

Abstract
The purpose of this study was to define the role of the Rho family of small GTPases in the β-adrenergic regulation of the Na,K-ATPase in alveolar epithelial cells (AEC). The β-adrenergic receptor agonist isoproterenol (ISO) increased the Na,K-ATPase protein abundance at the plasma membrane and activated RhoA in a time-dependent manner. AEC pretreated with mevastatin, a specific inhibitor of prenylation, or transfected with the dominant negative RhoAN19, prevented ISO-mediated Na,K-ATPase exocytosis to the plasma membrane. The ISO-mediated activation of RhoA in AEC occurred via β2-adrenergic receptors and involved Gs-PKA as demonstrated by incubation with the protein kinase A (PKA)-specific inhibitors H89 and PKI (peptide specific inhibitor), and Gi, as incubation with pertussis toxin or cells transfected with a minigene vector for Gi inhibited the ISO-mediated RhoA activation. However, cells transfected with minigene vectors for G12 and G13 did not prevent RhoA activation by ISO. Finally, the ISO-mediated Na,K-ATPase exocytosis was regulated by the Rho-associated kinase (ROCK), as preincubation with the specific inhibitor Y-27632 or transfection with dominant negative ROCK, prevented the increase in Na,K-ATPase at the plasma membrane. Accordingly, ISO regulates Na,K-ATPase exocytosis in AEC via the activation of β2-adrenergic receptor, Gs, PKA, Gi, RhoA, and ROCK.
INTRODUCTION
β-adrenergic receptors are members of the large family of seven membrane-spanning, GTP-binding protein-coupled receptors (GPCRs). In response to agonists, specific domains of the GPCRs interact with heterotrimeric GTP-binding proteins leading to the exchange of GTP for GDP, resulting in the dissociation of the heterotrimer into active Gαand Gβγ-subunits (Rockman et al., 2002). It is commonly assumed that β-adrenergic receptor agonists promote the activation of the stimulatory G protein, Gs, which in turn increases the activity of adenylyl cyclase, increasing the cellular levels of cAMP and the phosphorylation, via cAMP-dependent protein kinase A (PKA), of downstream proteins. In addition to Gs activation, β2-adrenergic receptors have been shown to be coupled to the pertussis toxin (PTX)-sensitive heterotrimeric G protein, Gi (Daaka et al., 1997; Post et al., 1999; Gosmanov et al., 2002).
β-adrenergic receptor agonists increase lung edema clearance by upregulating the Na,K-ATPase in the alveolar epithelium (Berthiaume et al., 1987; Suzuki et al., 1995; Saldias et al., 1998; Saldias et al., 1999, 2000; Sznajder et al., 2002; Ridge et al., 2003). The Na,K-ATPase located at the basolateral plasma membrane (BLM) of alveolar epithelial cells (AEC) generates, via the vectorial transport of Na+, the gradient necessary for the movement of water from the alveolar space into the pulmonary circulation (Rutschman et al., 1993; Saumon and Basset, 1993; Sznajder et al., 1995; Ridge et al., 2003). Activation of β-adrenergic receptors by isoproterenol (ISO) in AEC resulted in increased Na,K-ATPase activity due to the translocation of Na,K-ATPase molecules from intracellular compartments to the BLM via a process that is dependent on the actin cytoskeleton (Bertorello et al., 1999; Ridge et al., 2003).
Studies in secretory cells have shown that remodeling of the actomyosin cortex is a prerequisite for regulated exocytosis (Lang et al., 2000; Oheim and Stühmer, 2000). The Rho family of GTPases is thought to have a central role in vesicular trafficking pathways by controlling the organization of the actin cytoskeleton to spatially direct the transport of vesicles (Guo et al., 2001). Rho GTPases act as molecular switches cycling between GDPand GTP-bound states. When bound to GDP they are inactive; upstream events lead to the exchange of GDP for GTP and the protein switches into an active conformation (Van-Aelst and D'Souza-Schorey, 1997; Kjøller and Hall, 1999). Rho in its inactive state is cytosolic and complexed with GDIs (guanine nucleotide dissociation inhibitors; Fukumoto et al., 1990). Receptor-mediated activation leads to recruitment of Rho to the plasma membrane, where it gets anchored through a geranylgeranyl lipid residue that is attached to the C terminus. Once at the plasma membrane, through the action of GEFs (guanine nucleotide exchange factors), the GTP loading occurs (Cherfils and Chardin, 1999).
The present study was conducted to determine whether RhoA participated in the β-adrenergic receptor-mediated exocytosis of the Na,K-ATPase in AEC. The results demonstrate that ISO, via β2-adrenergic receptors, Gs-PKA and Gi, activates RhoA and that RhoA via Rho-associated kinase has an important regulatory role in the β-adrenergic-mediated Na,K-ATPase exocytosis in AEC.
MATERIALS AND METHODS
Reagents
86Rb+ was purchased from Amersham Pharmacia (Piscataway, NJ). PKA inhibitor peptide myristoylated, PTX, and Rho-associated kinase inhibitor, Y-27632, were from Calbiochem (La Jolla, CA). All other chemicals were purchased from Sigma (St. Louis, MO). The Na,K-ATPase α 1 mAb (clone 464.6) and antiphospho-MYPT1 (Thr696) polyclonal antibody were from Upstate Biotechnolgy (Lake Placid, NY). RhoA mAb (clone 26C4) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Myosin phosphatase target subunit (MYPT) polyclonal antibody was from BabCO (Berkeley Antibody Company, Richmond, CA). Secondary goat anti-mouse HRP and goat anti-rabbit HRP were from Bio-Rad (Hercules, CA).
Cell Culture
A549 cells (ATCC CCL 185, a human adenocarcinoma cell line) were grown in DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were seeded in 6or 10-cm plates, grown to confluence, and serum-starved 18–24 h before treatments. A549 cells has been proven to be a good model for the study of Na+-transport of AEC (Lazrak et al., 2000; Lecuona et al., 2000).
Permanent Transfection
A549 cells were plated in 6-cm plates at 2–3 × 105 cells/plate and transfected with 2 μg of plasmid DNA (dominant negative [dn] RhoA [RhoAN19], a gift of Dr. S. Gutkind, NIH; minigenes for Gi, G12, and G13, a gift from Dr. H. Hamm, Vanderbilt University) by using Superfect Reagent (Qiagen, Hilden, Germany), as indicated by the manufacturer. Transfectants were selected in the presence of 400 μg/ml geneticin (G418; Mediatech, Herndon, VA). Individual colonies were isolated using cloning cylinders (PGC Scientifics, Frederick, MD), and the resulting cell lines were propagated in complete DMEM supplemented with G418.
