

Abstract
In addition to tRNA and 5S RNA, Escherichia coli contains several other small, stable RNA species; these are M1, 10Sa, 6S, and 4.5S RNA. Although these RNAs are initially synthesized as precursor molecules, relatively little is known about their maturation. The data presented here show that 3′ exoribonucleolytic trimming is required for the final maturation of each of these molecules. As found previously with tRNA, but not 5S RNA, any one of a number of exoribonucleases can carry out the trimming reaction in vivo, although RNases T and PH are most effective. In their absence, large amounts of immature molecules accumulate for most of the RNAs, and these can be converted to the mature forms in vitro by the purified RNases. A model is proposed that identifies a structural feature present in all the small, stable RNAs of E. coli, and describes how this structure together with the RNases influences the common mechanism for 3′ maturation.
RNA molecules generally are synthesized as longer precursors that must undergo a series of processing reactions to remove extra residues and generate the mature, functional forms (1). While information about these RNA processing reactions and the RNases that catalyze them is accumulating, there still are major gaps in our knowledge of these pathways, particularly with regard to the less abundant classes of RNA.
In Escherichia coli, in addition to tRNA and 5S RNA, four other small, stable RNA species are known. These include M1 RNA (377 nucleotides), the catalytic subunit of RNase P (2); 10Sa or tmRNA (363 nucleotides), which is involved in tagging prematurely terminated polypeptides for ultimate degradation (3); 4.5S RNA (114 nucleotides), a component of the prokaryotic signal recognition particle needed for protein secretion (reviewed in ref. 4); and 6S RNA (181–184 nucleotides), part of an 11S ribonucleoprotein complex of unknown function (5, 6). Each of these RNAs is initially synthesized with additional residues at its 5′ and 3′ ends that are removed during the course of maturation. The endoribonucleases, RNase P, RNase III, and RNase E, are thought to be involved in generating the mature 5′ termini of some of these molecules, or in some instances, in removing portions of the 3′ trailer sequence (7–10). However, no information is available on how the mature 3′ termini are made.
In previous studies, we showed that the 3′ termini of tRNA and 5S RNA are generated by exoribonucleolytic trimming reactions (11–14). In the case of tRNA, multiple exoribonucleases can participate in the 3′ maturation process, with RNase PH and RNase T providing most of the processing activity (11–13, 15). In contrast, 3′ maturation of 5S RNA is completely dependent on RNase T (14). In this paper we demonstrate that exoribonucleolytic trimming reactions also are responsible for generating the mature 3′ termini of the other small, stable RNAs, and we define which RNases participate in the process. A general model for 3′ maturation of stable RNAs is also presented.
EXPERIMENTAL PROCEDURES
Bacterial Strains.
E. coli K12 strain CA244 (lacZ, trp, relA, spoT) (12) was considered wild type for these studies. Exoribonuclease-deficient derivatives of CA244 were described previously (11–15). Strain CA244cca− was constructed by introducing an interruption mutation of the cca gene encoding tRNA nucleotidyltransferase into strain CA244 by P1-mediated transduction (16). The mutations in PNPase, RNase D, RNase T, and RNase PH are interruption mutations and are devoid of the relevant activity. The mutations in RNase II and RNase BN have not been defined, but they lead to ≈98% loss of the relevant activity. All the strains used in this study are stable, though some multiple RNase-deficient cells grow slowly. Previous work (17) showed that inactivation of one or more RNases does not lead to overexpression of the remaining enzymes.
Materials.
[γ-32P]ATP was purchased from DuPont-New England Nuclear. Phage T4 polynucleotide kinase and Moloney murine leukemia virus reverse transcriptase were from GIBCO/BRL. RNasin was obtained from Promega. Sequenase 2.0 was a product of United States Biochemical. E. coli RNase T and RNase PH were purified as described (13, 18). Sequagel for DNA sequencing was purchased from National Diagnostics. The oligonucleotides used for Northern blot and primer extension analyses were prepared as follows: for M1 RNA, CTATGGAGCCCGGACTTTCCTC; 10Sa RNA, TTACGAGGCCAACCGCCCCTCG; 6S RNA, CGGACGGACCGAGCATGCTCAC; and 4.5S RNA, GCTGCTTCCTTCCGGACCTGAC. All other chemicals were reagent grade.
RNA Preparation.
Cells were grown in yeast-tryptone medium to an A550 ≈ 1. Total cellular RNA was isolated by phenol extraction as described (19). The RNA was used for the experiments presented without further fractionation.
Northern Blot Analysis.
Northern blot analysis was carried out according to the procedure described previously (14, 15) with slight modifications by using probes specific for the RNA species examined. Samples were loaded on 4% (for M1, 10Sa, or 6S RNAs) or 5% (for 4.5S RNA) polyacrylamide-urea (8.3 M) sequencing gels. Electrophoresis was carried out at 1,700 V until the xylene cyanol dye had migrated 60 cm (M1 or 10Sa RNAs) or 30 cm (6S or 4.5S RNAs). Generally, prehybridization, hybridization, and washing were carried out at 45°C.
