrow in Figure 1B, +TSA) demonstrated that in the presence of the deacetylase inhibitor cells entered mitosis with acetylated histones. Western blotting analysis on nuclear proteins confirmed the immunofluorescence results, showing an impressive accumulation of acetylated H3 histones in cells treated for 1 h with TSA alone or in combination with nocodazole (Figure 1C). Therefore, we could conclude that deacetylase activity was efficiently inhibited after 1-h treatment with 500 ng/ml TSA.
Figure 1.

Short-term treatments with the deacetylase inhibitor TSA decrease mitotic index and induce histone hyperacetylation in mitotic cells. (A) Asynchronously growing human fibroblast MRC-5 cells were treated for 1 h (solid bars) or 7 h (squared bars) with different concentrations of TSA and the mitotic frequency was evaluated on Giemsa-stained slides. The graph reports the mean ± SE of the results obtained in three independent experiments. (B) Immunofluorescence detection of Lys 9 acetylated H3 histone on interphase and mitotic (arrows) MRC-5 cells fixed after 7 h in the presence of 1% DMSO (–TSA) or 500 ng/ml TSA (+TSA). Similar results were obtained after 1-h incubation with 500 ng/ml TSA. Bar, 5 μm. (C) Anti Lys 9 acetyl H3 immunoblotting of nuclear proteins from MRC-5 cells incubated for 1 h with 0.1% DMSO (control) or 500 ng/ml TSA (TSA) or receiving 35 ng/ml nocodazole for 18 h (NOC) or 35 ng/ml nocodazole for 18 h and 500 ng/ml TSA for the last 2 h (NOC + TSA).
We next investigated whether acetylation of Lys 9 on histone H3 during prophase interfered with Ser 10 phosphorylation, the mitosis-specific modification of the H3 histone. In human fibroblasts, the appearance of anti-phospho H3 antibody reactivity was a very early marker of chromosome condensation because phospho H3 was already visible on nuclei that did not display chromosome condensation as detected by DAPI staining (Figure 2A, first row, arrow), and H3 histone was heavily phosphorylated in metaphase (Figure 2A, second row, arrow) and anaphase (Figure 2A, second row, arrowhead). In TSA-treated cells a decreased antibody reactivity was observed both on metaphase and anaphase chromosomes (Figure 2A, +TSA, third and forth row, respectively), suggesting a decreased phosphorylation of the H3 histone when hyperacetylated. When metaphases were observed at higher magnification, the phospho H3 antibody reactivity seemed confined to the periphery of the chromosome axis in TSA-treated cells compared with the intense staining all over chromatid arms in control cells (Figure 2B). The decreased phospho H3 reactivity was observed both in cells receiving TSA for 7 h (Figure 2A) and in cells treated for 1 h before fixation (Figure 2B).
Figure 2.

Lys 9 H3 acetylation decreases reactivity to the anti-Ser 10 phospho H3 antibody on immunocytochemical preparations. (A) Immunofluorescence detection of Ser 10 phospho H3 histone in different mitotic phases in MRC-5 cells treated for 7 h with 500 ng/ml TSA (+TSA) or receiving 0.1% DMSO (–TSA). First row, an early prophase cell (arrow) in control culture showing phospho H3 staining. Second row, metaphase (arrow) and anaphase (arrowhead) cells in control culture showing intense phospho H3 reactivity. Third and fourth row, metaphase (arrow) and anaphase (arrow) cells in TSA-treated cultures, in which phospho H3 reactivity is diminished. Bar, 10 μm. (B) Higher magnification of MRC-5 metaphase cells immunostained for the phospho-Ser10 H3 histone. A control metaphase cell (–TSA) and a cell treated with 500 ng/ml TSA for 1 h (+TSA) are shown. Bar, 5 μm. Note that the antibody reactivity is confined to the periphery of the chromatid.
We then examined whether deacetylase inhibition induced a nonspecific inhibition of mitotic kinase activities, which could explain both the decreased reactivity to the phospho H3 antibody and the reduced mitotic entry observed in TSA-treated cells. We performed double immunofluorescence staining with an anti-phospho H3 antibody and the MPM-2 antibody, which is known to recognize phosphoepitopes on several mitotic proteins (Ding et al., 1997). Mitotic chromosomes from control cultures were strongly positive for the phospho H3 antibody and the MPM-2 fluorescence was evenly distributed all over the mitotic cell (Figure 3A, –TSA). Remarkably, the presence of the MPM-2 reactive epitopes was clearly unaffected by the TSA treatment also in cells that completely lacked the phospho-histone staining (Figure 3A, +TSA). In >90% of control metaphases, reactivities to phospho H3 and MPM-2 antibodies were associated, whereas in cell cultures receiving TSA for 7 h ∼40% of MPM-2 positive metaphases were weakly positive or completely dull for phospho H3 staining (Figure 3B). A decreased phospho H3 staining in MPM-2–positive cells was also observed when nocodazole-treated cells were incubated with TSA (Figure 3B). These data excluded that histone acetylation at prophase might promote a general inhibition of mitotic kinases.
Figure 3.

Lys 9 H3 acetylation decreases reactivity to the anti-Ser 10 phospho H3 antibody on mitotic cells without affecting MPM-2 staining. (A) Immunofluorescence detection of Ser 10 phospho H3 histone and MPM-2 epitopes on metaphase MRC-5 cells treated with 0.1% DMSO (–TSA) or receiving 500 ng/ml TSA for 7 h (+TSA). The TSA-treated cell shows clear MPM-2 staining and no phospho H3 reactivity. Bar, 5 μm. (B) Analysis of phospo H3 reactivity in MPM-2–positive cells. MRC-5 cells were incubated for 7 h with 0.1% DMSO (control) or 500 ng/ml TSA (TSA) or received 35 ng/ml nocodazole for 7 h (NOC), or 35 ng/ml nocodazole and 500 ng/ml TSA for 7 h (NOC + TSA). MPM-2–positive metaphases were classified as weakly positive (example in Figure 2B, +TSA) or negative (example in Figure 3A, +TSA) for phospho H3 staining by eye. About 100 mitoses were scored per experimental point.