Transient Transfection
A549 cells were plated in 6-cm plates at 5 × 105 cells/plate and transfected with 5 μg of plasmid DNA (dominant negative [ROCK KD-IA]] and activated [ROCK Δ 3] forms of ROCK, a gift of Dr. S. Narumiya, Kyoto University) by using Lipofectin Reagent (Life Technologies, Gaithersburg, MD), as indicated by the manufacturer. Cells were used 48 h posttransfection.
1% Triton X-100–soluble Fraction
Cells were treated with different agonists/antagonists at 37°C, placed on ice, and washed twice with ice-cold phosphate-buffered saline (PBS). Cells were scraped in PBS, centrifuged, resuspended in homogenization buffer (1 mM EDTA, 1 mM EGTA, 10 mM Tris-HCl, pH 7.5, 1 μg/ml leupeptin, 100 μg/ml N-tosyl-l-phenylalanine chloromethyl ketone [TPCK] and 1 mM phenylmethylsulfonyl fluoride [PMSF]), and homogenized. Homogenates were centrifuged at 100,000 × g, 1 h, 4°C (TL ultracentrifuge, Beckman, Fullerton, CA; Rotor TLA 100.2), and the pellet containing the crude membrane fraction was resuspended in homogenization buffer + 1% Triton X-100 and centrifuged at 100,000 × g, 30 min, 4°C. The supernatant was considered as the 1% Triton X-100–soluble fraction.
Biotinylation of Cell Surface Proteins
Cells were treated with different agonists/antagonists at 37°C, placed on ice, and washed twice with ice-cold PBS, and surface proteins were labeled for 1 h using 0.5 mg/ml EZ-link NHS-SS-biotin (Pierce Chemical Co., Rockford, IL). After labeling, the cells were rinsed three times with PBS containing 50 mM glycine to quench unreacted biotin and then lysed in modified RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, and 1% sodium deoxycholate, 1 μg/ml leupeptin, 100 μg/ml TPCK, and 1 mM PMSF). Proteins, 150–300 μg, were incubated overnight at 4°C with end-over-end shaking in the presence of streptavidin beads (Pierce Chemical Co., Rockford, IL). Beads were thoroughly washed (Carranza et al., 1998), resuspended in 30 μl of Laemmli's sample buffer solution (Laemmli, 1970) and analyzed by Western blot.
Western Blot Analysis
Protein was quantified by Bradford assay (Bradford, 1976; Bio-Rad, Hercules, CA) and resolved in 10–15% polyacrylamide gels. Thereafter, proteins were transferred onto nitrocellulose membranes (Optitran, Schleicher & Schuell, Keene, NH) using a semidry transfer apparatus (Bio-Rad). Incubation with specific antibodies was performed overnight at 4°C. Blots were developed with a chemiluminescence detection kit (PerkinElmer Life Sciences, Boston, MA) used as recommended by the manufacturer. The bands were quantified by densitometric scan (Eagle Eye II, Stratagene, La Jolla, CA).
RhoA Pull-down Assay
The assay was performed using the Rho activation assay kit (Upstate Biotechnology, Lake Placid, NY) as recommended by the manufacturer. The assay is based in the method recently described by Ren et al. (1999), where cellular GTP-Rho is detected by Western blot after affinity precipitation with a fusion protein containing GST and the Rho binding domain of Rhotekin (GST-RBD).
86Rb+ Uptake
The assay was run at 37°C in a reciprocating bath at 100 rpm. Cells were preincubated in serum-free DMEM, containing HEPES with or without 100 μM ouabain and 10 μM ISO for 10 min, and then the medium was removed and fresh medium containing 1 μCi/ml 86Rb+, with or without 100 μM ouabain and 1 μM ISO, was added. After a 5-min incubation, uptake was terminated by aspirating the assay medium and washing the plates in 4°C MgCl2. Cells were air-dried and extracted with 0.1% NaOH, and 86Rb+ influx was quantified by liquid scintillation counting. Initial influx, expressed as micromoles of K+/μg of protein/min, is calculated as follows:
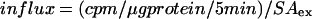 |
where SAex is the specific activity of the extracellular phase (cpm/μmol K+). Ouabain inhibitable fraction of the Na,K-ATPase was measured as 86Rb+ uptake in the presence of 100 μM ouabain.
Immunofluorescence
Cells, 2.5 × 105, were grown over glass coverslips, allowed to attach, and serum-starved for 18–24 h before adding 100 μM lysophosphatidic acid (LPA) for 20 min. Then cells were fixed with 2% formaldehyde in PBS for 20 min at room temperature. After permeabilization with 1% Triton X-100 and blocking with 0.2% BSA and normal goat serum, A549 cells were stained with rhodamine-phalloidin to study stress fiber formation. Cells were visualized under fluorescence microscope (Nikon Eclipse E800, Fryer Company, Huntley, IL).
Myosin Phosphatase Target Subunit Immunoprecipitation
Cells were treated with different agonists/antagonists at 37°C, placed on ice and washed twice with ice-cold PBS. Cells were scraped in immunoprecipitation buffer (20 mM Tris-HCl, 2 mM EGTA, 2 mM EDTA, 30 mM Na4P2O7, 30 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 10 μg/ml leupeptin, pH 7.4), frozen in liquid nitrogen, thawed, sonicated, frozen again, and centrifuged for 2 min at 14,000 × g. After protein determination, 0.2% SDS and 1% Triton X-100 was added to each sample. Equal amounts of protein (600–900 μg) were then incubated with anti-MYPT antibody for 2 h at 4°C. Protein A/G PLUS Agarose (Santa Cruz Biotechnology, Santa Cruz, CA) was added, and the samples were incubated overnight at 4°C. The samples were then washed three times with IP buffer supplemented with 0.2% SDS and 1% Triton X-100 and once with 250 mM Tris, pH 7.4. A Western blot was performed using phospho-MYPT (Thr696). Membranes were then stripped with stripping buffer (2% SDS, 100 mM 2-mercaptoethanol, 62.5 mM Tris, pH 6.8) and reprobed using an MYPT antibody.
