

Abstract
The E2F family of proteins is required to establish the correct cell-cycle-dependent transcription of genes that direct the process of cell division. All previously identified E2F proteins can act in a similar manner; depending on whether or not they are associated with the cell cycle inhibitors the retinoblastoma protein (pRB), p107, or p130, they can either repress or activate the transcription of E2F-responsive genes. We now report the cloning and characterization of another E2F family member, E2F-6, whose structure is reminiscent of the dominant inhibitors of other transcription factor families. The dimerization and DNA binding properties of E2F-6 are similar to those of the other E2F family members. However, it is not regulated by pRB, p107, or p130, and it is unable to activate transcription. Instead, it can act to repress the transcription of E2F responsive genes by countering the activity of the other E2F complexes via a pRB-, p107-, or p130-independent mechanism.
The retinoblastoma gene (RB-1) is one of the best studied of the tumor suppressor genes. Although it was originally identified by virtue of its absence in retinoblastomas, subsequent studies have shown that it is absent or mutated in at least one-third of all human tumors (1). The product of this gene [the retinoblastoma protein (pRB)] is also an essential target of the transforming proteins of the small DNA tumor viruses (2). In untransformed cells, the growth-suppressive properties of pRB are regulated by its cell-cycle-dependent phosphorylation (3). This phosphorylation is catalyzed by the cell-cycle-dependent kinase cyclin D⋅cdk4/6 (3). Consistent with this hypothesis, many human tumors contain activating mutations within either the cyclin D1 or cdk4 genes or have lost the cdk4/6-specific inhibitor p16 (4).
The growth-suppressive properties of pRB are dependent upon its ability to regulate a cellular transcription factor, E2F (3, 5). Many E2F-responsive genes have been identified, and their products are required for entry into, or passage through, the cell cycle. Consistent with its antiproliferative role, the pRB inhibits the transcriptional activity of E2F (6). Moreover, the resultant pRB–E2F complex actively represses the transcription of E2F-responsive genes by blocking the activity of adjacent transcription factors (7–9). Phosphorylation of the pRB causes it to dissociate from E2F, thereby switching E2F-responsive genes from the repressed to the induced state.
E2F is regulated by two additional proteins called p107 and p130 (10). These proteins share significant sequence similarity with the pRB (2, 10), and in overexpression assays they repress E2F in a similar manner to pRB (11, 12). However, genetic studies indicate that pRB, p107, and p130 have distinct properties in vivo. Whereas pRB is mutated in 30% of all human tumors, neither p107 nor p130 is a tumor suppressor (13). Moreover, analysis of mutant mouse strains indicates that pRB is essential for development, whereas loss of p107 or p130 does not alter viability or tumor incidence (14–18). It is unclear exactly how the different biological properties of these proteins relate to the manner in which they regulate E2F, but pRB, p107, and p130 are known to bind to E2F at defined but distinct stages of the cell cycle (10).
To date, at least seven human genes have been identified that encode components of the E2F transcriptional activity (10). These can be divided into two distinct groups, named E2F (E2F-1 through -5) and DP (DP-1 and -2). The products of these groups heterodimerize to give rise to high-affinity DNA binding activity and transcriptional activation (19–22). In vivo studies confirm that the endogenous E2F activity is generated from the combined properties of multiple E2F–DP complexes (22, 23). The individual E2F–DP species have different pRB, p107, and p130 binding properties. Although the DP subunit is essential, the E2F moiety mediates the specificity of this interaction. Complexes containing E2F-1, -2, or -3 associate with pRB but not p107 or p130 in vivo (24, 25). In contrast, E2F-4 and -5 complexes are capable of binding p107 and p130 (26–28). Consistent with these findings, sequence comparisons suggest that the family of E2F proteins can be subdivided into two distinct subgroups. The pRB-specific E2Fs (E2F-1 through -3) have an extended N-terminal domain that is absent in both E2F-4 or -5. There is also considerable variation in the sequence of the DNA binding, dimerization, and transactivation domains between members of the two E2F subgroups (E2F-1 through -3 versus E2F-4 and -5). These observations have led to the hypothesis that these two subgroups will play distinct roles in vivo that will at least partially account for the different biological consequences of loss of pRB, p107, or p130.
To examine the biochemical and functional properties of the endogenous E2Fs, we had previously developed specific antisera for each of the components of the E2F family (23). With these reagents, we have been able to demonstrate that the known E2F proteins are unable to account for all of the endogenous E2F–DP DNA binding activity (23). In this study, we describe the cloning and characterization of an additional E2F family member, E2F-6. The DNA binding and dimerization domains of E2F-6 are highly related to the corresponding domains of the previously identified family members, but this protein lacks the sequences necessary for either transactivation or pRB, p107, or p130 binding. We conclude that the E2F family contains a third subgroup of proteins whose structure is highly reminiscent of the dominant inhibitors of other transcription factor families.