Treatment with Purified RNases.
Total RNA was treated with purified RNase T or RNase PH as described (13, 14). Samples were incubated at 37°C for the indicated length of time. Three microliter samples were taken, diluted immediately into 7 μl of ice-cold gel loading buffer, and determined by Northern blot analysis. Activities of RNases T and PH were determined as described (13).
Primer Extension Analysis.
The protocol for primer extension was as described (14). The extension products were separated by electrophoresis on a 6% sequencing gel, and were detected by autoradiography.
RESULTS
To examine the role of exoribonucleases in the maturation of small, stable RNAs, we constructed mutant strains lacking various exoribonucleases, either alone or in combination, and used them to determine the RNA products that accumulated as a consequence of incomplete processing. Total RNA was isolated from wild type and the exoribonuclease-deficient cells and examined by Northern blot analysis using specific oligonucleotide probes complementary to the RNA species under study and by primer extension analysis to determine their 5′ termini. RNA samples also were treated with purified exoribonucleases in vitro to verify that they can act on the accumulated products to generate mature RNAs.
Maturation of M1 RNA.
Because of its large size, M1 RNA is difficult to analyze reproducibly at single nucleotide resolution by using Northern blots. However, by including an adjacent DNA sequencing ladder as a size marker, it was possible to determine the number of extra residues present in the M1 RNA products that accumulate in the mutant strains (DNA ladders are not shown). As seen in Fig. 1, mature M1 RNA (377 nucleotides) from wild-type cells (lanes 1, 7, and 13) is present as a single band, indicating efficient processing when sufficient RNase activity is available. Mature M1 RNA is likewise the only product in mutants lacking either RNase PH (Fig. 1, lane 2), or missing RNases D, II, and BN in combination (Fig. 1, lane 5). In contrast, substantial amounts of products up to three nucleotides longer than the mature form accumulate in a mutant strain lacking RNase T (Fig. 1, lane 3). Products larger than M1 RNA are not detected when RNase T is present, even when as many as four other exoribonucleases are absent (Fig. 1, lane 6). These data indicate that RNase T is the most important of the exoribonucleases for the maturation of M1 RNA.
Figure 1.
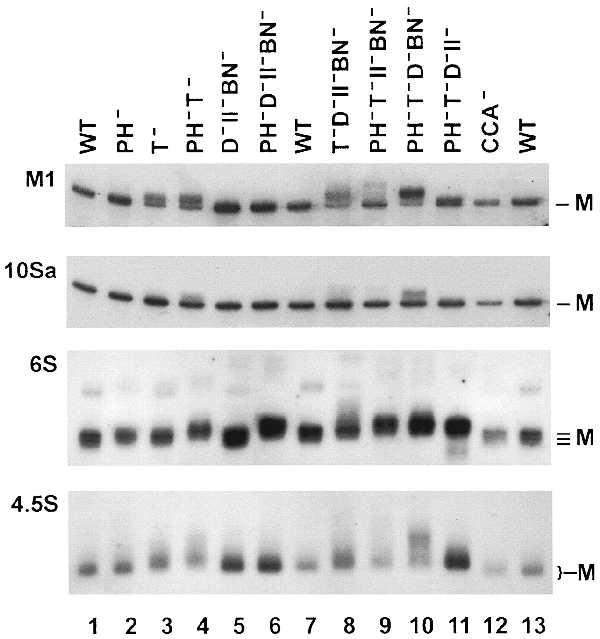
Northern blot analysis of small, stable RNAs accumulated in exoribonuclease-deficient cells. Electrophoresis and Northern blot analysis were carried out as described in Experimental Procedures. Five micrograms of total RNA from each of the indicated strains was used except for the CCA− strain in which less RNA was loaded. After electrophoresis, the bottom 10 cm of the gel, containing the RNA of interest, was transferred to a membrane and hybridized with a probe specific for the RNA. The lengths of RNA were determined by an adjacent DNA sequencing ladder. The identities of the RNAs examined are shown on the left. M indicates the size(s) of the mature RNA.
Because mature M1 RNA continues to be made in RNase T− cells, other exoribonucleases must also be able to participate in this process. However, RNase PH, which is known to be important for the 3′ maturation of tRNA (11–13, 15), is not very effective for M1 RNA processing. The absence of RNase PH by itself has no effect on the maturation of M1 RNA (Fig. 1, lane 2), and a RNase PH−T− double mutant (Fig. 1, lane 4) is no more defective than the RNase T− strain (Fig. 1, lane 3). Moreover, in a mutant strain lacking RNases T, D, II, and BN (PH+) (Fig. 1, lane 8), the majority of the products are defective and are two to four nucleotides longer than the mature form. The most dramatic defect is seen in cells missing RNases T, PH, D, and BN (II+) (Fig. 1, lane 10). In this strain the majority of M1 RNA is present as products up to five nucleotides longer than mature RNA. Somewhat more mature M1 RNA is made in the other quadruple mutants (Fig. 1, lanes 9 and 11). Products with up to six extra residues are found in small amounts in a strain lacking RNases T, PH, II, and BN (D+) (Fig. 1, lane 9), while in a RNase T−PH−D−II−(BN+) cell (Fig. 1, lane 11), about half the product is of mature size and the rest is just one nucleotide longer. These results suggest that RNases D and BN are each more effective than either RNase PH or II, but each exoribonuclease can generate some mature M1 RNA. This redundancy of processing activities has already been observed for the maturation of the 3′ end of tRNA molecules (12, 13, 15).