Statistical Analysis
Data are presented as means ± SEM. Comparisons were made using a one-way analysis of variance followed by a multiple comparison test (Dunnett) when the F statistic indicated significance. Results were considered significant when p < 0.05.
RESULTS
ISO-activated RhoA and Na,K-ATPase in A549 Cells
To determine whether RhoA was activated by β-adrenergic agonists, we performed a time course, incubating A549 cells with 10 μM ISO and assessed RhoA translocation to the 1% Triton X-100–soluble fraction by Western blot, a hallmark of RhoA activation (Fleming et al., 1996). As depicted in Figure 1A, ISO stimulated RhoA translocation to the Triton X-100–soluble fraction within 30 s of treatment. ISO-mediated activation of RhoA was also determined using a pull-down assay (Ren et al., 1999). As shown in Figure 1B, top panel, A549 cells treated with ISO had a time-dependent increased recovery of RhoA bound to GTP (active) compared with control. The total amount of RhoA in the cell lysates was unchanged in control vs. ISO-treated A549 cells, indicating the specificity of the increase in the pull-down assay (Figure 1B, bottom panel). The time course of RhoA activation paralleled the translocation of Na,K-ATPase from intracellular compartments to the plasma membrane as shown in Figure 1C.
Figure 1.

ISO activates RhoA and Na,K-ATPase in A549 cells. (A) A549 cells were incubated with 10 μM ISO for 0, 0.5, and 5 min; 1% Triton X-100–soluble fractions were obtained and RhoA translocation was evaluated by Western blot. Top: bars represent mean ± SEM (n = 6). *p < 0.05; **p < 0.01. Bottom: a representative Western blot. (B) A549 cells were incubated with 10 μM ISO and cell lysates were subjected to a pull-down assay with GST-RBD. A representative Western blot of RhoA bound to GTP (top) and total RhoA (bottom) is shown (n = 3). (C) A549 cells were incubated with 10 μM ISO, and Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins followed by streptavidin pull-down and Western blot analysis using a specific antibody. Top: bars represent mean ± SEM (n = 4). **p < 0.01. Bottom: a representative Western blot.
Mevastatin and dn-RhoA Prevented ISO-mediated Na,K-ATPase Exocytosis
3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors prevent RhoA isoprenylation, thus impairing RhoA membrane localization and activation (Laufs et al., 1999; Takeuchi et al., 2000). Therefore, to study the role of RhoA in Na,K-ATPase exocytosis, we pretreated A549 cells with the HMG-CoA reductase inhibitor mevastatin (10 μM, 16 h). Mevastatin treatment prevented both the ISO-mediated RhoA translocation to the 1% Triton X-100–soluble fraction (Figure 2A) and the ISO-mediated Na,K-ATPase exocytosis (Figure 2B).
Figure 2.

Mevastatin prevents ISO-mediated Na,K-ATPase exocytosis. A549 cells were incubated with 10 μM mevastatin for 16 h before ISO stimulation. (A) RhoA translocation to the 1% Triton X-100–soluble fraction was studied by Western blot using a specific antibody. Top: bars represent mean ± SEM (n = 4). *p <0.05; **p <0.01. Bottom: a representative Western blot. (B) Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins followed by streptavidin pull-down and Western blot analysis using a specific antibody. Top: bars represent mean ± SEM (n = 4). **p < 0.01. Bottom: a representative Western blot.
To further demonstrate a role for RhoA in ISO-mediated Na,K-ATPase regulation, we used A549 cells permanently transfected with a dn-RhoA. The dn-RhoA clones were characterized by assessing stress fiber formation induced by incubation with lysophosphatidic acid (LPA, 100 μM, 20 min), a known RhoA-mediated phenomenon (Ridley and Hall, 1992). As depicted in Figure 3A, formation of stress fibers was observed in control cells after LPA treatment, but not in the A549 cells expressing the dn-RhoA (RhoA N19).
Figure 3.

Dominant negative RhoA prevents isoproterenol-mediated Na,K-ATPase exocytosis. Permanently transfected A549 cells expressing either vector or dominant negative RhoA (RhoAN19) were grown in the presence of selection. (A) Actin stress fiber formation was assessed in cells fixed and stained with rhodamin-palloidin and evaluated by using fluorescence microscopy. Representative photomicrographs of control cells (1), and control cells (2), and cells transfected with RhoAN19 (3) treated with 100 μM lysophosphatidic acid (LPA) for 20 min are shown. (B) Na,K-ATPase activity, measured as 86Rb+ uptake, in control cells and cells transfected with the vector or RhoAN19, after incubation with 10 μM ISO for 15 min. Top: bars represent mean ± SEM (n = 4). ***p < 0.001. (C) Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins followed by streptavidin pull-down and Western blot analysis using a specific antibody. Top: bars represent mean ± SEM (n = 4). **p < 0.01. Bottom: a representative Western blot.
A549 cells permanently expressing dn-RhoA had the same basal Na,K-ATPase activity (assessed by 86Rb+ uptake) as control A549 cells or cells transfected with the empty vector, but failed to increase Na,K-ATPase activity or exocytosis of the Na,K-ATPase to the plasma membrane upon ISO stimulation (Figure 3, B and C).
ISO-activated RhoA via β2-Adrenergic Receptors, cAMP-PKA, and Gi
To determine which β-adrenergic receptor was mediating RhoA stimulation, we pretreated A549 cells with specific β1-adrenergic receptor (atenolol, 100 μM, 30 min) and β2-adrenergic receptor (ICI 118,551, 10 μM, 30 min) antagonists. As shown in Figure 4, ISO-mediated RhoA translocation to the 1% Triton X-100–soluble fraction was prevented in cells pretreated with the β2--adrenergic receptor antagonist ICI 118,551, but not in cells pretreated with the β1-adrenergic receptor antagonist atenolol.
Figure 4.

ISO-mediated RhoA activation occurs via β2-adrenergic receptors. A549 cells were incubated with 100 μM atenolol (β1-adrenergic receptor antagonist) or 10 μM ICI 118,551 (β2-adrenergic receptor antagonist) before adding 10 μM ISO for 15 min. 1% Triton X-100–soluble fraction was isolated and Western blot against RhoA was performed. (A) Bars represent mean ± SEM (n = 4). **p < 0.01. (B) A representative Western blot.