The reduced H3 phosphorylation, if not due to a general inhibition of kinase activity, might depend on an increased dephosphorylation. For this reason, we decided to analyze the association of the δ isoform of the PP1 phosphatase to mitotic chromosomes. PP1-δ is a major mitotic chromatin-associated protein and its phosphatase activity counteracts the aurora B kinase activity on the H3 histone. We used an antibody specific to PP1-δ and showed that during interphase PP1-δ displays a homogeneous nuclear distribution both in control and in TSA-treated cells (Figure 4A). However, a stronger signal for PP1-δ was evident on interphase nuclei, when cells were treated with the deacetylase inhibitor (Figure 4A, +TSA). The observation of mitotic cells showed that PP1-δ was specifically associated to condensed chromosomes during mitosis and that chromosomes from TSA-treated cultures displayed a much stronger staining compared with untreated cultures (Figure 4B).
Figure 4.

PP1-δ is recruited on interphase and mitotic chromatin containing hyperacetylated histones. (A) Immunofluorescence detection of PP1-δ on interphase MRC-5 cells treated for 1 h with 0.1% DMSO (–TSA) or receiving 500 ng/ml TSA (+TSA). PP1-δ antibody stains heavily nuclei in TSA-treated cultures. Bar, 10 μm. (B) Immunofluorescence detection of PP1-δ on mitotic chromosomes in MRC-5 cells treated for 1 h with 0.1% DMSO (–TSA) or receiving 500 ng/ml TSA (+TSA). The metaphase chromosomes from a TSA-treated culture show an intense PP1-δ staining. Bar, 5 μm.
We finally tested directly the phosphorylation state of the H3 histone by using Western blot analysis. Surprisingly, when nuclear proteins were assayed by Western blot the intracellular levels of Ser 10 phosphorylated H3 histone were similar in samples obtained from asynchronously growing control MRC-5 cells or cells exposed for 1 or 7 h to the deacetylase inhibitor (Figure 5, MRC-5). Comparable levels of phospho H3 histone were also observed in MRC-5 cells arrested in mitosis by a 7-h nocodazole treatment and in cells receiving the deacetylase inhibitor together with nocodazole (Figure 5, MRC-5). To investigate H3 phosphorylation in a mitotic cell population, HeLa cells were synchronized in S phase by a thymidine/aphidicolin block, and H3 phosphorylation was assayed 13 h after release, the time when most cells entered mitosis, with or without a preceding incubation with TSA. In this case, intracellular levels of phospho H3 histone also did not show significant variations (Figure 5, HeLa), corroborating the finding obtained in asynchronous human fibroblasts.
Figure 5.
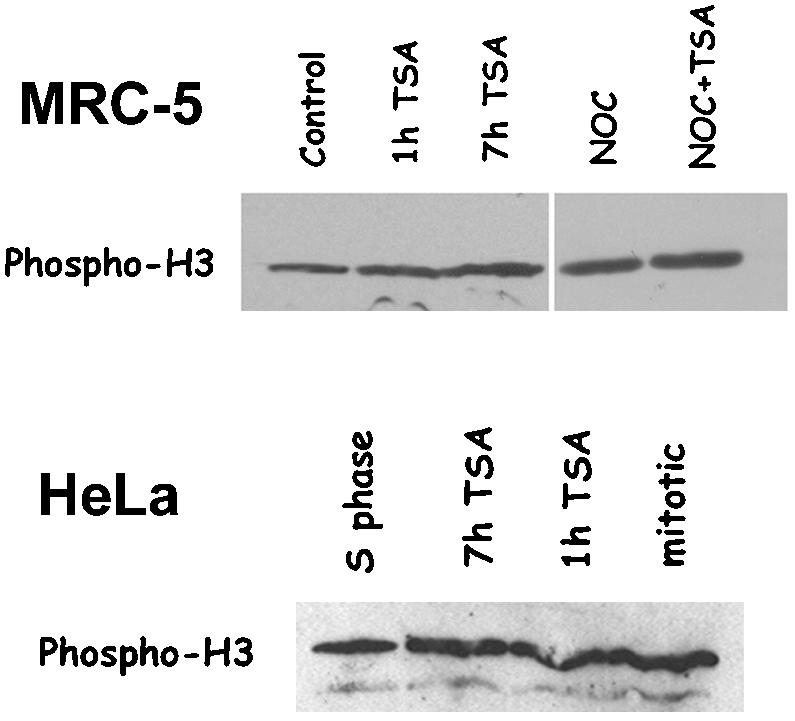
Lys 9 H3 acetylation does not interfere with Ser 10 H3 phosphorylation. Anti-Ser 10 phospho H3 immunoblotting of nuclear proteins in asynchronously growing MRC-5 cells (MRC-5) or synchronized HeLa cells (HeLa). MRC-5 cells were either incubated for 7 h with 0.1% DMSO (control) or 500 ng/ml TSA for 1 h (1h TSA) or 7 h (7 h TSA), or else received 35 ng/ml nocodazole for 7 h (NOC) or 35 ng/ml nocodazole and 500 ng/ml TSA for 7 h (NOC + TSA). Nuclear proteins were prepared, immunoblotted with the anti-Ser 10 phospho H3 antibody, and revealed by chemiluminescence. HeLa cells were synchronized in S phase by a thymidine/aphidicolin block. Nuclear proteins were prepared and immunoblotted with the anti-Ser 10 phospho H3 antibody at the end of the aphidicolin treatment (S phase), after 13-h release in complete medium (mitotic), or after 13-h release in complete medium, including a final 1 h (1 h TSA) or7h(7h TSA) treatment with 500 ng/ml TSA. In both MRC-5 and HeLa cells, intracellular levels of phospho H3 histone did not show significant variations after TSA.