M1 RNA is known to be transcribed from two different start sites (7). The upstream start site results in a transcript that requires processing to generate the mature 5′ end. Consequently, it was necessary to determine whether the M1 products that accumulate in the RNase− strains contain extra residues at their 5′ termini. To answer this question, we examined the 5′ end of M1 RNA from the most defective strain (RNase T−PH−D−BN−) by using primer extension analysis (Fig. 2). The extension products obtained with RNA from the wild-type and the RNase-deficient strains are identical and correspond to the mature 5′ end of M1 RNA (Fig. 2, lanes 4–6). Inasmuch as the mutant strain accumulates a longer form of M1 RNA, as shown by Northern blot analysis, it is apparent that this longer species must contain extra residues at its 3′ terminus. Identical results were obtained with RNA isolated from RNase T− cells (data not shown).
Figure 2.
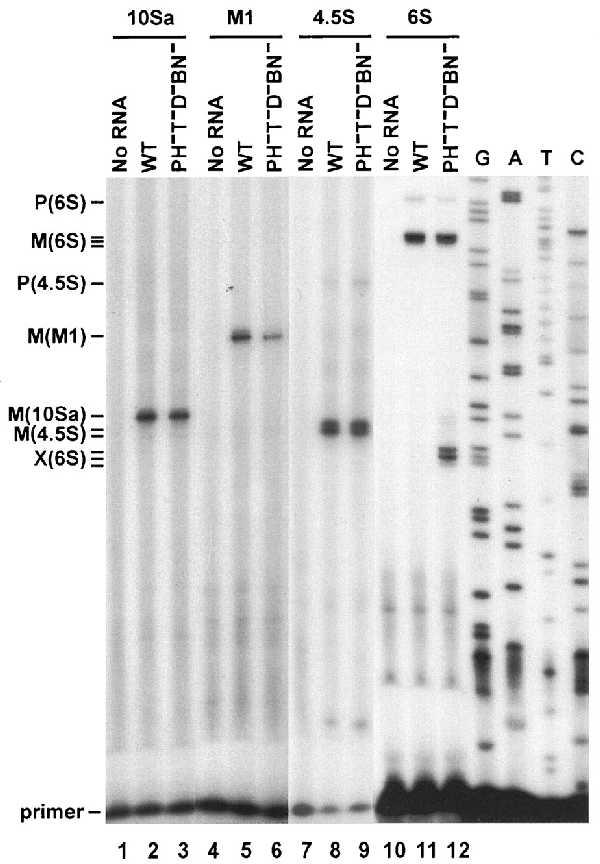
Primer extension analysis of RNA from wild type and a RNase PH−T−D−BN− mutant cell. The analysis was carried out as described in Experimental Procedures by using 20 μg of total RNA from each strain and 2 pmol of 32P-labeled primers. The RNA analyzed is shown at the top. The mature 5′ end(s) is marked M, and the 5′ end of the nascent transcript are marked P. Unknown products are marked by X. Phage M13 mp18 DNA was used to generate the ladder.
The maturation of M1 RNA was also examined in vitro by using purified exoribonucleases. RNA was treated with purified RNase T or RNase PH (at amounts equivalent to their relative levels in cell extracts) and subjected to Northern blot analysis (Fig. 3). The products of M1 RNA that accumulate in RNase T−PH−D−BN−(II+) mutant cells are converted to mature size very rapidly (within 3 min) after mixing with RNase T (Fig. 3, lane 6), and the mature product remains unaffected even after 10 min of incubation (Fig. 3, lane 7), indicating that RNase T acts effectively and accurately in vitro. Treatment with RNase PH also results in shortening of the M1 RNA products (Fig. 3, lanes 9 and 10); however, this enzyme is much less efficient than RNase T. Nevertheless, increased RNase PH and prolonged incubation (30 min) can convert M1 RNA completely to the mature form (lane 12). Mature M1 RNA from the wild-type cell remains unchanged by this treatment. Thus, RNase PH can process M1 accurately. Because both RNase T and PH trim from the 3′ terminus, the extra residues removed must have been present at this end. These in vitro data support the conclusion that RNase T is the most important exoribonuclease for 3′ maturation of M1 RNA.
Figure 3.