MATERIALS AND METHODS
cDNA Identification and Characterization.
GenBank, EMBL, and DDBJ databases were searched with the protein sequence QKRRIYDITNVLEG by using the tblastn program. The identified E2F-6 human and mouse expressed sequence tags (ESTs) were obtained from Research Genetics (Huntsville, AL). A human fetal brain cDNA library (Stratagene) was screened with 1.6-kb EcoRI fragment of a human EST labeled with [α-32P]dCTP by random priming. Hybridization was performed at 42°C in 5× SSC/5× Denhardt’s solution/30% formamide/0.5% SDS/dextran sulfate (50 μg/ml)/salmon sperm DNA (150 μg/ml). Filters were washed at 55°C for three 20-min periods in 1× SSC/0.1% SDS. Positive clones were identified by autoradiography. Exonuclease III (New England Biolabs) digestion was used to generate nested deletions of both EST and cDNA clones, which were then sequenced with Sequenase 2.0 (United States Biochemical).
Plasmid Construction.
The human E2F-6 ORF was amplified by PCR with the primers 6.6 (GTTAGGATCCATGCGGCACGAGAAGTTACCCAG) and 6.10 (CTCAGGATCCATCAGTTGCTTACTTCAAG) and Vent polymerase. The PCR product was digested with BamHI and subcloned into pHACMV-neo-Bam to generate pCMV-HA-E2F-6. The plasmids pE2F4-CAT, pRSV-luciferase, pCMV-E2F-1, pCMV-E2F-4, pCMV-DP-1, pCMV-HA-DP-1, pCMV-HA-DP2, pCMV-RB, and pCMV-p107 have been described (20, 22, 23). The 6× His-tagged E2F-6 vector was constructed by amplifying the E2F-6 ORF with primers 6.6 and 6.12 (CACTAAGCTTATCAGTTGCTTACTTCAAGCA). The PCR product was digested with BamHI and EcoRI and subcloned into pQE30 (Qiagen, Chatsworth, CA).
Northern Blot Analysis.
The cell-line Northern blot [containing poly(A)+ RNA from 293 (adenocarcinoma), HeLa (cervical carcinoma), ML-1 (myeloid leukemia), T98G (neuroblastoma), MCF7 (human breast cancer cell line), or C33-A (cervical carcinoma)] or a human tissue blot [containing 2 μg (per lane) of poly(A)+ RNA isolated from the indicated tissues (CLONTECH)] was screened with probes corresponding to the full-length E2F-6 ORF or a 1,052-bp XmnI–SacI fragment from the 3′ untranslated region. These fragments were labeled with [α-32P]dCTP by using the Prime-It II kit (Stratagene). The blots were hybridized for 18 h at 65°C in 0.5 M sodium phosphate, pH 7.5/1 mM EDTA, pH 8.0/5% SDS/1% BSA and washed three times with 1× SSC/0.1% SDS at 65°C before autoradiography.
Polyclonal Antibody Production and Western Blotting.
The full-length 6× His-tagged human E2F-6 protein (amino acids 1–275) was expressed in bacteria, purified over a Ni2+-nitrilotriacetic acid-agarose resin (Qiagen), and used to immunize mice. Western blotting was performed as described by Moberg et al. (23).
Transient Transfection and in Vitro Assays.
Cells were grown under standard conditions in DMEM supplemented with 10% fetal calf serum. Transient transfections were performed as described (29). For the immunoprecipitation and gel retardation assays, transfections were conducted with 10 μg of each of the indicated plasmids plus pCMV-neo-Bam to give a total of 30 μg. Gel shift assays were carried out as described (29) with unlabeled cell extracts normalized for total protein concentration. For immunoprecipitations, the cells were labeled with 250 μCi of [35S]methionine Express labeling mix (NEN) in methionine-free medium (GIBCO/BRL) for 3.5 h. Immunoprecipitations were performed as described (25) with the following antibodies: 12CA5 [anti-hemagglutinin (HA) tag], KH20 [anti-E2F-1 (30)], LLF4–1 [anti-E2F-4 (31)], sc-610x [anti-DP-1 (Santa Cruz Biotechnology)], sc-829x [anti-DP-2 (Santa Cruz Biotechnology)]. Precipitates were resolved on a 10% SDS polyacrylamide gels by PAGE and detected by fluorography. For transactivation assays, C33-A cells were transfected in duplicate with 4 μg of E2F4-CAT, 2 μg of pRSV-luciferase (as an internal control for transfection efficiency), 14 μg of carrier DNA (pBKS+), and the indicated amounts of the pCMV-E2F expression vectors. These transfections were performed in the presence or absence of 3 μg of pCMV-HA-DP2. Within each experiment, the total concentration of CMV expression vector was kept constant by the addition of pCMV-neo-Bam. chloramphenicol acetyltransferase (CAT) and luciferase assays were conducted as described in Lees et al. (25).