The β2-adrenergic receptor is known to couple to Gs, stimulate adenylyl cyclase, and activate the cyclic-AMP-dependent protein kinase (PKA) (Daaka et al., 1997; Post et al., 1999). PKA has been described to play a role in the β-adrenergic regulation of Na,K-ATPase in AEC (Bertorello et al., 1999). Thus, to determine whether the ISO-mediated activation of RhoA occurred via PKA, we pretreated the cells with 1 μM H89 (specific PKA inhibitor) or with a myristoylated peptide that specifically inhibits PKA (PKI, 10 μM) for 30 min before treatment with 10 μM ISO for 5 min. As shown in Figure 5A, incubation with H89 (top panel) or with PKI (bottom panel) blocked the ISO-mediated RhoA translocation to the 1% Triton X-100–soluble fraction. This result was further confirmed by the use of the adenylyl cyclase activator forskolin (50 μM, 5 min), which translocated RhoA to the 1% Triton X-100–soluble fraction (Figure 5B).
Figure 5.

RhoA is activated via Gs-PKA and Gi heterotrimeric G proteins. (A) A549 cells were incubated with 1 μM H89 or 10 μM PKI for 30 min before adding 10 μM ISO. 1% Triton X-100–soluble fractions were isolated, and Western blot against RhoA was performed. Top: bars represent mean ± SEM (n = 3). **p < 0.01. Bottom: a representative Western blot. (B) A549 cells were incubated with 100 ng/ml PTX for 16 h before adding 10 μM ISO or 50 μM forskolin for 5 min. 1% Triton X-100–soluble fractions were isolated, and Western blot against RhoA was performed. A representative Western blot is shown (n = 3). (C) A549 cells were permanently transfected with empty vector or minigene vectors expressing Gα COOH-terminal peptides for Gi, G12, and G13. Cells were incubated with 10 μM ISO for 5 min, 1% Triton X-100–soluble fractions were isolated, and Western blot against RhoA was performed. A representative Western blot is shown (n = 3).
β2-adrenergic receptors are coupled to the PTX-sensitive heterotrimeric G protein, Gi (Daaka et al., 1997; Post et al., 1999; Gosmanov et al., 2002). To examine whether Gi proteins play a role in ISO-mediated RhoA activation, we pretreated A549 cells with 100 ng/ml PTX for 16 h before stimulating with ISO or forskolin for 5 min. As shown in Figure 5B, PTX prevented RhoA translocation to the 1% Triton X-100–soluble fraction of both ISOand forskolin-stimulated cells.
Several RhoA-regulated processes are mediated by heterotrimeric G proteins of the G12/13 family (Buhl et al., 1995; Fromm et al., 1997; Hart et al., 1998; Mao et al., 1998). Thus, to determine whether G12/13 were involved in ISO-mediated RhoA activation, we generated A549 cells permanently transfected with minigene vectors expressing Gα COOH-terminal peptides for Gi (to confirm the PTX data), G12 and G13 (Gilchrist et al., 2002a, 2002b). As shown in Figure 5C, ISO-mediated activation of RhoA was not affected in A549 cells expressing the minigene for G12 and G13. However, in cells expressing the minigene for Gi, RhoA activation by ISO was prevented. Taken together these results suggest a role for β2-adrenergic receptor, Gs, PKA and Gi in the upstream pathway leading to RhoA stimulation by ISO.
Downstream Effectors of RhoA
We also studied whether two well-known downstream effectors of RhoA, Rho-associated kinase (ROCK) and phospholipase D (PLD), had a role in the ISO-mediated Na,K-ATPase exocytosis. To study ROCK involvement, A549 cells were treated with the ROCK-specific inhibitor Y-27632 (10 μM, 30 min) before incubation with 10 μM ISO for 15 min. As shown in Figure 6A, incubation with Y-27632 prevented ISO-induced Na,K-ATPase exocytosis. To further confirm the involvement of ROCK in the ISO-induced Na,K-ATPase exocytosis, we transiently transfected A549 cells with plasmids coding for dominant negative (ROCK KD-IA) and activated (ROCK Δ 3) forms of ROCK (Ishizaki et al., 1997). As shown in Figure 6B, cells expressing ROCK KD-IA were unable to induce the translocation of the Na,K-ATPase to the plasma membrane after ISO stimulation, whereas cells expressing the activated form of ROCK had an increased amount of Na,K-ATPase at the plasma membrane in control conditions that could not be further increased after ISO treatment. These data strongly suggest a role for ROCK as a downstream effector of RhoA in the ISO-induced Na,K-ATPase exocytosis in AEC.
Figure 6.

ROCK/MYPT are involved in the β-adrenergic receptor induced Na,K-ATPase exocytosis.(A) A549 cells were incubated with 10 μM Y-27632 before incubating with 10 μM ISO for 15 min. Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins followed by streptavidin pull-down and Western blot analysis using a specific antibody. Top: bars represent mean ± SEM (n = 3). *p < 0.05. Bottom: a representative Western blot. (B) A549 cells were transiently transfected with vector, dominant negative ROCK (ROCK KD-IA), or activated ROCK (ROCKΔ 3) and Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins. A representative Western blot is shown (n = 3). (C) A549 cells were incubated with 10 μM ISO in the presence or absence of Y-27632. The cell lysates were immunoprecipitated with polyclonal MYPT antibody. The resulting immunoprecipitates were then immunoblotted with either phospho-MYPT (Thr696) antibody or pan-MYPT antibody. A representative Western blot is shown (n = 3).
Activated ROCK regulates the phosphorylation of myosin light chain in part by the inactivation of myosin phosphatase through the phosphorylation of its regulatory subunit MYPT (Kimura et al., 1996). To determine whether MYPT was a direct target for ROCK in the β-adrenergic regulation of AEC, A549 cells were incubated with 10 μM ISO in the presence or absence of the ROCK inhibitor Y-27632, and phosphorylation of MYPT at Thr696 (Feng et al., 1999) was examined by immunoprecipitation of MYPT and Western blot with an specific phospho-antibody against Thr696. As shown in Figure 6C ISO increased the phosphorylation of MYPT at Thr696, which was prevented by preincubation with the ROCK inhibitor Y-27632, suggesting an involvement of the myosin phosphatase downstream ROCK.