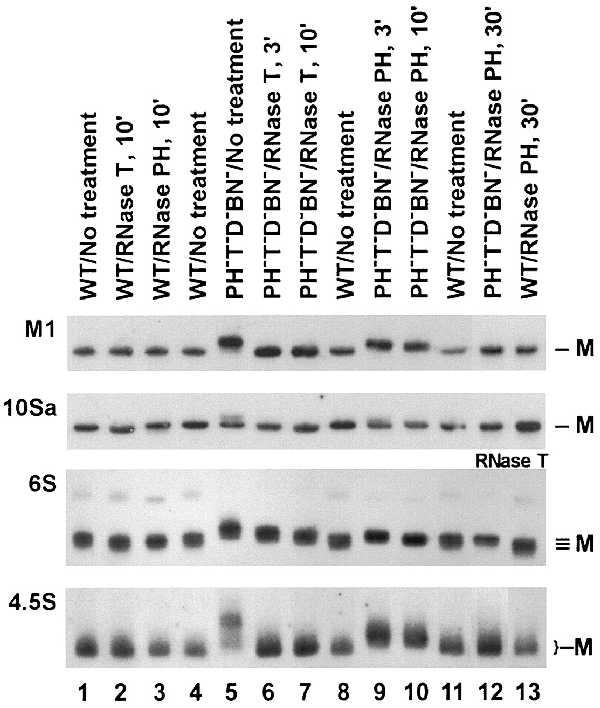
Maturation of RNA in vitro by purified exoribonucleases. Twenty micrograms of total RNA were treated as described in Experimental Procedures with either RNase T (0.5 μg) or RNase PH (0.15 μg) in 15 μl. Additional RNase PH (0.6 μg) was used in lanes 12 and 13 except for 6S RNA, where RNase T was used. The source of the RNA sample, the enzyme used and the length of incubation are indicated at the top. The RNA under study is indicated at the left. After incubation, the RNA samples were subjected to Northern blot analysis as in Fig. 1. M indicates the size of the mature RNAs.
Maturation of 10Sa RNA.
As pointed out earlier, 10Sa RNA (363 nucleotides) is similar in size to M1 RNA. Accordingly, the experimental conditions for its analysis were the same as those for M1 RNA. Only mature 10Sa RNA is present in wild-type cells (Fig. 1, lanes 1, 7, and 13). In a RNase T− cell, a small amount of a larger product can be seen (Fig. 1, lane 3). However, larger products in increased amounts accumulate in mutant strains lacking RNase T in combination with deficiencies in other RNases (Fig. 1, lanes 4, 8–10). When RNase T is present, products larger than the mature form of 10Sa are not observed indicating that RNase T is an important enzyme for the processing of 10Sa RNA. Because removal of other RNases results in accumulation of more and longer immature forms, other exoribonucleases must contribute to the maturation of 10Sa RNA. The highest level of immature 10Sa RNA, containing up to five extra residues, is found in a RNase T−PH−D−BN−(II+) strain (Fig. 1, lane 10). The other quadruple mutants, RNase T−D−II−BN−(PH+) and RNase T−PH−II−BN−(D+) (Fig. 1, lanes 8 and 9, respectively), accumulate fewer, but longer, products (up to six extra residues); only a small amount of immature product is seen in a RNase T−PH−D−II−(BN+) strain, suggesting that RNase BN is relatively effective on 10Sa precursors.
Primer extension analysis of 10Sa RNA revealed only a single band corresponding to the mature 5′ end of the molecule (Fig. 2, lanes 2 and 3). The size of the extension products for the wild-type and RNase-deficient strain are the same, despite the presence of longer products in the RNase T−PH−D−BN−(II+) sample as seen by Northern blot analysis. This shows that the extra residues on 10Sa RNA are at the 3′ end.
Both purified RNase T and RNase PH can remove extra residues from the immature 10Sa RNA products in vitro (Fig. 3). Extra residues are removed rapidly after mixing with purified RNase T (Fig. 3, lane 6). With RNase PH treatment, the longer products gradually shorten with time (Fig. 3, lanes 9 and 10). Additional RNase PH and extended incubation time are required to convert all the products to mature size (Fig. 3, lane 12). Interestingly, a product of 10Sa one nucleotide shorter than the mature size is generated by RNase T treatment upon incubating for 10 min (Fig. 3, lanes 2 and 7). A similar pattern was observed when tRNA or 5S RNA were treated with RNase T in vitro (13, 14). Removal of the 3′ end of tRNA also occurs in vivo by a process referred to as end-turnover. Defective tRNAs generated by this process are repaired by tRNA nucleotidyltransferase (16), and in a cca− cell lacking tRNA nucleotidyltransferase defective tRNAs can be detected because repair does not occur (16). Because 10Sa RNA has a tRNA-like structure and function, it was possible that the same end-turnover reaction might occur in vivo with this molecule. However, the expected 10Sa RNA shorter than mature size was not detected in a cca− strain (Fig. 1, lane 12). This result suggests that the end-turnover reaction does not extend to 10Sa RNA, despite the fact that RNase T removes one additional residue in vitro.