RESULTS
Isolation of cDNAs Encoding an E2F Family Member.
At the start of this study, five genes had been identified that encode members of the E2F family of proteins. We have shown previously that these proteins account for a significant proportion of the endogenous E2F–DP complexes, but there must be at least one additional E2F (23). The greatest homology between the known E2F family members maps to the C-terminal half of the DNA binding domain. This contains a stretch of 15 amino acids (QKRRIYDITNVLEGI) that is invariant in the previously identified E2Fs. In an attempt to identify additional E2F family members, we searched the EST database for cDNA clones that encode this motif. As expected, we were able to identify multiple ESTs derived from E2F-1, -2, -3, -4, or -5. In addition, this search identified one mouse (GenBank accession no. AA041604/AA050073) and one human (GenBank accession no. AA127210) EST that did not correspond to the known E2Fs. These clones were highly related to one another at the nucleotide level, suggesting that they were mouse and human homologues of the same gene. The QKRRIYDITNVLEGI motif was highly, but not completely, conserved in one of the predicted translation products and almost all of the variation between these two clones was in the third base position of this ORF. These data strongly suggested that this was the correct ORF and that these cDNAs were excellent candidates to encode another E2F.
With both the EST database and standard library screening techniques, we identified multiple overlapping cDNAs that encompassed 2,027 bp of the human gene. This sequence starts within the ORF and extends to the poly(A)+ tail. None of the human cDNAs diverged from this assembled sequence. We also identified two noncontiguous mouse cDNAs. The first is a 638-bp clone that shares significant homology with the 3′ untranslated region of the human gene. The second includes 216 bp of 5′ untranslated region followed by the initiating ATG (as judged by the presence of a good Kozak consensus and an upstream in-frame termination codon) and 536 bp of ORF. Sequence comparison suggested that the human cDNA lacks only a short region (18 bp) of 5′ coding sequence (data not shown). The overlapping regions of the human and mouse sequence are highly conserved at both the nucleotide (83%) and predicted amino acid (92%) level (Fig. 1A). We therefore concluded that we had identified the mouse and human orthologues of another gene, hereafter designated E2F-6, that share significant homology with the known E2Fs.
Figure 1.

E2F-6 is another member of the E2F family. (A) Amino acid sequence comparison of human versus murine E2F-6. (B) Amino acid sequence comparison of the human E2F proteins. Domains responsible for DNA binding, dimerization (leucine zipper, marked box), and pocket protein binding are indicated. Conserved residues are denoted in boldface type.
The previously identified E2F proteins have been divided into two distinct subgroups (E2F-1 through -3 versus E2F-4 and -5) on the basis of differences in their amino acid sequence. The central portion of the E2F-6 protein shared considerable homology with the the domains of E2F-1 through -5 that are known to mediate their dimerization (both the leucine zipper domain and region of homology known as the marked box) and DNA binding properties (Fig. 1B). Within these domains, many of the residues that are absolutely conserved between E2F-1 through -5 are also maintained in E2F-6. However, at a significant proportion of these conserved residues, E2F-6 had alternative codon usage from that found in the other E2F genes (data not shown). Moreover, when we examined the amino acid positions that are known to distinguish E2F-1 through -3 from E2F-4 and -5, the corresponding residue in E2F-6 rarely fit into either subgroup (see Fig. 1B). The distinction between E2F-6 and the other E2F family members was further underscored by the degree of sequence variation outside of the DNA binding and dimerization domains. The N-terminal domain of E2F-6 was of intermediate length, relative to those of the two E2F subgroups, and resembled neither subclass. More importantly, the E2F-6 protein terminated just 42 amino acids beyond the marked box motif (the C-terminal portion of the dimerization domain). As a result this protein lacks the sequences that are know to mediate either the transcriptional activation or pRB, p107, or p130 binding properties of the other E2F proteins. Thus, these findings suggest that E2F-6 represents a third subclass of the E2F protein family that is likely to display distinct properties from the previously identified members.
E2F-6 Is Widely Expressed in Vivo.
E2F-1 through -5 are all expressed in a wide variety of tissues (25–28, 30, 32, 33). We were therefore interested to establish the expression pattern of E2F-6. Initially, we isolated poly(A)+ RNA from the indicated human cell lines and screened them for the presence of the E2F-6 mRNA by Northern blotting using a probe derived from the coding sequence (Fig. 2A). In these and every other cell line examined, this probe hybridized with similar stringency to two distinct messages. To date, each of the E2F-6 cDNAs that we have identified corresponds to a single common transcript. It was therefore unclear why the E2F-6 probe detected two different mRNAs. To address this issue, we rescreened these Northern blots with a probe derived from the 3′ untranslated region. This second probe also hybridized to the same two transcripts (data not shown), indicating that they must both contain at least some of the sequences from both the coding and 3′ untranslated region of our E2F-6 cDNA.