Together, the above-mentioned results suggested that Lys 9 acetylation does not interfere with Ser 10 phosphorylation but rather indicated that the presence of Lys 9 acetylated/Ser 10 phosphorylated tails may modify DNA/histone interaction. An altered DNA/histone association might decrease the accessibility of the N-tails to the antibody recognizing Ser 10 phosphorylated H3 on mitotic preparations, thus reducing the phospho H3 staining of mitotic chromosomes. Reduced chromatin accessibility might also increase the loading on DNA of the chromatin-associated kinases and phosphatases that regulate H3 phosphorylation. This might explain the recruitment of higher concentrations of PP1-δ on the chromosomes to maintain the correct phosphorylation/dephosphorylation equilibrium. The combined data indicate that cells entering mitosis with hyperacetylated histones display altered chromatin conformation.
Cells Entering Mitosis in the Presence of TSA Show Altered Chromosome Condensation and Impaired Mitotic Progression
The above-mentioned results led us to speculate that chromatin condensation into mitotic chromosomes might be affected by the presence of hyperacetylated histones. To avoid possible artifacts due to fixation procedures, we decided to investigate mitotic chromosome condensation after deacetylase inhibition in living cells. To this aim, we used a stable transfectant PtK1 cell line expressing a histone 2B-GFP chimeric protein (Cimini et al., 2002). The H2B-GFP protein is incorporated into nucleosomes, and the resulting bright fluorescence of chromosomes makes chromosome structure in living mitotic cells very easy to observe. H2B-GFP PtK1 cells were exposed to TSA for 30 min or 5 h and then followed through mitosis by using fluorescence microscopy (Figure 6). The TSA treatment induced a clear undercondensation of mitotic chromosomes in all mitotic stages. Already in prophase, the long thread of condensing chromosomes did not look homogeneously fluorescent as in control cells (Figure 6, pro-, –TSA), but showed areas of low GFP fluorescence alternated with globular fluorescent spots (Figure 6, pro-, +TSA). A decreased compaction of the chromatid thread was also clear in prometaphase (Figure 6, prometa-, +TSA) and metaphase (Figure 6, meta-, +TSA) and was maintained up to telophase (Figure 6, telo-, +TSA). At all mitotic stages, chromosomes seemed composed of multiple globular subdomains, suggesting that they did not fold to the regular level of compaction. To investigate whether this condensation defect interfered with the regular progression of mitosis in living cells, we performed phase contrast timelapse microscopy to follow mitosis in TSA-treated PtK1 cells. In our observations, prometaphase length was defined as the time cells spent between nuclear envelope breakdown and congression of chromosomes in a tight rod-like metaphase plate, anaphase onset was marked by sister chromatid separation, and beginning of telophase by cleavage furrow ingression. PtK1 cells were observed under an inverted microscope 30 min or 5 h after TSA addition and mitotic progression of treated cells was followed thereafter. Time-lapse analysis of ∼30 cells per experimental point showed a statistically significant lengthening of the time cells spent in prometaphase already for the 30-min TSA treatment (Table 1). After 5 h growth in TSA-containing medium prometaphase length was almost doubled and metaphase length was also statistically increased. This corresponded to a lengthening of the total time spent in mitosis by TSA-treated cells (Table 1). Interestingly, a fraction of cells arrested in prometaphase for >3 h in cultures progressing through mitosis in TSA-containing medium. For the 5-h TSA treatment, 20% of observed cells did not exit prometaphase during the time of observation. These results show that histone underacetylation in euchromatic and heterochromatic regions is required for proper chromosome condensation and regular mitotic progression.
Figure 6.

Chromosome condensation is impaired in living cells when histones are maintained acetylated in mitosis. H2B-GFP–expressing PtK1 cells were treated for 5 h with 500 ng/ml TSA (+TSA) or 0.1% DMSO (–TSA) and then observed for H2B-GFP by fluorescence microscopy. Cells at different mitotic stages are shown: prophase (Pro-), prometaphase (prometa-), metaphase (meta-), and telophase (telo-). At all mitotic stages, chromosomes in TSA-treated cells were less compact and showed areas of low GFP fluorescence alternated with globular fluorescent spots. Bar, 5 μm.
Table 1.
Living cell analysis of the length (mean ± s.d.) in minutes of different stages of mitosis in PtK1 cells progressing through mitosis in TSA-containing medium
| N | Prometaphase | Metaphase | Anaphase | Telophase/Cytokinesis | Total | |
|---|---|---|---|---|---|---|
| 5 h DMSO | 29 | 18.8 ± 9.4 | 21.6 ± 11.6 | 17.1 ± 6.2 | 17.8 ± 9.6 | 75.3 ± 25.1 |
| 30 min TSA | 30 | 26.9 ± 17.9a | 24.4 ± 21.5 | 15.1 ± 7.6 | 22.0 ± 16.3 | 87.5 ± 38.6c |
| 5 h TSA | 29 | 40.4 ± 15.9b | 27.8 ± 10.8a | 18.7 ± 8.4 | 24.4 ± 16.0 | 108.9 ± 20.9bd |
Cells were exposed to TSA for the indicated times before observation and kept in TSA-containing medium during the experiment. N = total number of cells analyzed.
Student t test, p < 0.05; b, Student t test, p < 0.01, when values were compared to DMSO treatment.
1 cell: prometaphase arrest (> 3 h); 1 cell: metaphase arrest (> 3 h); 3 cells: absence of cytokinesis.
6 cells: prometaphase arrest (> 3 h); 2 cells: absence of cytokinesis.