Maturation of 6S RNA.
Mature 6S RNA (181–184 nucleotides) displays length heterogeneity at both its 5′ and 3′ termini (5). Consistent with this heterogeneity, Northern blot analysis of RNA from wild-type cells revealed several closely spaced bands differing from each other by a single nucleotide (Fig. 1, lanes 1, 7, and 13). In addition, a minor band nine nucleotides longer than the longest mature 6S RNA was also detected. This product, which was reported previously, contains the complete 5′ fragment of the primary transcript (20). The multiple forms of 6S RNA made it more difficult to analyze the products that accumulated in the mutants. Nevertheless, by careful comparisons of the length of each band, it was possible to identify immature RNAs present in the mutant cells.
Thus, in mutant cells lacking either RNase T or PH, products one to two residues longer than the longest mature 6S RNA can be seen (Fig. 1, lanes 2 and 3). In the RNase T−PH− double mutant, molecules up to three nucleotides longer accumulate (Fig. 1, lane 4), and the shortest forms are absent. Unfortunately, because of the overlapping lengths of the products from mutant and wild-type cells, it was not possible to determine whether any mature 6S RNA is made in RNase T−PH− mutant cells. In contrast, longer products are not seen in a mutant strain lacking RNases D, II, and BN (Fig. 1, lane 5). These results indicate that RNase T and PH are most important for the maturation of 6S RNA. In quadruple mutant strains lacking four of the five exoribonucleases under study, products of different lengths and intensities accumulate, supporting the notion that no single enzyme is sufficient for 6S RNA maturation. For example, in the RNase T−D−II−BN−(PH+) mutant strain a major product of the same size and with the same 5′ end (data not shown) as the longest mature 6S RNA is made, but additional products up to six nucleotides longer also can be seen (Fig. 1, lane 8). The other quadruple mutants accumulate products that are primarily two nucleotides longer than the RNA from wild-type cells (Fig. 1, lanes 6, 9–11). These data suggest that RNase PH might be more active than the other exoribonucleases in removing residues close to the 3′ end of 6S RNA. Interestingly, the RNase PH−T−D−II−(BN+) mutant strain also generates some minor products that are up to three nucleotides shorter than the shortest mature 6S RNA (Fig. 1, lane 11). The structure and source of these molecules has not been examined.
The longer product containing nine additional nucleotides at the 5′ end of 6S RNA was detected in all the mutant cells (Fig. 1). Moreover, in cells in which immature 6S RNA is seen, these minor bands also are longer than in wild type. Based on this information, it appears that small amounts of 6S RNA precursors having extra residues at either or both ends can accumulate. The presence of products with 5′ extra residues indicates that removal of the 5′ fragment from 6S RNA is relatively slow.
As expected from the length heterogeneity, primer extension experiments detected several 5′ ends on 6S RNA (Fig. 2). Three extension products are seen in both the wild-type and the multiple RNase-deficient samples that correspond to the various mature 5′ ends known to be present in 6S RNA (Fig. 2, lanes 10–12). The minor product, 9 nucleotides longer, which represents the 5′ end of the nascent transcript also is seen. Inasmuch as no difference is observed between the two RNA samples with regard to their 5′ termini, any extra residues detected in the mutant cell samples by Northern blot analysis must represent changes at the 3′ terminus.
Surprisingly, major primer extension products of the RNase T−PH−D−BN−(II+) strain are reproducibly found at a position 38–40 nucleotides downstream of the longest mature 5′ end (Fig. 2, lane 12). Because these shorter 6S RNA products were not detected by Northern blot analysis (data not shown), they must be due to a change in RNA structure that blocks the passage of reverse transcriptase. The nature of this structural alteration and the RNase deficiency that leads to it are currently under investigation.
Treatment of the 6S RNA immature products with purified RNases results in their conversion to mature size (Fig. 3). As with M1 RNA and 10Sa RNA, the 6S RNA products can be processed in vitro by both RNase T and RNase PH. However, in contrast to the other RNAs, RNase T treatment does not remove extra residues from 6S RNA very efficiently. Immature products remain even after 10 min of incubation, and are completely converted to mature size only after 30 min (Fig. 3, lanes 6, 7, and 12). On the other hand, RNase PH is more efficient, converting all the RNA species to mature size during 10 min of incubation (Fig. 3, lane 10). This observation agrees with the Northern analyses, which suggested that RNase PH is more effective than RNase T for 6S RNA maturation.
Another interesting feature of 6S RNA is that the sequence at its 3′ end can be either CC, CA, or C (5). While the two C residues are encoded by the ssrS gene for 6S RNA, the A residue would have to be added posttranscriptionally. One possible route for generating a CA end would be the addition of an A residue to the shorter 3′ end by tRNA nucleotidyltransferase. However, there was no indication that the RNA in a cca− cell differs from that of wild type (Fig. 1, lanes 12 and 13).
Maturation of 4.5S RNA.