PLD catalyzes the hydrolysis of phosphatidylcholine to generate phosphatidic acid, and choline and plays an important role in regulated exocytosis. Primary alcohols are known to drive the generation of alcoholic phosphatidic acid from phosphatidylcholine, thus inhibiting the formation of phosphatidic acid itself (Jones et al., 1999). Therefore, A549 cells were treated with 1% 1-butanol for 15 min, before stimulation with ISO. As shown in Figure 7A, Na,K-ATPase translocation from intracellular compartments to the plasma membrane was not inhibited by incubation with 1%-butanol nor with 1% ethanol (our unpublished results). These results suggest that PLD does not have a role in the ISO-mediated Na,K-ATPase exocytosis.
Figure 7.

Role of Phospholipase D and Na+-H+ exchangers on ISO-induced Na,K-ATPase exocytosis. A549 cells were pretreated with (A) 1% 1-butanol, 15 min (to study PLD) and (B) 10 μM EIPA, 30 min (to study NHE), before incubating with 10 μM ISO for 15 min. Na,K-ATPase exocytosis was studied by biotin labeling of surface proteins followed by streptavidin pull-down and Western blot analysis using a specific antibody. Top: bars represent mean ± SEM (n = 3). *p < 0.05; **p < 0.01. Bottom: a representative Western blot.
ISO-mediated Na,K-ATPase Exocytosis Was Independent of Na+-H+ Exchanger Regulation
One of the mechanisms by which the Na,K-ATPase function can be modulated is by the intracellular Na+ concentrations ([Na+i]; Ewart and Klip, 1995). RhoA has been shown to regulate the Na+-H+ exchangers (NHE; Hooley et al., 1996; Tominaga et al., 1998). Therefore we conducted experiments to determine whether the effect of ISO on Na,K-ATPase was mediated by NHE stimulation. Thus, we incubated A549 cells with the amiloride analog EIPA (5-(N-ethyl-N-isopropyl) amiloride; 10 μM, 30 min) prior to stimulation with 10 μM ISO for 15 min and observed that EIPA did not prevent the effect of ISO on Na,K-ATPase exocytosis (Figure 7B). These data suggest that RhoA has a direct effect on the Na,K-ATPase exocytosis that is not due to NHE stimulation.
DISCUSSION
We present data supporting a role for the GTP-binding protein RhoA in the β-adrenergic receptor-mediated Na,K-ATPase exocytosis in AEC. We have identified components of the pathway leading to RhoA activation by β-adrenergic receptor stimulation and the downstream effectors involved in the Na,K-ATPase translocation from intracellular compartments to the plasma membrane resulting in increased Na,K-ATPase activity.
We have demonstrated, by studying translocation to the membrane fraction and pull-down with GST fusion proteins, that RhoA is activated by the β-adrenergic receptor agonist ISO in AEC. Also, we report that RhoA activation is necessary for Na,K-ATPase exocytosis. First, we utilized cholesterol-lowering agents (statins) that inhibit RhoA function by preventing its geranylgeranylation and membrane-targeting (Laufs et al., 1999; Takeuchi et al., 2000). Using this approach, we demonstrated that activation of RhoA is required in ISO-mediated Na,K-ATPase exocytosis. Second, we used permanently transfected A549 cells expressing dominant negative RhoA, an approach widely used to study the involvement of small GTP-binding proteins in different cellular events (Kjøller and Hall, 1999; see Figures 2 and 3), which also prevented the ISO-mediated exocytosis of the Na,K-ATPase. Taken together, these results suggest an important role for RhoA in the ISO-mediated increase in Na,K-ATPase activity by regulating the exocytosis of the Na,K-ATPase to the plasma membrane
Our data suggest that ISO, via β2-adrenergic receptors, cAMP/PKA and Gi heterotrimeric G protein activates RhoA in AEC (see Figures 4 and 5). Classically, cAMP-dependent PKA activation results in phosphorylation of RhoA in its C-terminal region leading to its inactivation (Lang et al., 1996). However, it has been reported that RhoA can be activated by ISO/cAMP (Yamauchi et al., 2001), which is in agreement with our data. We reason that, Gs/cAMP-activated PKA does not phosphorylate RhoA directly; therefore, we studied whether another heterotrimeric G proteins could be involved in the ISO-mediated RhoA activation. β2-adrenergic receptors have been shown to be coupled to the PTX-sensitive heterotrimeric G protein, Gi (Daaka et al., 1997; Post et al., 1999; Gosmanov et al., 2002). To study Gi involvement we used two different strategies: incubation with PTX and AEC transfected with a minigene vector expressing Gαi COOH-terminal peptide that selectively blocks signal transduction through Gi proteins (Gilchrist et al., 1999, 2002a, 2002b). We report that both Gs/cAMP/PKA and Gi are upstream of RhoA activation. Importantly, incubation with PTX also inhibited FSK-mediated RhoA stimulation (see Figure 5). These data lead us to reason, as recently reported (Lefkowitz, 1998), that receptor-dependent activation of Gs stimulates adenylyl cyclase, generates cAMP, and activates PKA, leading to phosphorylation of the β2-adrenergic receptor, switching its coupling specificity from Gs to Gi. Then, Gi could regulate RhoA activation as previously reported (Laudanna et al., 1996; Liu et al., 2001). Traditionally, the heterotrimeric G proteins of the G12/13 family have been reported to modulate RhoA activation (Buhl et al., 1995; Fromm et al., 1997; Hart et al., 1998; Mao et al., 1998). Therefore, to further study the role of heterotrimeric G proteins in the ISO-mediated RhoA activation, we used AEC transfected with minigene vectors expressing Gα12 and Gα13 COOH-terminal peptides, selectively blocking signal transduction through the G12/13 family of heterotrimeric G proteins (Gilchrist et al., 1999, 2002a, 2002b). Using this approach, we found that G12 and G13 are not involved in the stimulation of RhoA by β-adrenergic agonists (see Figure 5C).