Centromeric Heterochromatin Is Modified When Histone Deacetylation Is Inhibited in Mitosis
Centromeric regions are defined by a compact heterochromatic structure that is characterized by chemical modifications of both DNA and histones. Lys9 methylation on H3 histone has been shown to recruit the heterochromatin-associated protein HP1, which plays a crucial role in maintaining the compact heterochromatic structure at centromeres (Eissenberg and Elgin, 2000). Recently, long-term TSA treatments in mouse cells have been reported to disrupt interphase nuclear domains enriched in HP1 proteins (Taddei et al., 2001). Having observed a defective chromosome condensation after treatment with the deacetylase inhibitor, we decided to investigate whether a less compact chromatin structure was present also at the centromere of these under-condensed chromosomes and whether short-term TSA treatments affected HP1 recruitment to the centromeric regions of mitotic chromosomes. Human primary fibroblasts and PtK1 cells were treated for 1 or 7 h with the deacetylase inhibitor, and cultures were processed for immunofluorescence after a mild hypotonic treatment to detect constitutive kinetochore proteins by CREST staining (Earnshaw and Rothfield, 1985) and HP1 protein by using an antibody directed against the HP1α isoform. HP1α protein was associated to chromatin during mitosis and preferentially concentrated at the centromeric regions of mitotic chromosomes in untreated cells (Figure 7, –TSA, Remboutsika et al., 1999). Combined immunodetection of kinetochore (CREST) and HP1 proteins showed that the region of HP1 accumulation at centromeres was much larger than the CREST signal and overlapped the inner centromeric area between the two CREST-stained sister kinetochores (Figure 7, –TSA, right column). In TSA-treated cells, the HP1-chromatin association was maintained but the preferential accumulation at the centromeric heterochromatin was diminished (Figure 7, +TSA). Mean HP1 fluorescence intensity on kinetochore regions was quantified in at least 51 kinetochores per experimental point and results for PtK1 and MRC-5 cells are shown in Figure 7B. Statistically significant reductions in the mean HP1 fluorescence intensities at kinetochores were observed both in MRC-5 and PtK1 cells treated for 1 or 7 h with TSA. Differences ranged between ∼25% (PtK1, 7 h) and 50% (MRC-5, 1 h) reduction compared with their respective controls (p < 0.001 for all comparisons).
Figure 7.

Heterochromatin protein 1 accumulation at centromeres is impaired in cells briefly exposed to TSA. (A) Immunofluorescence detection of HP1α and kinetochore proteins by using 2HP 1H5 antibody and CREST serum on PtK1 metaphase cells from cultures treated for 7 h with 0.1% DMSO (–TSA) or 500 ng/ml TSA (+TSA). DNA was counterstained using DAPI (blue). The right column shows an overlay of CREST, HP1, and DAPI staining. HP1 is detected at centromeres in a region wider than the one defined by the CREST antibody in the control cell (top, middle panel). In the TSA-treated cell the centromeric accumulation is lost (bottom, middle panel). Bar, 5 μm. (B) Quantitative analysis of HP1 fluorescence on MRC-5 and PtK1 cells treated for 7 h with 0.1% DMSO (red bars) or receiving 500 ng/ml TSA for 1 h (white bars) or 7 h (gray bars). The values shown are the mean + SE of kinetochore fluorescence intensities (obtained as described in MATERIALS AND METHODS). Numbers above bars represent numbers of kinetochores measured per each experimental condition. p < 0.001 when TSA-treated samples were compared with their respective controls by the Student's t test.
Inhibition of Histone Deacetylation Produces Chromosome Segregation Defects in Anaphase Cells
To assess whether the persistence of acetylated histones on mitotic chromosomes interfered with chromosome segregation in mitosis we performed an analysis of chromosome segregation defects in human primary fibroblasts. The analysis of ∼1000 anaphase MRC-5 cells by DAPI and CREST staining showed that the TSA treatment significantly induced lagging chromosomes (χ2 test, p < 0.01), i.e., chromosomes left behind at the spindle equator during anaphase (Figure 8A, left column, and B). Both lagging of single chromatids showing one CREST signal and lagging of paired sisters with two CREST signals were observed (Figure 8B). We could identify such lagging as paired sisters, because the size of the two paired chromatids was the same and the two CREST signals were close to each other (an example is shown in Figure 8A). However, the predominant segregation defect was constituted by bridges of chromatin between the two reforming daughter nuclei (Figure 8B; p < 0.001, χ2 test). Chromatin bridges in control cells always looked like DAPI-stained material stretched between the two groups of migrated chromosomes (Figure 8B, no CREST signal). Conversely, a substantial fraction of chromatin bridges in TSA-treated cells showed kinetochore signals at both ends of the DAPI-stained connecting material (example in Figure 8A; B, CREST distal). Alternatively, stretched kinetochore signals could be observed along the DNA extending in between the two groups of segregating chromosomes (Figure 8B, CREST central), suggesting that the DNA bridge between the two connected centromeres was much shorter than in control cells or that the two centromeres could not split. Similar results were obtained in cells that received the drug either for 1 or 7 h, suggesting that the critical time window for TSA action was just at the beginning of mitosis, when chromosome condensation is achieved.
Figure 8.

Chromosome segregation defects in TSA-treated MRC-5 cells derive from defective sister chromatid separation. (A) Immunofluorescence detection of kinetochores and DNA staining on anaphase cells, showing lagging chromosomes or chromatin bridges. The lagging chromosome shows two paired CREST signals (arrow). The chromatin bridge shows two extended CREST signals at the ends of the bridge (arrows). Bar, 5 μm. (B) Quantitative analysis of the induction of chromosome segregation defects after TSA treatment. Lagging chromosomes were classified as showing one CREST signal (single sisters) or two paired CREST signals (paired sisters) both in DMSO- and TSA-treated cells. Chromatin bridges, both in TSA-treated cells and DMSO-treated cells, were classified as follows: without identifiable CREST signal (without CREST signal); with CREST signal at opposite ends of the bridge (CREST distal); with CREST signal along the chromatin bridge (CREST central). The graph shows results obtained scoring 937 anaphase cells in DMSO-treated cells and 921 anaphase cells in TSA-treated cultures. Data from 1-h and 7-h treatments with TSA were pooled because no appreciable differences were observed. (C) Fluorescence in situ hybridization with chromosome 16-specific alphoid probe (fluorescein isothiocyanate, green) and chromosome 1 classical satellite probe (rhodamine, red) on anaphase MRC-5 cells after treatment with TSA. DNA was counterstained with DAPI (blue). A regular distribution of the two chromosomes is expected to give two green signals and two red signals on each group of segregated chromosomes (normal cell). Some chromatin bridges on anaphase cells showed two extended red (chromatin 1 bridge) or green (chromosome 16 bridge) signals at the opposite ends of the connecting chromatin. In other cases, the centromeric signal totally overlapped the chromatin bridge (centromeric bridge). Anaphases (519) were scored in TSA-treated cells and 11 chromatin bridges involving either chromosome 1 or chromosome 16 were recorded. Two of 11 were centromeric bridges.