As shown in Fig. 1, 4.5S RNA (114 nucleotides) does not resolve to a single, sharp band by Northern blot analysis. The blurred bands, with significant trailing as well, were observed with RNA from every strain examined including wild type (Fig. 1, lanes 1, 7, and 13). These results suggest either that 4.5S RNA is heterogeneous in length, that it is modified heterogeneously or that even under the denaturing conditions of gel electrophoresis it exists in multiple conformations. Despite this complication, the pattern of 4.5S RNA was reproducible, and the length of the products could still be estimated from a DNA sequencing ladder run next to the RNA samples (data not shown).
In RNase T− cells, a portion of the major 4.5S RNA products is shifted to a size one or two nucleotides longer than RNA from the wild type (Fig. 1, lane 3). The products are even longer when both RNase T and PH are absent (Fig. 1, lane 4). A more dramatic effect is observed with RNA from the RNase PH−T−D−BN−(II+) mutant (Fig. 1, lane 10). In this case, the majority of 4.5S RNA molecules are clearly longer than wild type and contain as many as six additional residues. Although it was not possible to get as detailed a picture for 4.5S RNA as with the other RNAs examined, it appeared that RNase T is relatively more important for maturation than the other RNases, and that RNase II is the least effective. Nevertheless, it is clear, even in this situation, that exoribonucleolytic trimming is necessary for maturation of 4.5S RNA.
Moreover, as with the other RNAs examined here, primer extension analysis of the 4.5S RNA products showed the same 5′ ends for the wild type and the RNase T−PH−D−BN−(II+) mutant (Fig. 2, lanes 8 and 9), indicating that any increased size in 4.5S RNA in the mutant cells is due to extra residues at the 3′ end. Interestingly, in addition to the extension product corresponding to the mature 5′ end of 4.5S RNA, we also detected a major product one nucleotide shorter. This product is not due to a heterogeneous primer (data not shown), and it is present at the same level in each strain. It is not yet clear whether this second product represents a subpopulation of 4.5S RNA with a shorter 5′ end, or whether it is due to premature termination by reverse transcriptase. An additional minor product 24 nucleotides longer than the mature 5′ end also can be seen (Fig. 2, lanes 8 and 9). This product apparently results from the presence of a small amount of the initial transcript containing the uncleaved 5′ fragment. It can also be observed by Northern blotting when the film is overexposed (data not shown). It should also be noted that because the blurred Northern blot pattern is not seen by primer extension, factors other than a heterogeneous 5′ terminus are probably responsible for that effect.
The importance of exoribonucleases for maturation of the 3′ end of 4.5S RNA was also shown by the action of these enzymes in vitro (Fig. 3). Treatment with purified RNase T rapidly and accurately converts the immature 4.5S RNA to mature size (Fig. 3, lanes 5–7). RNase PH is relatively ineffective by comparison. With this enzyme the longer products gradually shorten during 10 min of incubation (Fig. 3, lanes 9 and 10), but complete maturation of 4.5S RNA is achieved only with additional RNase PH and longer treatments (Fig. 3, lane 12).
DISCUSSION
Although complete processing pathways for M1, 10Sa, 6S, and 4.5S RNAs remain to be elucidated, it is clear from the data presented here that exoribonuclease action is a required step for the final 3′ maturation of each RNA. Taken together with what is already known about the maturation of tRNAs and 5S RNA (11–15), it appears that 3′ exoribonucleolytic trimming is a universal feature of small, stable RNA processing in E. coli. It is also likely that a downstream endonucleolytic cleavage precedes the 3′ trimming events in all of the RNAs. This cleavage might serve either to remove a downstream, functional portion of the transcript or to eliminate a stem-loop transcription termination signal. Based on primer extension analysis, it was also observed that 5′ maturation of each of the RNAs can be completed despite a block to processing at the 3′ end. From this information it is possible to propose a general scheme for stable RNA maturation that includes 3′ endonucleolytic cleavage, complete 5′ maturation and 3′ exonucleolytic trimming as the final maturation event.
For some of the RNAs examined here, elimination of multiple exoribonucleases, leads to almost a complete absence of the mature species. Inasmuch as M1 and 4.5S RNAs are essential to cell survival (21, 22), these data imply that the incompletely processed molecules may retain function (23). A similar situation was reported earlier for 5S RNA. In that case a precursor with two extra nucleotides at its 3′ end could be assembled into ribosomes and functioned essentially normally (14). This, of course, raises the interesting question of why the mature forms of several RNAs are shorter than is necessary to generate a functional molecule (see below).
An unexpected feature of M1 RNA maturation was the finding that molecules with as many as six extra 3′ residues accumulated when multiple exoribonucleases were absent (Fig. 1). Yet, in vitro RNase E was shown to cleave the M1 RNA precursor one or two nucleotides downstream of the mature 3′ end (7, 8, 24). One possibility to explain this discrepancy is that the in vitro and in vivo RNase E cleavage sites differ. It is also possible that RNase E may actually cleave the M1 RNA precursor in vitro further downstream than was suggested, but exoribonucleases present in the relatively crude RNase E preparation rapidly trimmed the product to the +1 or +2 form. Alternatively, in the absence of the exoribonucleases needed to generate the mature 3′ terminus, the RNase E cleavage product may be lengthened by a synthetic activity, such as poly(A) polymerase. Interestingly, in earlier work that examined 5S RNA maturation, products longer than those expected from the presumed RNase E cleavage site were also found (14). Studies to clarify these surprising observations are currently underway.