As potential downstream effectors of RhoA, we focused on PLD and Rho-associated kinase. PLD is an important effector in receptor-mediated signal transduction pathways, and it has been described to play a role in the exocytosis of the GLUT4 glucose transporter in adipocytes (Emoto et al., 2000; Bandyopadhyay et al., 2001). However, PLD did not regulate the ISO-mediated Na,K-ATPase exocytosis (see Figure 7). In contrast, we provide evidence that ISO-mediated Na,K-ATPase exocytosis was prevented in A549 cells pretreated with the ROCK inhibitor Y27632 and in cells expressing dn-ROCK, suggesting that the pathway leading to the ISO-mediated Na,K-ATPase exocytosis occurs via RhoA/Rho-associated kinase (see Figure 6). Rho-associated kinase is known to play a role in the regulation of acto-myosin contraction (Kimura et al., 1996; Maekawa et al., 1999), and we found that the regulatory subunit of myosin phosphatase is phosphorylated by ROCK (i.e., inhibiting its activity). Thus, we propose that ROCK/MYPT may be important during Na,K-ATPase exocytosis because it has been reported that vesicle-associated myosin is capable of actinbased vesicular transport (Evans et al., 1998; Lang et al., 2000).
Na,K-ATPase regulation may be affected not only by translocation of Na,K-ATPase molecules from intracellular compartments to the plasma membrane, but also by changes in intracellular Na+ concentrations (Bertorello and Katz, 1993; Ewart and Klip, 1995; Blanco and Mercer, 1998; Feraille and Doucet, 2001). NHE regulate intracellular Na+ concentrations and several NHE isoforms have been shown to be modulated by RhoA (Tominaga and Barber, 1998; Szaszi et al., 2000). Therefore, we studied whether a specific NHE inhibitor prevented ISO-mediated Na,K-ATPase exocytosis. We provide evidence that entry of Na+ in the cell by NHE was not involved in the ISO-mediated Na,K-ATPase exocytosis, which suggests that RhoA has a direct effect on the Na,K-ATPase exocytosis rather that an indirect effect due to increased intracellular [Na+]i concentration. This is in agreement with a previous report where the regulation of Na,K-ATPase by ISO was independent to the Na+ entry via apical Na+-channels (Bertorello et al., 1999).
Reorganization of the actin cytoskeleton is a prerequisite for exocytosis, so as to enable docking and fusion of vesicles/secretory granules with the plasma membrane (Lang et al., 2000). The role of RhoA in the exocytosis of proteins is cell type dependent. Several reports have described a role for RhoA in this process, such as in the stimulation of exocytosis of secretory granules in mast cells (Norman et al., 1994, 1996; Sullivan et al., 1999), exocytosis in pancreatic acini (Nozu et al., 1999), and, as recently reported, in the translocation of the Na,K-ATPase itself in renal epithelial cells (Maeda et al., 2002). However, other reports indicate an inhibitory role for RhoA, as reported in the translocation of aquaporin-2 induced by vasopressin in renal cells (Klussmann et al., 2001).
In summary, we present here evidence for a positive role of RhoA/Rho-associated kinase in the β2-adrenergic receptor-mediated Na,K-ATPase exocytosis and increased activity in AEC. As depicted in Figure 8, we propose a model for the pathway leading to the modulation of Na,K-ATPase activity by β2-adrenergic receptor agonists. We propose that activation of β2-adrenergic receptors leads to Gs/cAMP/PKA and Gi activation, which in turn activates RhoA and its downstream effector ROCK leading to the organization of the actin cytoskeleton necessary for Na,K-ATPase exocytosis and consequent increase in Na,K-ATPase activity.
Figure 8.

Proposed model of RhoA-mediated Na,K-ATPase exocytosis and increased catalytic activity by β-adrenergic agonists in AEC.
Acknowledgments
The authors thank Renee Vena for her excellent technical support. This research was supported by National Institutes of Health Grant HL 48129. K.M.R. is a Parker B. Francis Fellow.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–12–0781. Article and publication date are available at www.molbiolcell.org/cgi/doi/0.1091/mbc.E02-12-0781.
References
- Bandyopadhyay, G., Sajan, M.P., Kanoh, Y., Standaert, M.L., Quon, M.J., Reed, B.C., Dikic, I., and Farese, R.V. (2001). Glucose activates protein kinase C-zeta/lambda through proline-rich tyrosine kinase-2, extracellular signal-regulated kinase, and phospholipase D: a novel mechanism for activating glucose transporter translocation. J. Biol. Chem. 276, 35537–35545. [DOI] [PubMed] [Google Scholar]
- Berthiaume, Y., Staub, N.C., and Matthay, M.A. (1987). Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J. Clin. Invest. 79, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorello, A.M., and Katz, A.I. (1993). Short-term regulation of renal Na-K-ATPase activity: physiological relevance and cellular mechanisms. Am. J. Physiol. 265, F743–F755. [DOI] [PubMed] [Google Scholar]
- Bertorello, A.M., Ridge, K.M., Chibalin, A.V., Katz, A.I., and Sznajder, J.I. (1999). Isoproterenol increases Na+-K+-ATPase activity by membrane insertion of alpha-subunits in lung alveolar cells. Am. J. Physiol. 276, L20–L27. [DOI] [PubMed] [Google Scholar]
- Blanco, G., and Mercer, R.W. (1998). Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am. J. Physiol. 275, F633–F650. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Buhl, A.M., Johnson, N.L., Dhanasekaran, N., and Johnson, G.L. (1995). Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631–24634. [DOI] [PubMed] [Google Scholar]
- Carranza, M.L., Rousselot, M., Chibalin, A.V., Bertorello, A.M., Favre, H., and Feraille, E. (1998). Protein kinase A induces recruitment of active Na+, K+-ATPase units to the plasma membrane of rat proximal convoluted tubule cells. J. Physiol. 511, 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils, J., and Chardin, P. (1999). GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem. Sci. 24, 306–311. [DOI] [PubMed] [Google Scholar]