Figure 2.
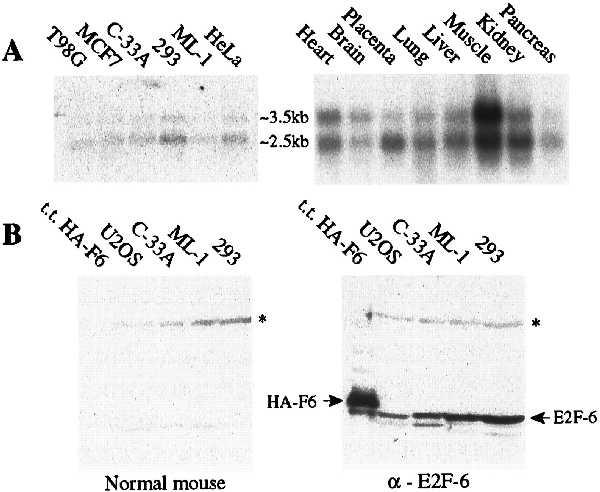
Expression patterns of E2F-6. (A) Northern blot analysis of poly(A)+ RNA isolated from the indicated cell lines and tissues (human tissue blot from CLONTECH), screened with a probe derived from the full-length E2F-6 coding sequence. (B) Western blot analysis of extracts derived from C33-A cells transiently transfected with pCMV–HA–E2F-6 (200 ng) and from untransfected U2OS, C33-A, ML-1, and 293 cells (50 μg). The positions of HA–E2F-6 and endogenous E2F-6 proteins are indicated by arrows. The band running beneath HA–E2F-6 is a degradation product of this transfected protein (data not shown). The asterisk denotes a nonspecific band.
We also examined the expression of E2F-6 in human tissues. For these experiments, we screened a human tissue blot (CLONTECH) with the probe corresponding to the E2F-6 coding region (Fig. 2A). As with cultured cells, we detected two E2F-6 transcripts in every tissue examined. We therefore conclude that E2F-6 will be expressed in most if not all cell and tissue types. At this time, we are unable to explain the structural difference(s) between the two mRNAs.
Given the existence of the two E2F-6 transcripts, we could not rule out the possibility that alternate splicing could give rise to protein products with two distinct C termini, one that corresponds to that encoded by our identified cDNA clones and one that more closely resembles the domain structure of the previously identified E2F proteins. To address this issue, we raised polyclonal antiserum against the predicted human E2F-6 protein. Control experiments confirmed that the antiserum recognized epitopes throughout the E2F-6 protein but did not cross-react to any of the other known E2Fs (data not shown). When tested on Western blots, the antiserum detected a single 35-kDa protein in all tested cell lines that migrated slightly faster than an HA-tagged version of the E2F-6 protein produced by transient transfection (Fig. 2A and data not shown). We therefore conclude that the E2F-6 protein is expressed in vivo and it exists predominantly in the form predicted by the cDNA clones.
E2F-6 Displays Low-Affinity E2F DNA Binding Activity.
Given the unusual structure of E2F-6, we wished to establish whether this protein retained any of the properties of the known E2F family members. All previously identified E2Fs can bind to either of the human DPs and this heterodimerization is known to be a prerequisite for the high-affinity DNA binding. We therefore initiated our analysis by comparing the ability of E2F-1, -4, and -6 to bind to known E2F-associated proteins. For these experiments, C33-A cells were transiently transfected with eukaryotic expression vectors encoding E2F-1, E2F-4, or an HA-tagged version of E2F-6 in the presence or absence of the DP (either DP-1 or DP-2) or pocket (pRB, p107, or p130) proteins. The transfectants were labeled with [35S]methionine and then subjected to immunoprecipitation with the indicated antibodies (Fig. 3A). Although there was considerable variation in the efficiency of the individual transfections, these experiments allowed us to assess the DP and pocket protein binding properties of the individual E2F proteins. A monoclonal antibody specific for the HA tag was able to recover either DP-1 or DP-2 in approximately stochiometric amounts with HA–E2F-6 (Fig. 3A, lanes 18 and 21). Moreover, HA–E2F-6 seemed to bind to these proteins as well as either E2F-1 or E2F-4 (Fig. 3A, compare lanes 18 and 21 with lanes 3 and 11 and lanes 6 and 14). This confirmed that E2F-6 is able to heterodimerize with the DP proteins despite the sequence variation within its presumed dimerization domain. Consistent with previous studies, the E2F-1–DP-1 (Fig. 3A, lanes 4 and 5) and E2F-4–DP-1 (Fig. 3A, lanes 12 and 13) complexes were able to associate with pRB and p107 when overexpressed. In contrast, we did not detect any interaction between the HA–E2F-6–DP complexes and any of the three pocket proteins (Fig. 3A, lanes 19 and 20 and data not shown). This finding is consistent with the absence of a pocket protein binding motif within the E2F-6 protein sequence.