As was observed previously for the maturation of tRNAs and 5S RNA (12–15), either RNase T or RNase PH appear to be the most effective exoribonucleases for the 3′ processing of the RNAs studied here. In vivo and in vitro studies both showed that RNase T was the most important exoribonuclease for the 3′ maturation of M1, 10Sa and 4.5S RNAs, whereas RNase PH was the more effective enzyme for 6S RNA. However, in the absence of both RNase T and RNase PH, or even of as many as four exoribonucleases, E. coli survives. Thus, while the absence of RNase D, II, and BN generally does not lead to the accumulation of immature RNAs, each of these latter enzymes also can provide sufficient exoribonuclease activity to allow cells to grow, albeit quite slowly in some instances (17). Overall, the 3′ maturation of M1, 10Sa, 4.5S, and 6S RNAs closely resembles that of tRNA in that multiple exoribonucleases can participate in the 3′ trimming reactions. This contrasts with 5S RNA processing in which RNase T is uniquely required for the process (14). A possible explanation for this diverse state of affairs is discussed below.
Analysis of the deduced secondary structures of the stable RNAs revealed an interesting feature common to all of them; i.e., the 5′ and 3′ ends pair with each other to form a stable, double-stranded stem generally followed by four unpaired 3′ nucleotides (Fig. 4). 5S RNA differs in having only one unpaired 3′ residue. The extra 3′ residues in the immediate RNA precursor extend the 3′ unpaired region further, and it is these residues that are removed by exoribonucleolytic trimming in the final maturation step. Secondary structures are known to impede exonucleolytic reactions (reviewed in ref. 25), and it is tempting to speculate that this structural arrangement (stable stem followed by a single-stranded tail) may serve as both a recognition signal for binding exoribonucleases and as physical barrier to limit the extent of trimming. For most exoribonucleases this barrier serves to stop the trimming reaction when the single-stranded tail is four nucleotides long. However, RNase T appears to differ from the other RNases in that it can approach closer to the double-stranded stem. This property would explain why RNase T is the only exoribonuclease that can mature the 3′ terminus of 5S RNA (14) and why it is the only enzyme that participates in the end-turnover of the -CCA sequence of tRNA. It is also consistent with the fact that RNase T is generally the most active RNase for removing the extra residues closest to the mature 3′ termini of the other stable RNAs. It does not act on long 3′ trailer sequences or on single-stranded substrates (13, 18, 26). RNase T is known to have homology with DNases that trim mismatched residues immediately 3′ to a base-paired region (27), and apparently it has retained similar catalytic properties for RNA substrates.
Figure 4.
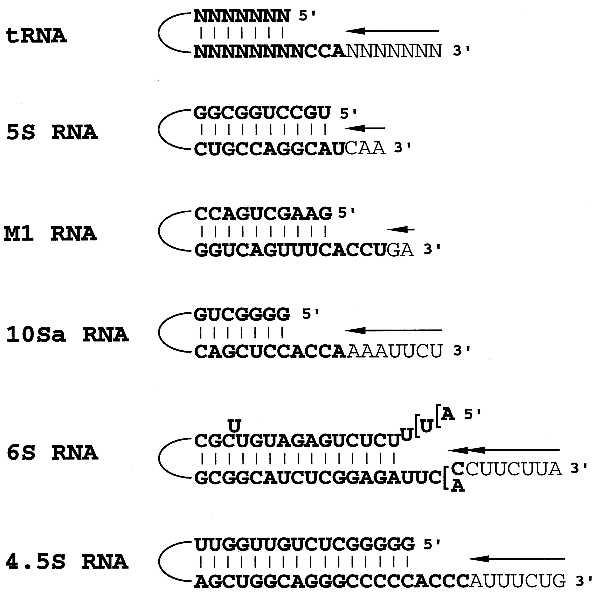
The terminal structures of the small, stable RNAs of E. coli. The sequence at the 3′ and 5′ ends of each RNA are shown together with their base pairing potential (lines for GC, AU, and GU pairs). Nucleotides present in the mature RNAs are shown in boldface type, while those to be removed from the 3′ ends during maturation are shown in lightface type. For those RNAs in which a 3′ endo cleavage already has been suggested (5S RNA and M1 RNA), only those extra residues that would remain after the cleavage are shown. For the other RNAs, seven extra residues are presented.