To analyze more in detail such chromatin bridges, we applied fluorescence in situ hybridization with centromeric probes on anaphase cells. Chromosome 1 classical satellite and chromosome 16 alphoid DNA sequences were labeled with different fluorochromes to detect the segregation of these chromosomes at anaphase. As shown in Figure 8C (normal cell) the regular distribution of chromosomes at anaphase would elicit 2 signals for each chromosome in each group of segregated chromosomes. Chromatin bridges in TSA-treated cultures often displayed two centromeric signals for one of the chromosomes under study at opposite ends of the bridge, whereas the two other centromeric signals were correctly localized at the two poles (Figure 8C, Chrom 1 bridge, Chrom 16 bridge), indicating that chromatin bridges were sister chromatids that remained connected during anaphase. In other cases, chromatin bridges in TSA-treated cells extended over the centromeric signals and the centromeres were highly stretched (Figure 8C, centromeric bridge). Together, these results show that sister chromatids could not separate when cells entered mitosis with hyperacetylated histones. Thus, the presence of acetylated histones in mitosis induced both aberrant chromosome numbers (i.e., aneuploidy) and defects in chromosome structure.
DISCUSSION
Condensation of DNA into chromosomes, resolution of the two sister chromatids and physical separation of the two sisters are the mitotic chromatin events essential for proper chromosome segregation during cell division. These events require dramatic changes in chromatin structure that are achieved through histone tail modifications by chromatin-modifying enzymatic complexes and by the action of several other enzymatic activities such as condensins and cohesins, topoisomerase II, and the securin/separin pathway (reviewed in Hans and Dimitrov, 2001; Losada and Hirano, 2001; Nasmyth, 2002). Our results discover new aspects of the interplay between histone modifications, sister chromatid condensation and cohesion, and chromosome segregation, and they provide new mechanistic insights into the origin of genome instability in mammalian cells.
Chromosome Structure Is Profoundly Altered When Histones Are Maintained Acetylatetd in Mitosis
In cells undergoing mitosis with hyperacetylated chromatin, the presence of H3 Lys 9 acetylation did not inhibit H3 Ser 10 phosphorylation. This result is consistent with the coexistence of Ser 10 phosphorylation and Lys 9 acetylation on histone H3 upon gene activation (Clayton et al., 2000), the increased kinase activity of recombinant Ipl1/aurora kinase on acetylated H3 substrates (Rea et al., 2000), and our own data on phospho H3 levels in nuclear proteins after treatment with the deacetylase inhibitor (Figure 5). However, phospho H3 antibody reactivity of TSA-treated mitotic chromosomes was greatly reduced in immunofluorescence studies (Figure 2), indicating that the presence of acetylated histones in mitotic chromosomes decreases the accessibility of histone N-tails to the antibody for Ser 10 phospho H3. The results presented suggest that the decreased antibody reactivity depends on a modified association of hyperacetylated histone tails to DNA and a consequent distortion in chromatin structure. A reduction in H3 N-tail/DNA binding is associated with mitotic H3 phosphorylation and chromosome condensation and may be involved in promoting the binding of condensation factors such as topoisomerase II and the condensin complex to the modified chromatin (Sauve' et al., 1999). Our data support the idea that this chromatin remodeling process is strongly altered when histones are hyperacetylated during mitotic condensation, producing mitotic chromosomes with altered three-dimensional structure. This conclusion is corroborated by the stronger association of the human chromatin-associated phosphatase PP1-δ to acetylated mitotic chromosomes (Figure 4). Indeed, a modified chromatin conformation may stimulate phosphatase loading on H3 phosphorylated chromatin to maintain an adequate balance of kinase and phosphatase activities during mitosis (Murnion et al., 2001, Sugiyama et al., 2002).
Consequent to the modifications in chromatin conformation identified by antibody staining and protein association, we showed that inhibition of histone deacetylation shortly before mitosis produces defects in chromosome condensation in living cells (Figure 6). This indicates that association of condensation factors on chromatin is dependent on a favorable chromatin structure, which requires both Lys 9 deacetylation and Ser 10 phosphorylation. In line with this idea is the observation that Aurora B-depleted Drosophila cultured cells show altered chromosome condensation and defective association of the condensin Barren protein together with reduced H3 phosphorylation (Giet and Glover, 2001). The impaired association of HP1 protein to the centromeric region observed in Ptk1 and human cells after TSA treatment (Figure 7) demonstrates that centromeric chromatin structure is also altered when histones are maintained acetylated in mitosis. This may be explained by the fact that acetylation of Lys 9 residues prevents methylation by the SUV39H1 methyltransferase (Rea et al., 2000, Nakayama et al., 2001). Given the strong specificity of the HP1/Swi6 chromodomain for Lys 9 methylated H3 (Bannister et al., 2001; Jacobs et al., 2001; Nakayama et al., 2001), HP1 association to heterochromatin containing Lys 9 acetylated H3 may be defective. Our data show that acetylation-dependent defective accumulation of HP1 onto heterochromatin occurs not only after several day exposure to TSA, as shown by Taddei et al. (2001), but also when histones are maintained hyperacetylated specifically during prophase. These results highlight the dynamism of histone modifications and chromatin structure during mitosis, suggesting that Lys 9 H3 histone methylation still occurs during chromosome condensation. This is consistent with the high level of H3 methyltransferase activity observed in HeLa mitotic cells (Rice et al., 2002).