- Daaka, Y., Luttrell, L.M., and Lefkowitz, R.J. (1997). Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91. [DOI] [PubMed] [Google Scholar]
- Emoto, M., Klarlund, J.K., Waters, S.B., Hu, V., Buxton, J.M., Chawla, A., and Czech, M.P. (2000). A role for phospholipase D in Glut4 glucose transporter translocation. J. Biol. Chem. 275, 7144–7151. [DOI] [PubMed] [Google Scholar]
- Evans, L., Lee, A., Bridgman, P., and Mooseker, M. (1998). Vesicle-associated brain myosin-V can be activated to catalyze actin-based transport. J. Cell Sci. 111, 2055–2066. [DOI] [PubMed] [Google Scholar]
- Ewart, H.S., and Klip, A. (1995). Hormonal regulation of the Na+-K+-ATPase: mechanisms underlying rapid and sustained changes in pump activity. Am. J. Physiol. 269, C295–C311. [DOI] [PubMed] [Google Scholar]
- Feng, J., Ito, M., Ichikawa, K., Isaka, N., Nishikawa, M., Hartshorne, D.J., and Nakano, T. (1999). Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 274, 37385–37390. [DOI] [PubMed] [Google Scholar]
- Feraille, E., and Doucet, A. (2001). Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol. Rev. 81, 345–418. [DOI] [PubMed] [Google Scholar]
- Fleming, I.N., Elliott, C.M., and Exton, J.H. (1996). Differential translocation of Rho family GTPases by lysophosphatidic acid, endothelin-1, and platelet-derived growth factor. J. Biol. Chem. 271, 33067–33073. [DOI] [PubMed] [Google Scholar]
- Fromm, C., Coso, O.A., Montaner, S., Xu, N., and Gutkind, J.S. (1997). The small GTP-binding protein Rho links G protein-coupled receptors and Gα12 to the serum response element and to cellular transformation. Proc. Natl. Acad. Sci. USA 94, 10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto, Y., Kaibuchi, K., Hori, Y., Fujioka, H., Araki, S., Ueda, T., Kikuchi, A., and Takai, Y. (1990). Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 5, 1321–1328. [PubMed] [Google Scholar]
- Gilchrist, A., Bunemann, M., Li, A., Hosey, M., and Hamm, H.E. (1999). A dominant-negative strategy for studying roles of G proteins in vivo. J. Biol. Chem. 274, 6610–6616. [DOI] [PubMed] [Google Scholar]
- Gilchrist, A., Li, A., and Hamm, H.E. (2002a). Design and use of C-terminal minigene vectors for studying role of heterotrimeric G proteins. Methods Enzymol. 344, 58–69. [DOI] [PubMed] [Google Scholar]
- Gilchrist, A., Li, A., and Hamm, H.E. (2002b). G alpha COOH-terminal minigene vectors dissect heterotrimeric G protein signaling. Sci. STKE 118, PL1. [DOI] [PubMed] [Google Scholar]
- Gosmanov, A.R., Wong, J.A., and Thomason, D.B. (2002). Duality of G protein-coupled mechanisms for beta-adrenergic activation of NKCC activity in skeletal muscle. Am. J. Physiol. Cell Physiol. 283, C1025–C1032. [DOI] [PubMed] [Google Scholar]
- Guo, W., Tamanoi, F., and Novick, P. (2001). Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3, 353–360. [DOI] [PubMed] [Google Scholar]
- Hart, M.J., Jiang, X., Kozasa, T., Roscoe, W., Singer, W.D., Gilman, A.G., Sternweis, P.C., and Bollag, G. (1998). Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha 13. Science 280, 2112–2114. [DOI] [PubMed] [Google Scholar]
- Hooley, R., Yu, C.Y., Symons, M., and Barber, D.L. (1996). Gα13 stimulates Na+-H+ exchange through distinct Cdc42-dependent and RhoA-dependent pathways. J. Biol. Chem. 271, 6152–6158. [DOI] [PubMed] [Google Scholar]
- Ishizaki, T., Naito, M., Fujisawa, K., Maekawa, M., Watanabe, N., Saito, Y., and Narumiya, S. (1997). p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404, 118–124. [DOI] [PubMed] [Google Scholar]
- Jones, D., Morgan, C., and Cockcroft, S. (1999). Phospholipase D and membrane traffic. Potential roles in regulated exocytosis, membrane delivery and vesicle budding. Biochim. Biophys. Acta 1439, 229–244. [DOI] [PubMed] [Google Scholar]
- Kimura, K. et al. (1996). Regulation of myosin phosphatase by Rho and Rho-Associated kinase (Rho-kinase). Science 273, 245–248. [DOI] [PubMed] [Google Scholar]
- Kjøller, L., and Hall, A. (1999). Signaling to Rho GTPases. Exp. Cell Res. 253, 166–179. [DOI] [PubMed] [Google Scholar]
- Klussmann, E., Tamma, G., Lorenz, D., Wiesner, B., Maric, K., Hofmann, F., Aktories, K., Valenti, G., and Rosenthal, W. (2001). An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 276, 20451–20457. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lang, P., Gesbert, F., Delespine-Carmagnat, M., Stancou, R., Pouchelet, M., and Bertoglio, J. (1996). Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 15, 510–519. [PMC free article] [PubMed] [Google Scholar]
- Lang, T., Wacker, I., Wunderlich, I., Rohrbach, A., Giese, G., Soldati, T., and Almers, W. (2000). Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys. J. 78, 2863–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudanna, C., Campbell, J.J., and Butcher, E.C. (1996). Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science 271, 981–983. [DOI] [PubMed] [Google Scholar]
- Laufs, U., Marra, D., Node, K., and Liao, J.K. (1999). 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing Rho GTPase-induced down-regulation of p27Kip1. J. Biol. Chem. 274, 21926–21931. [DOI] [PubMed] [Google Scholar]
- Lazrak, A., Samanta, A., and Matalon, S. (2000). Biophysical properties and molecular characterization of amiloride-sensitive sodium channels in A549 cells. Am. J. Physiol. Lung Cell Mol. Physiol. 278, L848–L857. [DOI] [PubMed] [Google Scholar]
- Lecuona, E., Garcia, A., and Sznajder, J.I. (2000). A novel role for protein phosphatase 2A in the dopaminergic regulation of Na, K-ATPase. FEBS Lett. 481, 217–220. [DOI] [PubMed] [Google Scholar]
- Lefkowitz, R.J. (1998). G Protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 273, 18677–18680. [DOI] [PubMed] [Google Scholar]