These considerations suggest that the 3′ termini of stable RNAs are determined both by their secondary structures and the inherent properties of the RNases that carry out the trimming reactions. For those RNAs, such as tRNAs, which might be susceptible to additional, unwanted trimming of 3′ residues, other protective mechanisms have evolved. Normally, aminoacylation would protect the 3′ end, but when a tRNA is uncharged and this protective mechanism fails, a shortened tRNA can be repaired by tRNA nucleotidyltransferase (16). The mature 3′ termini of other RNAs may be protected by their inclusion within ribonucleoprotein particles because repair reactions have not been observed.
The data presented here extend our knowledge of RNA maturation to an additional group of RNA molecules. The commonality of their 3′ processing with that of tRNAs and 5S RNA suggests that general strategies for RNA maturation may have evolved that apply to all species of stable RNA.
Acknowledgments
This work was supported by Grant GM16317 from the National Institutes of Health.
References
- 1.Deutscher M P. In: Nucleases. Linn S M, Lloyd R S, Roberts R J, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 377–406. [Google Scholar]
- 2.Reed R E, Baer M F, Guerrier-Takada C, Donis-Keller H, Altman S. Cell. 1982;30:627–636. doi: 10.1016/0092-8674(82)90259-8. [DOI] [PubMed] [Google Scholar]
- 3.Tu G F, Reid G E, Zhang J G, Moritz R L, Simpson R J. J Biol Chem. 1995;270:9322–9326. doi: 10.1074/jbc.270.16.9322. [DOI] [PubMed] [Google Scholar]
- 4.Wolin S L. Cell. 1994;77:787–790. doi: 10.1016/0092-8674(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 5.Brownlee G G. Nat New Biol. 1971;229:147–149. doi: 10.1038/newbio229147a0. [DOI] [PubMed] [Google Scholar]
- 6.Lee S Y, Bailey S C, Apirion D. J Bacteriol. 1978;133:1015–1023. doi: 10.1128/jb.133.2.1015-1023.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundberg U, Altman S. RNA. 1995;1:327–334. [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Kim H, Park I, Lee Y. J Biol Chem. 1996;271:19330–19337. doi: 10.1074/jbc.271.32.19330. [DOI] [PubMed] [Google Scholar]
- 9.Makarov E M, Apirion D. Biochem Intl. 1992;26:1115–1124. [PubMed] [Google Scholar]
- 10.Peck-Miller K A, Altman S. J Mol Biol. 1991;221:1–5. doi: 10.1016/0022-2836(91)80194-y. [DOI] [PubMed] [Google Scholar]
- 11.Kelly K O, Reuven N B, Li Z, Deutscher M P. J Biol Chem. 1992;267:16015–16018. [PubMed] [Google Scholar]
- 12.Reuven N B, Deutscher M P. FASEB J. 1993;7:143–148. doi: 10.1096/fasebj.7.1.8422961. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Deutscher M P. J Biol Chem. 1994;269:6064–6071. [PubMed] [Google Scholar]
- 14.Li Z, Deutscher M P. Proc Natl Acad Sci USA. 1995;92:6883–6886. doi: 10.1073/pnas.92.15.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Deutscher M P. Cell. 1996;86:503–512. doi: 10.1016/s0092-8674(00)80123-3. [DOI] [PubMed] [Google Scholar]
- 16.Zhu L, Deutscher M P. EMBO J. 1987;6:2473–2477. doi: 10.1002/j.1460-2075.1987.tb02528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly K O, Deutscher M P. J Bacteriol. 1992;174:6682–6684. doi: 10.1128/jb.174.20.6682-6684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deutscher M P, Marlor C W. J Biol Chem. 1985;260:7067–7071. [PubMed] [Google Scholar]
- 19.Deutscher M P, Hilderman R H. J Bacteriol. 1974;118:621–627. doi: 10.1128/jb.118.2.621-627.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin B E, Baillie D L. FEBS Lett. 1973;34:273–279. doi: 10.1016/0014-5793(73)80811-7. [DOI] [PubMed] [Google Scholar]
- 21.Sakano H, Yamada S, Ikemura T, Shimura Y, Ozeki H. Nucleic Acids Res. 1974;1:355–371. doi: 10.1093/nar/1.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourgaize D B, Fournier M J. Nature (London) 1987;325:281–284. doi: 10.1038/325281a0. [DOI] [PubMed] [Google Scholar]
- 23.Guerrier-Takada C, Altman S. Science. 1984;223:285–286. doi: 10.1126/science.6199841. [DOI] [PubMed] [Google Scholar]
- 24.Gurevitz M, Jain S K, Apirion D. Proc Natl Acad Sci USA. 1983;80:4450–4454. doi: 10.1073/pnas.80.14.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen S N. Cell. 1995;80:829–832. doi: 10.1016/0092-8674(95)90284-8. [DOI] [PubMed] [Google Scholar]
- 26.Salavati, R. & Altman, S. (1998) Antisense and Nucleic Acid Drug Development, in press. [DOI] [PubMed]
- 27.Koonin E V, Deutscher M P. Nucleic Acids Res. 1993;21:2521–2522. doi: 10.1093/nar/21.10.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]