Figure 3.
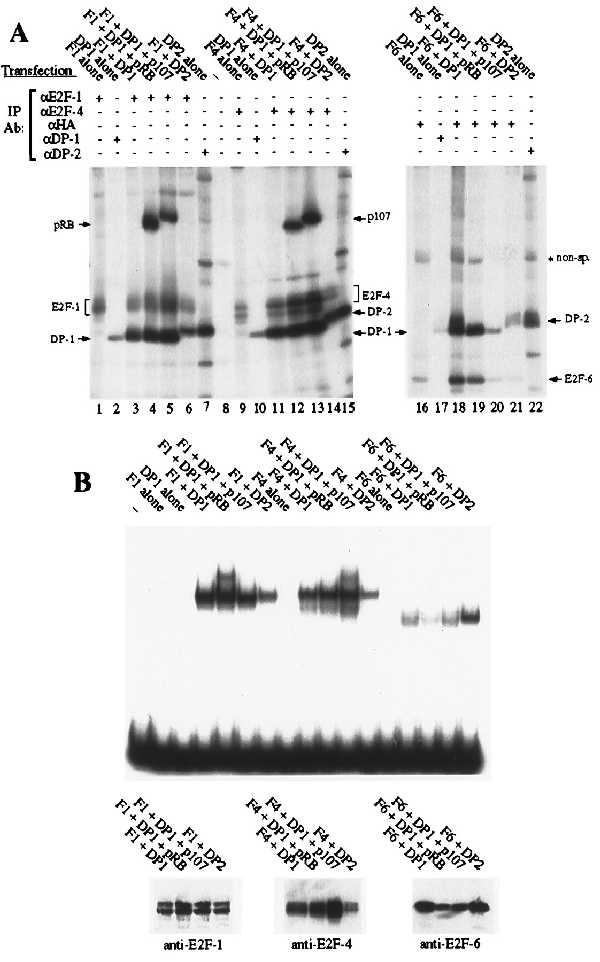
Dimerization and DNA binding properties of E2F-6 (A) C33-A cells were transiently transfected with expression vectors encoding E2F-1, E2F-4, or HA–E2F-6 in the presence or absence of DP (DP-1 or DP-2) or pocket proteins (pRB, p107) and immunoprecipitated with the indicated antibodies. (B) C33-A cells were transiently transfected with the identical combinations of expression vectors as in A. Gel shift assays were carried out on the indicated unlabeled cell extracts (1.5 μg of total protein per lane) by using the consensus E2F site from the adenoviral E2 promoter (TTTCGCGCCCTTT). Western blot assays were carried out on the same unlabeled cell extracts (300 ng of total protein per lane) by using monoclonal antibodies against E2F-1 (KH95), E2F-4 (LLF4–1), or the HA tag (12CA5). Gel retardation assays have also been conducted by using the additional E2F sites TTTCCCGCCTTT, TTTCCCGCCAAA, TTTCCCGCGTGT, or ATTCCCGCGCTTT with similar differences in affinity.
After establishing that E2F-6 can associate with the DP proteins, we next examined the DNA binding activity of these heterodimers. Transient transfection was used to generate the relevant E2F complexes, exactly as described above, and the resultant cell extracts were measured for total protein content. These were then tested in gel retardation assays with the consensus E2F site and equivalent input levels of total protein (Fig. 3B). Consistent with previous studies, E2F-1 and E2F-4 were unable to bind to DNA in the absence of a cotransfected DP protein (Fig. 3B, lanes 3 and 8). In a similar manner, we were unable to detect any increase in the levels of E2F DNA binding activity in cells that were transfected with HA–E2F-6 alone (Fig. 3B, lane 13). In the same assay, E2F-1, -4, and -6 were able to bind to DNA when associated with either DP-1 (Fig. 3B, lanes 4, 9, and 14) or DP-2 (Fig. 3B, lanes 7, 12, and 17). However, we consistently detected less E2F DNA binding activity in extracts derived from cells transfected with HA–E2F-6 rather than E2F-1 or E2F-4 (Fig. 3B, compare lanes 14–17 with lanes 4–7 and 9–12). Given this finding, we also assessed the expression levels of these E2Fs in Western blots (Fig. 3B). Because the blots were probed with different antibodies, we cannot make definitive conclusions about the relative levels of these proteins. However, these antibodies have similar avidities for their respective antigens (unpublished observations), suggesting that E2F-1 and E2F-4 are not expressed at significantly higher levels than HA–E2F-6. This suggests that HA–E2F-6 containing complexes have a lower affinity for the consensus E2F site relative to complexes containing either E2F-1 or -4. This low DNA binding activity was observed with multiple independent transfections and was not improved by the removal of the HA tag (data not shown). Given these observations, we also tested the E2Fs for their ability to bind to other E2F binding sites (data not shown). The DNA binding activity of E2F-6–DP did increase when we used the probe TTTCCCGCC(A/T)(A/T)(A/T). However, this site was previously identified by site selection assays as the preferred recognition sequence of complexes containing E2F-1, -2, -3, or -4, and these species also have a higher affinity for this sequence than for any other E2F site (B.F., unpublished data). In fact, relative to the other E2F species, E2F-6–DP complexes bound less well to each of the five probes tested. We therefore conclude that E2F-6 has a lower affinity for DNA, at least when associated with either of the known DP proteins.