- Liu, F., Verin, A.D., Wang, P., Day, R., Wersto, R.P., Chrest, F.J., English, D.K., and Garcia, J.G.N. (2001). Differential regulation of sphingosine-1-phosphate-and VEGF-induced endothelial cell chemotaxis. Am. J. Respir. Cell Mol. Biol. 24, 711–719. [DOI] [PubMed] [Google Scholar]
- Maeda, A., Amano, M., Fukata, Y., and Kaibuchi, K. (2002). Translocation of Na+, K+-ATPase is induced by Rho small GTPase in renal epithelial cells. Biochem. Biophys. Res. Commun. 297, 1231–1237. [DOI] [PubMed] [Google Scholar]
- Maekawa, M. et al. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898. [DOI] [PubMed] [Google Scholar]
- Mao, J., Yuan, H., Xie, W., and Wu, D. (1998). Guanine nucleotide exchange factor GEF115 specifically mediates activation of Rho and serum response factor by the G protein α subunit Gα13. Proc. Natl. Acad. Sci. USA 95, 12973–12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, J., Price, L., Ridley, A., Hall, A., and Koffer, A. (1994). Actin filament organization in activated mast cells is regulated by heterotrimeric and small GTP-binding proteins. J. Cell Biol. 126, 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, J., Price, L., Ridley, A., and Koffer, A. (1996). The small GTP-binding proteins, Rac and Rho, regulate cytoskeletal organization and exocytosis in mast cells by parallel pathways. Mol. Biol. Cell 7, 1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozu, F., Tsunoda, Y., Ibitayo, A.I., Bitar, K.N., and Owyang, C. (1999). Involvement of RhoA and its interaction with protein kinase C and Src in CCK-stimulated pancreatic acini. Am. J. Physiol. 276, G915–G923. [DOI] [PubMed] [Google Scholar]
- Oheim, M., and Stühmer, W. (2000). Tracking chromaffin granules on their way through the actin cortex. Eur. Biophys. J. 29, 67–89. [DOI] [PubMed] [Google Scholar]
- Post, S.R., Hammond, H.K., and Insel, P.A. (1999). β-Adrenergic receptors and receptor signaling in heart failure. Annu. Rev. Pharmacol. Toxicol. 39, 343–360. [DOI] [PubMed] [Google Scholar]
- Ren, X.D., Kiosses, W.B., and Schwartz, M.A. (1999). Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18, 578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge, K.M. et al. (2003). Alveolar type 1 cells express the α2 Na, K-ATPase which contributes to lung liquid clearance. Circ. Res. 92, 453–460. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., and Hall, A. (1992). The small GTP-binding protein regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399. [DOI] [PubMed] [Google Scholar]
- Rockman, H.A., Koch, W.J., and Lefkowitz, R.J. (2002). Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212. [DOI] [PubMed] [Google Scholar]
- Rutschman, D.H., Olivera, W., and Sznajder, J.I. (1993). Active transport and passive liquid movement in isolated perfused rat lungs. J. Appl. Physiol. 75, 1574–1580. [DOI] [PubMed] [Google Scholar]
- Saldias, F., Lecuona, E., Friedman, E., Barnard, M.L., Ridge, K.M., and Sznajder, J.I. (1998). Modulation of lung liquid clearance by isoproterenol in rat lungs. Am. J. Physiol. 274, L694–L701. [DOI] [PubMed] [Google Scholar]
- Saldias, F.J., Comellas, A., Ridge, K.M., Lecuona, E., and Sznajder, J.I. (1999). Isoproterenol improves ability of lung to clear edema in rats exposed to hyperoxia. J. Appl. Physiol. 86, 30–35. [DOI] [PubMed] [Google Scholar]
- Saldias, F.J., Lecuona, E., Comellas, A.P., Ridge, K.M., Rutschman, D.H., and Sznajder, J.I. (2000). β-Adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am. J. Respir. Crit. Care Med. 162, 282–287. [DOI] [PubMed] [Google Scholar]
- Saumon, G., and Basset, G. (1993). Electrolyte and fluid transport across the mature alveolar epithelium. J. Appl. Physiol. 74, 1–15. [DOI] [PubMed] [Google Scholar]
- Sullivan, R., Price, L.S., and Koffer, A. (1999). Rho controls cortical F-actin disassembly in addition to, but independently of, secretion in mast cells. J. Biol. Chem. 274, 38140–38146. [DOI] [PubMed] [Google Scholar]
- Suzuki, S., Zuege, D., and Berthiaume, Y. (1995). Sodium-independent modulation of Na(+)-K(+)-ATPase activity by beta-adrenergic agonist in alveolar type II cells. Am. J. Physiol. 268, L983–L990. [DOI] [PubMed] [Google Scholar]
- Szaszi, K., Kurashima, K., Kapus, A., Paulsen, A., Kaibuchi, K., Grinstein, S., and Orlowski, J. (2000). RhoA and Rho kinase regulate the epithelial Na+/H+ exchanger NHE3. Role of myosin light chain phosphorylation. J. Biol. Chem. 275, 28599–28606. [DOI] [PubMed] [Google Scholar]
- Sznajder, J.I., Factor, P., and Ingbar, D.H. (2002). Lung edema clearance: role of Na+-K+-ATPase. J. Appl. Physiol. 93, 1860–1866. [DOI] [PubMed] [Google Scholar]
- Sznajder, J.I., Olivera, W.G., Ridge, K.M., and Rutschman, D.H. (1995). Mechanisms of lung liquid clearance during hyperoxia in isolated rat lungs. Am. J. Respir. Crit. Care Med. 151, 1519–1525. [DOI] [PubMed] [Google Scholar]
- Takeuchi, S., Kawashima, S., Rikitake, Y., Ueyama, T., Inoue, N., Hirata, K.I., and Yokoyama, M. (2000). Cerivastatin suppresses lipopolysaccharide-induced ICAM-1 expression through inhibition of Rho GTPase in BAEC. Biochem. Biophys. Res. Commun. 269, 97–102. [DOI] [PubMed] [Google Scholar]
- Tominaga, T., and Barber, D.L. (1998). Na-H exchange acts downstream of RhoA to regulate integrin-induced cell adhesion and spreading. Mol. Biol. Cell 9, 2287–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga, T., Ishizaki, T., Narumiya, S., and Barber, D.L. (1998). p160ROCK mediates RhoA activation of Na-H exchange. EMBO J. 17, 4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van-Aelst, L., and D'Souza-Schorey, C. (1997). Rho GTPases and signaling networks. Genes Dev. 11, 2295–2322. [DOI] [PubMed] [Google Scholar]
- Yamauchi, J., Hirasawa, A., Miyamoto, Y., Itoh, H., and Tsujimoto, G. (2001). β2-Adrenergic receptor/cyclic adenosine monophosphate (cAMP) leads to JNK activation through Rho family small GTPases. Biochem. Biophys. Res. Commun. 284, 1199–1203. [DOI] [PubMed] [Google Scholar]