In our live cell observation of mitotic progression, a significant lengthening of prometaphase and metaphase was observed. Furthermore, 20% of observed cells were blocked in prometaphase in the presence of TSA (Table 1). This raises the possibility that an improper chromosome condensation can affect kinetochore–microtubule interaction, inducing mitotic checkpoint activation and cell cycle arrest in prometaphase. Furthermore, the metaphase lengthening observed in TSA-treated cells, indicates that even when all chromosomes correctly align at the metaphase plate, a mitotic delay is induced in response to the defect in chromosome condensation. This suggests the intriguing possibility that the mitotic checkpoint can detect not only kinetochore-microtubule attachment but also chromosome three-dimensional architecture during mitosis. Conformational defects in the centromeric region, due to histone hyperacetylation-dependent HP1 dispersal might be the architectural defects monitored by the mitotic checkpoint after deacetylase inhibitors.
Chromosomal passenger proteins might be good candidates as molecular players that connect the chromosome condensation process with mitotic progression. These proteins localize along the whole chromosome length during prophase and move to the inner kinetochore domain during prometaphase. It can be speculated that inhibition of chromosome condensation by histone hyperacetylation may interfere with the dissociation of these proteins from chromosome arms, inhibiting their accumulation and function at kinetochores. The passenger protein Aurora B kinase has been shown to be responsible for the tension-dependent activation of the mitotic checkpoint and has been suggested to promote sister kinetochore biorientation (reviewed in Shannon and Salmon, 2002, Tanaka, 2002). This protein may be an excellent candidate as mediator of the mitotic delay induced by histone hyperacetylation, thus integrating the chromosome condensation process with kinetochore-microtubule attachment and mitotic progression.
Histone Hyperacetylation Prevents Sister Chromatid Resolution and Centromere Separation
Cells reaching anaphase in the presence of the deacetylase inhibitor TSA showed increased frequencies of chromosome segregation defects, such as lagging chromosomes and chromatin bridges (Figure 8). These mitotic abnormalities were present also for exposure times that affected histone deacetylation only during late G2/prophase, suggesting a critical time window when histone deacetylation is required for proper chromosome condensation and segregation.
Both chromatin bridges derived from persistent cohesion along sister chromatid arms and chromatin bridges due to persistent cohesion at the centromere were observed in our FISH studies. However, the majority of chromosomes producing chromatin bridges showed persistent cohesion along sister chromatid arms but proper centromere separation, demonstrating that inhibition of histone deacetylation in mitosis affects chromosome cohesion along chromatid arms more than at the centromere. It is now becoming clear that chromatin dynamics in mitosis is regulated by the coordinated action of cohesin and condensin activities and that condensin loading on DNA promotes cohesin dissociation (Losada and Hirano, 2001; Nasmyth, 2002). It has been proposed that chromosome arm cohesion in mammalian cells is removed in prophase through a separin-independent mechanism and that centromeric cohesion is released by Scc1 cleavage at the metaphase to anaphase transition (Waizenegger et al., 2000). Our data provide cytological evidence that the release of centromeric cohesion is independent from sister chromatid resolution and that this last process is strongly influenced by histone hyperacetylation. It can be speculated that impaired association of condensation factors on hyperacetylated chromatin may alter the dissociation of cohesins during early mitotic stages, resulting in persistent cohesion along chromatid arms. The separin-dependent release of centromeric cohesins at anaphase onset would promote the formation of chromatin bridges between sister chromatids due to poor sister chromatid arm resolution, as already reported for condensin mutants (Bhat et al., 1996; Ouspenski et al., 2000, Steffensen et al., 2001) or after inhibition of sister chromatid decatenation by topoisomerase II inhibitors (Cimini et al., 1997). On the other hand, a lack of sister chromatid separation at the centromere level is clearly indicated by the observation of chromatin bridges spanning the centromeric region. It can be hypothesized that the disentangling of centromeric DNA by topoisomerase II may be defective when the centromeric structure is distort due to histone hyperacetylation. In line with this, induction of polyploidy through a lack of centromere separation has been found in Suv39h histone methyltransferase-deficient mice (Peters et al., 2001).
Among TSA-induced lagging chromosomes, either single chromatid or paired sister chromatids with two CREST signals were observed. Previous studies in PtK1 and human cells have demonstrated that lagging chromosomes at anaphase are single sisters that cannot migrate because their kinetochore is attached to microtubules coming from opposite spindle poles (merotelic kinetochore orientation) (Cimini et al., 2001, 2002). Our results suggest that when HP1 is not correctly assembled on the centromeric heterochromatin in human cells, the less constrained centromeric organization may promote the attachment of individual kinetochores to microtubules coming from both spindle poles, thus producing anaphase-lagging chromosomes. A similar mechanism for the origin of lagging chromosomes has been proposed from work in yeast cells (Bernard et al., 2001; Bernard and Allshire, 2002). Disruption of centromeric architecture by loss of heterochromatin components may also promote the assembly of sister kinetochores that have distort reciprocal orientations, which would allow both of them to become merotelically oriented. When merotelic kinetochore orientation and persistent sister chromatid cohesion at the centromere level occur at the same time, the possibility that sister chromatids separate at anaphase will be further reduced. This would explain the observation of lagging chromosomes representing both sisters with closely paired CREST signals. It can be hypothesized that the final disentangling of the two sister chromatids at anaphase onset requires both loss of cohesion mediated by SCC1 degradation and tension at kinetochores induced by opposite traction forces. When both sister kinetochores are merotelically oriented, the tension in opposite directions at the centromere may be strongly reduced, inhibiting centromere separation.