E2F-6 Is Unable to Activate the Transcription of E2F-Responsive Genes.
E2F-6 lacks the C-terminal sequences that are known to mediate the transcriptional activity of E2F-1 through -5. However, its shortened C-terminal domain is highly charged and contains a significant proportion of acidic residues (22%), suggesting that it might activate transcription via this alternative motif. To test this hypothesis, we transiently transfected C33-A cells with a chimeric reporter construct, E2F4-CAT, in which the expression of the CAT gene is controlled by a minimal promoter containing four consensus E2F sites upstream of the E1B TATA box (20) and increasing amounts of the eukaryotic expression vectors encoding E2F-1, E2F-4, or HA–E2F-6 (Fig. 4A). These transfections were conducted in either the absence or presence of CMV-DP-2 as indicated. E2F-1 and E2F-4 both substantially increased the activation of this reporter. In contrast, E2F-6 did not bring about any increase in the level of E2F transcriptional activity, either in the absence or presence of cotransfected DP proteins (Fig. 4A). Instead, the increasing input levels of CMV-E2F-6 steadily inhibited the ability of the endogenous E2F complexes to activate this reporter (Fig. 4B). We did not see any inhibition of reporter activity when the E2F sites were deleted, indicating this repression was specific for E2F-responsive reporters (data not shown). This inhibition was observed in either the absence or presence of an excess of exogenously expressed DP protein (Fig. 4B and data not shown).
Figure 4.
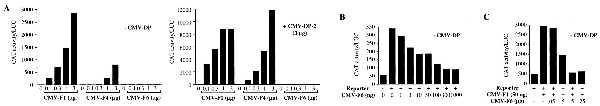
Effects of E2F-6 on transactivation of an E2F4-CAT reporter C33-A cells were transiently transfected in duplicate with 4 μg of E2F4-CAT, 2 μg of Rous sarcoma virus–luciferase (as an internal control), 14 μg of carrier DNA (pBKS+ and pCMV-neo-Bam) in the absence or presence of 3 μg of pCMV-HA-DP2 plus pCMV-E2F expression vectors as indicated. CAT and luciferase activity were determined 24 h after transfection. The values shown are the average of duplicate transfectants for representative experiments.
After finding that E2F-6 can block the activity of the endogenous E2F complexes, we wanted to establish whether it could also inhibit a cotransfected E2F. For this experiment, C33-A cells were transiently transfected with the E2F4-CAT reporter in either the absence or presence of CMV-E2F-1 (50 ng) and increasing amounts of CMV-E2F-6 (Fig. 4C). At this input level, E2F-1 increased the activation of this reporter by approximately 6-fold. This was effectively inhibited by increasing input levels of CMV-E2F-6. Exactly as described above, this repression was observed in either the absence or presence of cotransfected DP (data not shown). We therefore conclude that E2F-6 is unable to activate transcription and, at least when overexpressed, can inhibit the transcriptional activity of the other E2F species.
DISCUSSION
E2F plays a pivotal role in the regulation of cellular proliferation by controlling the expression of genes that are essential for either entry into, or passage through, the cell cycle. This activity is extremely complex, arising from the combined action of multiple E2F–DP heterodimers. Although there are differences in their cell cycle regulation, the previously identified E2F–DP species can all display the same transcriptional properties. In the presence of an associated pocket protein, they each behave as transcriptional repressors. Once released from the pocket proteins, these same E2F–DP species can activate transcription. Although the DP moiety is required for activity, the transcriptional activation and pocket protein binding properties of the E2F–DP complexes are entirely dependent upon sequences within the E2F protein.
Herein we describe the cloning and characterization of the human and murine orthologues of a gene that contains the most conserved domains of the known E2F family members, including the core sequences required for DNA binding and dimerization. Functional studies confirm that the human protein can dimerize with either DP-1 or DP-2 and the resultant complexes can bind to DNA in a sequence-specific manner. These findings confirm that this protein is a genuine member of the E2F family, and we have, therefore, named it E2F-6. Despite this designation, our data indicate that this protein functions in a different manner from the other E2F family members. (i) E2F-6-containing complexes bind to DNA significantly less well than the other E2F–DP species. Although we have yet to calculate dissociation constants for these interactions, our preliminary studies suggest that E2F-6-containing complexes recognize the same target sequences as the other E2F species but they bind to these sites with at least a 5-fold lower affinity. (ii) E2F-6 lacks the domains that are known to mediate the transactivation and pocket protein binding properties of the other E2Fs and it is unable to perform either of these functions. (iii) Overexpressed E2F-6 is able to block the transcriptional activity of either cotransfected E2F-1 or the endogenous E2F complexes. Although the mechanism of this repression is unclear, our data indicate that transcriptional inhibition is not due solely to the sequestration of DP from the other E2F family members by abnormally high levels of E2F-6.
Many other transcription factor families include one or more members that behave as transcriptional inhibitors but there is considerable variation in the mechanism by which repression is mediated. The Id proteins of the myogenic basic helix–loop–helix family, the I-POU protein of the POU domain family, and the CHOP protein of the c/EBP-like family lack the sequences necessary for high-affinity DNA binding and, therefore, function in a DNA-independent manner (34–36). Instead, they are able to inhibit transcription by binding to the essential heterodimeric partners of the positively acting family members and forming nonfunctional complexes. There are also several examples of inhibitory proteins that repress transcription in a DNA-binding-dependent manner. In this case, inhibition can be mediated by either exclusion of other family members from the DNA or through the recruitment of cellular factors that actively inhibit transcription. One example of this latter case is the Mad–Max complex that has recently been shown to mediate repression in a sequence-specific manner through its ability to recruit histone deacetylase activity to its target genes (37).
Because the E2F proteins function as part of a heterodimeric complex, it is possible to envisage that the repressive effects of E2F-6 could be mediated by any of the mechanisms described above. Although we cannot rule out any of these models, two observations suggest that E2F-6 is unlikely to act solely through the sequestration of the DP proteins. (ii) Unlike the other transcriptional repressors that act by this mechanism, E2F-6 retains the ability to bind to DNA. (ii) At least as observed in our overexpression assays, addition of excess DP protein does not prevent E2F-6 from inhibiting the activity of the other E2F complexes. This strongly suggests that the observed repression is dependent upon an additional function(s) of the E2F-6 protein. DP binding might still be required for this repression as an essential component of an actively repressive complex.
Clearly, the experiments described above do not allow us to address the mechanism of action of the endogenous E2F-6 species. In fact, it is still unclear whether or not E2F-6 functions as a repressor in vivo. It is possible to envisage alternate models for E2F-6 function. For example, the DP proteins are known to be present in excess relative to the E2Fs in vivo and E2F-6 could exist to chaperone this pool of “unbound” DP. If correct, the “chaperone” model yields two clear predictions about the endogenous E2F-6 protein. (i) It should exist at sufficiently high levels in vivo to be able to bind to any DP protein that is not associated with E2F-1 through -5. (ii) E2F-6 should have a lower affinity/avidity for the DPs than any of the other E2F family members to ensure the efficient transfer of the DP from E2F-6 to E2F-1 through -5. Clearly, the “repressor” model of E2F-6 action also yields testable predictions. Regardless of whether or not its repressive properties are dependent or independent of its ability to bind to DNA, E2F-6 should have a similar or greater affinity for the DP proteins than the other E2Fs. Depending on its mechanism of action, it could also be present at either greater (if it acts through the sequestration of DP) or similar/lower (if it regulates transcription in a sequence specific manner) levels than the other E2Fs in vivo. A direct comparison of the relative affinity/avidity of the individual E2F family members for DP should help us to distinguish between these “chaperone” and “repressor” models, as will information about the relative abundance and DNA binding activity of the endogenous E2F-6 protein. Although we have yet to establish the true physiological role of E2F-6, its broad expression pattern suggests that it makes an important contribution to the regulation of E2F activity in vivo.
Acknowledgments
We are grateful to members of the Lees laboratory for helpful discussion throughout the course of this study and to Patrick Humbert and Steve Bell for critical reading of the manuscript. This work was supported by a grant from the National Institutes of Health. J.A.L. is a recipient of the Professor Irwin Sizer and Helen Sizer Career Development Professorship.
ABBREVIATIONS
- pRB
retinoblastoma protein
- EST
expressed sequence tag
- CAT
chloramphenicol acetyltransferase
- HA
hemagglutinin
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF041381).