In conclusion, our results highlight the fact that both persistence of cohesion along chromatid arms and at centromeres are responsible for defects in chromosome segregation and chromosome structure when histones are maintained acetylated during mitosis. Furthermore, defective kinetochore–microtubule interactions leading to lagging chromosomes seem to be favored when centromeric heterochromatin possess a less constrained structure due to HP1 depletion.
Acknowledgments
We thank Prof. Pierre Chambon and Dr. Regine Losson (Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Strasbourg) for the HP1α antibody, Dr. Emma Villa-Moruzzi for the PP1-δ antibody, Dr. Clara Nervi for the anti Lys9 acetyl-H3 antibody, and Dr. Paulo P. Magalhaes for the H2B-GFP vector. We thank Drs. Maurizia Caruso and Patrizia Filetici for critical reading of the manuscript.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03–01–0860. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-01-0860.
References
- Adams, R.R., Maiato, H., Earnshaw, W.C., and Carmera, M. (2001). Essential role of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction and chromosome segregation. J. Cell Biol. 153, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen, P.R., Lacroix, F.B., Villa-Moruzzi, E., and Margolis, R.L. (1998). Differential subcellular localization of protein phosphatase-1α, γ1 and δ isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 141, 1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister, A.J., Zegerman, P., Partridge, J.F., Miska, E.A., Thomas, J.O., Allshire, R.C., and Kouzarides, T. (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Belayev, N.D., Keohane, A.M., Turner, B.M. (1996). Differential underacetylation of histones H2A, H3, and H4 on the inactive X chromosome in human female cells. Hum. Genet. 97, 573–578. [DOI] [PubMed] [Google Scholar]
- Bernard, P., and Allshire, R. (2002). Centromeres become unstuck without heterochromatin. Trends Cell Biol. 12, 419–424. [DOI] [PubMed] [Google Scholar]
- Bernard, P., Maure, J.F., Partridge, J.F., Genier, S., Javerzat, J.P., and Allshire, R.C. (2001). Requirement of heterochromatin for cohesion at centromeres. Science 294, 2539–2542. [DOI] [PubMed] [Google Scholar]
- Bhat, M.A., Philp, A.V., Glover, D.M., and Bellen, H.J. (1996). Chromatid segregation at anaphase requires the barren product, a novel chromosome-associated protein that interacts with topoisomerase II. Cel1 87, 1103–1114. [DOI] [PubMed] [Google Scholar]
- Cheung, W.L., Briggs, S.D., and Allis, C.D. (2000). Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 12, 326–333. [DOI] [PubMed] [Google Scholar]
- Cimini, D., Antoccia, A., Tanzarella, C., and Degrassi, F. (1997). Topoisomerase II inhibition in mitosis produces numerical and structural chromosomal aberrations in human fibroblasts. Cytogenet. Cell Genet. 76, 61–67. [DOI] [PubMed] [Google Scholar]
- Cimini, D., Fioravanti, D., Salmon, E.D., and Degrassi, F. (2002). Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J. Cell Sci. 115, 507–515. [DOI] [PubMed] [Google Scholar]
- Cimini, D., Howell, B., Maddox, P., Khodjakov, A., Degrassi, F., and Salmon, E.D. (2001). Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., Tanzarella, C., and Degrassi, F. (1999). Differences in malsegregation rates obtained by scoring ana-telophase or binucleate cells. Mutagenesis 14, 563–568. [DOI] [PubMed] [Google Scholar]
- Clayton, A.L., Rose, S., Barratt, M.J., and Mahadevan, L.C. (2000). Phosphoacetylation of histone H3 on c-fos-and c-jun-associated nucleosomes upon gene activation. EMBO J. 19, 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke, H.J., and Hindley, J. (1979). Cloning of human satellite III DNA: different components are on different chromosomes. Nucleic Acids Res. 6, 3177–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio, C., Fimia, G.M., Loury, R., Kimura, M., Okano, Y., Zhou, H., Sen, S., Allis, C.D., and Sassone-Corsi, P. (2002). Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 22, 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, M., Feng, Y., and Vandré, D. (1997). Partial characterization of the MPM-2 phosphoepitope. Exp. Cell Res. 231, 3–13. [DOI] [PubMed] [Google Scholar]
- Earnshaw, W.C., and Rothfield, N. (1985). Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma 91, 313–321. [DOI] [PubMed] [Google Scholar]
- Eissenberg, J.C., and Elgin, S.C. (2000). The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev. 10, 204–210. [DOI] [PubMed] [Google Scholar]
- Ekwall, K., Olsson, T., Turner, B.M., Cranston, G., and Allshire, R.C. (1997). Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 91, 1021–1032. [DOI] [PubMed] [Google Scholar]
- Giet, R., and Glover, D.M. (2001). Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 152, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein, M. (1997). Histone acetylation in chromatin structure and transcription. Nature 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Hans, F., and Dimitrov, S. (2001). Histone H3 phosphorylation and cell division. Oncogene 20, 3021–3027. [DOI] [PubMed] [Google Scholar]
- Hendzel, M.J., Wei, Y., Mancini, M.A., Van Hooser, A., Ranalli, T., Brinkley, B.R., Bazett-Jones, D.P., and Allis, C.D. (1997). Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106, 348–360. [DOI] [PubMed] [Google Scholar]
- Hsu, J.-Y., et al. (2000). Mitotic phosphorylation of histone H3 is governed by Ipl1/auroraB kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102, 279–291. [DOI] [PubMed] [Google Scholar]