References
- 1.Weinberg R A. Cancer Surv. 1992;12:43–57. [PubMed] [Google Scholar]
- 2.Dyson N, Harlow E. Cancer Surv. 1992;12:161–195. [PubMed] [Google Scholar]
- 3.Bartek J, Bartkova J, Lukas J. Curr Opin Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- 4.Pollock P M, Pearson J V, Hayward N K. Genes Chromosomes Cancer. 1996;15:77–88. doi: 10.1002/(SICI)1098-2264(199602)15:2<77::AID-GCC1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Nevins J R. Nature (London) 1992;358:375–376. doi: 10.1038/358375a0. [DOI] [PubMed] [Google Scholar]
- 6.Hiebert S W, Chellappan S P, Horowitz J M, Nevins J R. Genes Dev. 1992;6:177–185. doi: 10.1101/gad.6.2.177. [DOI] [PubMed] [Google Scholar]
- 7.Weintraub S J, Prater C A, Dean D C. Nature (London) 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- 8.Bremner R, Cohen B L, Sopta M, Hamel P A, Ingles C J, Gallie B L, Phillips R A. Mol Cell Biol. 1995;15:3256–65. doi: 10.1128/mcb.15.6.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weintraub S J, Chow K N B, Luo R X, Zhang S H, He S, Dean D C. Nature (London) 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- 10.Beijersbergen R L, Bernards R. Biochim Biophys Acta. 1996;1287:103–120. doi: 10.1016/0304-419x(96)00002-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D M, Dyson N, Harlow E. Genes Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]
- 12.Starostik P, Chow K N, Dean D C. Mol Cell Biol. 1996;16:3606–3614. doi: 10.1128/mcb.16.7.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg R A. Cell. 1996;85:457–459. doi: 10.1016/s0092-8674(00)81244-1. [DOI] [PubMed] [Google Scholar]
- 14.Clarke A R, Maandag E R, van Roon M, van der Lugt N M T, van der Valk M, Hooper M L, Berns A, te Riele H. Nature (London) 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 15.Jacks T, Fazeli A, Schmitt E M, Bronson R T, Goodell M A, Weinberg R A. Nature (London) 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 16.Lee E Y, Chang C Y, Hu N, Wang Y C, Lai C C, Horrup K, Lee W H, Bradley A. Nature (London) 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 17.Cobrinik D, Lee M H, Hannon G, Mulligan G, Bronson R T, Dyson N, Harlow E, Beach D, Weinberg R A, Jacks T. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 18.Lee M H, Williams B O, Mulligan G, Mukai S, Bronson R T, Dyson N, Harlow E, Jacks T. Genes Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- 19.Bandara L R, Buck V M, Zamanian M, Johnston L H, La Thangue N B. EMBO J. 1993;12:4317–4324. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helin K, Wu C, Fattaey A R, Lees J A, Dynlacht B D, Ngwu C, Harlow E. Genes Dev. 1993;7:1850–1861. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]
- 21.Krek W, Livingston D M, Shirodkar S. Science. 1993;262:1557–1560. doi: 10.1126/science.8248803. [DOI] [PubMed] [Google Scholar]
- 22.Wu C L, Zukerberg L R, Ngwu C, Harlow E, Lees J A. Mol Cell Biol. 1995;15:2536–2546. doi: 10.1128/mcb.15.5.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moberg K, Starz M A, Lees J A. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyson N, Dembski M, Fattaey A, Ngwu C, Ewen M, Helin K. J Virol. 1993;67:7641–7647. doi: 10.1128/jvi.67.12.7641-7647.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lees J A, Saito M, Vidal M, Valentine M, Look T, Harlow E, Dyson N, Helin K. Mol Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beijersbergen R L, Kerkhoven R M, Zhu L, Carlee L, Voorhoeve P M, Bernards R. Genes Dev. 1994;8:2680–2690. doi: 10.1101/gad.8.22.2680. [DOI] [PubMed] [Google Scholar]
- 27.Ginsberg D, Vairo G, Chittenden T, Xiao Z X, Xu G, Wydner K L, DeCaprio J A, Lawrence J B, Livingston D M. Genes Dev. 1994;8:2665–2679. doi: 10.1101/gad.8.22.2665. [DOI] [PubMed] [Google Scholar]
- 28.Hijmans E M, Voorhoeve P M, Beijersbergen R L, van ’T Veer L J, Bernards A. Mol Cell Biol. 1995;15:3082–3089. doi: 10.1128/mcb.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Greene & Wiley; 1988. [Google Scholar]
- 30.Helin K, Lees J A, Vidal M, Dyson N, Harlow E, Fattaey A. Cell. 1992;70:337–350. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- 31.Verona R, Moberg K, Estes S, Starz M, Vernon J P, Lees J A. Mol Cell Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivey-Hoyle M, Conroy R, Huber H E, Goodhart P J, Oliff A, Heimbrook D C. Mol Cell Biol. 1993;13:7802–7812. doi: 10.1128/mcb.13.12.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaelin W G, Jr, Krek W, Sellers W R, DeCaprio J A, Ajchenbaum F, Fuchs C S, Chittenden T, Li Y, Farnham P J, Blanar M A, et al. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- 34.Benezra R, Davis R L, Lockshon D, Turner D L, Weintraub H. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 35.Treacy M N, He X, Rosenfeld M. Nature (London) 1991;350:577–584. doi: 10.1038/350577a0. [DOI] [PubMed] [Google Scholar]
- 36.Ron D, Habener J F. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 37.Pazin M J, Kadonaga J T. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]