- Jacobs, S.A., Tavernam, S.D., Briggs, S.D., Li, J., Eissenberg, J.C., Allis, C.D., and Khorasanizadeh, S. (2001). Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 20, 5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, P., Mitchell, A., Turner, B., and Perry, P. (1992). Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma 101, 322–332. [DOI] [PubMed] [Google Scholar]
- Kosugi, H., et al. (1999). Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia 13, 1316–1324. [DOI] [PubMed] [Google Scholar]
- Kruhlak, M.J., Hendzel, M.J., Fischle, W., Bertos, N.R., Hameed, S., Yang, X.-J., Verdi D.E., and Bazett-Jones, D.P. (2001). Regulation of global acetylation in mitosis through loss of histone acetylatransferases and deacetylases from chromatin. J. Biol. Chem. 276, 38307–38319. [DOI] [PubMed] [Google Scholar]
- Losada, A., and Hirano, T. (2001). Shaping the metaphase chromosome: coordination of cohesion and condensation. Bioessays 23, 924–935. [DOI] [PubMed] [Google Scholar]
- Murnion, M.E., Adams, R.R., Callister, D.M., Allis, C.D., Earnshaw, W.C., and Swedlow, J.R. (2001). Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J. Biol. Chem. 276, 26656–26665. [DOI] [PubMed] [Google Scholar]
- Nakayama, J., Rice, J.C., Strahl, B.D., Allis, C.D., and Grewal, S.I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K. (2002). Segregating sister genomes: the molecular biology of chromosome separation. Science 297, 559–565. [DOI] [PubMed] [Google Scholar]
- Ouspenski, I.I., Cabello, O.A., and Brinkley, B.R. (2000). Chromosome condensation factor Brn1p is required for chromatid separation in mitosis. Mol. Biol. Cell 11, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A.H., et al. (2001). Loss of suv39h Histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337. [DOI] [PubMed] [Google Scholar]
- Qiu, L., Burgess, A., Fairlie, D.P., Leonard, H., Parson, P.O., and Gabrielli, B.O. (2000). Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 11, 2069–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea, S., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–598. [DOI] [PubMed] [Google Scholar]
- Remboutsika, E., Lutz, Y., Oansmuller, A., Vonesch, J.L., Losson, R., and Chambon, P. (1999). The putative nuclear receptor mediator TIF1alpha is tightly associated with euchromatin. J. Cell Sci. 112, 1671–1683. [DOI] [PubMed] [Google Scholar]
- Rice, J.C., Nishioka, K., Sarma, K., Steward, R., Reinberg, D., and Allis, C.D. (2002). Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 16, 2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon, V.M., Sandhoff, T.W., Rifkind, R.A., and Marks, P.A. (2000). Histone deacetylase inhibitor selectively induces p21 WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 97, 10014–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, N., Dicker, A., Popa, C., Jones, S., and Dahler, A. (1999). Histone deacetylase inhibitors as potential anti-skin cancer agents. Cancer Res. 59, 399–404. [PubMed] [Google Scholar]
- Schmiesing, J.A., Gregson, H.C., Zhou, S., and Yokomori, K. (2000). A human condensin complex containing hCAP-C-hCAP-E and CNAP1, a homolog of Xenopus XCAP-D2, colocalizes with phosphorylated histone H3 during the early stage of mitotic chromosome condensation. Mol. Cell. Biol. 20, 6996–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, K.B., and Salmon, E.D. (2002). Chromosome dynamics: new light on Aurora B kinase function. Curr. Biol. 12, R458–R460. [DOI] [PubMed] [Google Scholar]
- Speliotes, E.K., Uren, A., Vaux, D., and Horvitz, H.R. (2000). The survivin-like C. elegans BIR-I protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol. Cell 6, 211–223. [DOI] [PubMed] [Google Scholar]
- Steffensen, S., Coelho, P.A., Cobbe, N., Vass, S., Costa, M., Hassan, B., Prokopenko, S.N., Bellen, H., Heck, M.M., and Sunkel, C.E. (2001). A role for Drosophila SMC4 in the resolution of sister chromatids in mitosis. Curr. Biol. 11, 295–307. [DOI] [PubMed] [Google Scholar]
- Suavé, D.M., Anderson, H.J., Ray, J.M., James, W.M., and Roberge, M. (1999). Phosphorylation-induced rearrangement of the histone H3 NH2-terminal domain during mitotic condensation. J. Cell Biol. 145, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, K., Sugiura, K., Hara, T., Sugimoto, K., Shima, H., Ronda, K., Furukawa, K., Yamashita, S., and Urano, T. (2002). Aurora-B associated protein phosphatases as negative regulators of kinase activation. Oncogene 21, 3103–3111. [DOI] [PubMed] [Google Scholar]
- Taddei, A., Maison, C., Roche, D., and Almouzni, O. (2001). Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol. 3, 114–119. [DOI] [PubMed] [Google Scholar]
- Tanaka, T.U. (2002). Bi-orienting chromosomes on the mitotic spindle. Curr. Opin. Cell Biol. 14, 365–371. [DOI] [PubMed] [Google Scholar]
- Van Lint, C., Emiliani, S., Ott, M., and Verdin, E. (1996). Transcriptional activation and chromatin remodeling of the mv-1 promoter in response to histone acetylation. EMBO J. 15, 1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Wade, P.A., Pruss, D., and Wolffe, A. (1997). Histone acetylation: chromatin in action. Trends Bioch. Sci. 22, 128–132. [DOI] [PubMed] [Google Scholar]
- Waizenegger, I.C., Hauf, S., Meinke, A., and Peters, J.M. (2000). Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103, 399–410. [DOI] [PubMed] [Google Scholar]
- Wei, Y., Mizzen, C.A., Cook, R.G., Gorovsky, M.A., and Allis, C.D. (1998). Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc. Natl. Acad. Sci. USA 95, 7480–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y., Yu, L., Bowen, J., Gorovsky, M.A., and Allis, C.D. (1999). Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 97, 99–109. [DOI] [PubMed] [Google Scholar]
- Yoshida, M., Horinouchi, S., and Beppu, T. (1995). Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessay 17, 423–430. [DOI] [PubMed] [Google Scholar]