

Abstract
The protein energy landscape theory is used to obtain optimal energy functions for protein structure prediction via simulated annealing. The analysis here takes advantage of a more complete statistical characterization of the protein energy landscape and thereby improves on previous approximations. This schema partially takes into account correlations in the energy landscape. It also incorporates the relationships between folding dynamics and characteristic energy scales that control the collapse of the proteins and modulate rigidity of short-range interactions. Simulated annealing for the optimal energy functions, which are associative memory hamiltonians using a database of folding patterns, generally leads to quantitatively correct structures. In some cases the algorithm achieves “creativity,” i.e., structures result that are better than any homolog in the database.
Keywords: protein folding, energy landscape
The prediction of protein structure from sequence is a practical art. As such, although there is much freedom in the detailed way protein structure prediction can be done, there are necessarily several constraints in the design of prediction algorithms. Anfinsen’s thermodynamic hypothesis (1), inferred from in vitro refolding experiments, suggests that protein structures might be predicted from sequence by minimizing an appropriate free-energy function. Currently, such functions must be simple in form so as to allow efficient computational search of the configuration space. While in the laboratory the folded protein is at a minimum of the exact free energy, there is no mathematical guarantee that a simplified energy function exists with good approximate structures as global minima. Nevertheless the search for such a useful energy function can be guided by the “wisdom” of the already huge database of known protein structures and by an understanding of the physics and chemistry of protein folding. To satisfy the Anfinsen hypothesis, an energy function at the very least for the proteins already in the database, must have global minimum structures near to the observed ones. This is not the only practical constraint. For computational efficiency, the global minimum should be rapidly found by the conformational search algorithm. Here theory can help. If the search method imitates real folding, as does simulated annealing by molecular dynamics or Monte Carlo, the energy landscape theory of folding kinetics can be used to predict which energy functions allow rapid folding and which ones permit only very slow folding, i.e., inefficient computational search (2–4). Several studies have used ideas from the energy landscape theory to discuss the optimization of energy functions for the inference of simplified potentials from a database of known structures (5–9).
In this paper we extend our previous work by showing how optimized energy functions can be determined that take advantage of a more complete statistical characterization of the protein energy landscape throughout its extent. Efficient folding requires avoiding traps on the energy landscape, so a fast-folding protein’s landscape must resemble a funnel leading toward the global minimum (2, 4, 10, 11). In such a landscape, the time to fold can be approximated as τ = τ0eF‡/kT. Here τ0 is the time it takes to explore a local region of configuration space and sample a configurationally distinct set of minima and F‡ is the thermodynamic free energy barrier between the set of unfolded states and the folded configurations. These quantities are related to statistical characteristics of the energy landscape. For fixed temperature, the thermodynamic barrier, F‡ strongly decreases with the ground state energy itself, characterized by the folding temperature Tf below which the global minimum is thermodynamically stable. The barrier also depends on other features, e.g., the extent of partial ordering in the denatured state, whether it is collapsed or not and whether the microscopic forces are pairwise additive, giving low barriers, or many-body, giving larger barriers (12). The reconfiguration time, τ0, increases with the ruggedness of the landscape, quantified by the magnitude of the statistical fluctuations of energy between local minima, ΔE2 that in turn depends on the degree of collapse. It also depends on local rigidity of the chain. τ0 depends weakly on the ground state energy, unless the folding search encounters strong topological problems, i.e., quasiknots in the chain. In general the reconfiguration time depends on proximity to the ideal glass transition temperature Tg at which typically the smaller traps become kinetically competitive with the global minimum.
When the folding temperature Tf exceeds Tg, folding of longer chains occurs for Tg < T < Tf, in a time that scales only polynomially with the chain length, whereas for T ≈ Tf < Tg, the longest search time grows exponentially of some power of the chain length N (13–15). Maximizing the ratio Tf/Tg leads to a large temperature range where the energy landscape is dominated by a folding funnel and where the search procedure allows the global minimum to be found rapidly.
By using simple statistical mechanical approximations based on the random energy model, both Tf and Tg for a given set of energy parameters in protein sequence can be determined. Optimal parameters then can be found by maximizing the ratio Tf/Tg appropriately averaged over a database of known structures. This yields the most efficient folding energy parameters. Implementing this “decoding” algorithm is an easier extremization problem than “forward” folding of a random sequence. The mathematical expression maximized in the simplest decoding is equivalent to the energy gap criterion later used to design foldable sequences, assuming the energy function is known (16, 17). As problems, decoding and design are mathematically “dual” to each other. In the early attempts at using the optimization decoding strategy, the ensemble of the denatured states was taken as given and independent of the potential. The statistical ruggedness of the landscape controlling the reconfiguration time is not uniform, however, so the actual denatured states that compete with the ground state depend on values of other order parameters such as the degree of collapse. The denatured configuration space sampled and the appropriate landscape statistics depend on those characteristics of the energy function that control the character of the thermodynamically occupied states. Folding efficiency therefore indirectly varies with parameters of the energy function that influence collapse and the type of secondary structures formed in the molten globule. To see this, note that expanded configurations have few contacts and little tendency to lead to traps whereas collapsed configurations have many opportunities to form opportunistic incorrect contacts. Therefore folding speed depends also on another characteristic temperature, Tc, defined as the temperature at which a nonspecific collapsed molten globule can stably form from the random coil. Increasing the tendency to collapse has two contradictory effects on folding speed (15, 18, 19). The thermodynamic free energy barrier is reduced entropically by collapse whereas the capability of collapsed structures to form bad contacts increases trapping. Klimov and Thirumalai (20, 21) have emphasized the ratio Tf/Tc as playing a role in determining the folding rates—in addition to the well-established dependence on Tf/Tg. Like generic collapse, rigidifying secondary structure in the denatured state has contradictory effects on folding kinetics. Increasing local interactions and hydrogen bonding raises Tf, thereby lowering the effective thermodynamic barrier, but again slows reconfigurational motions by introducing barriers to even local rearrangements (22, 23).
Quantitative treatment of how various landscape characteristics change folding speed is an ongoing activity (24–29). Using such quantitative theories for optimizing structure-prediction algorithms may be a task with a long future. Here we continue on this route by developing a practical approach to optimizing energy functions that as in earlier work achieves a funnel-like energy landscape but also takes into account these weaker determinants of folding speed to some extent. We do this first by producing quantitative measures of those statistical quantities that characterize the collapse and partial order of the denatured states. We then use these as additional constraints when optimizing the Tf over Tg ratio. Specifically, we use Lagrange multipliers constraining the collapse temperature and short-range sequence ruggedness. By doing so we ensure that the attempt to obtain good discrimination against a set of traps in one part of configuration space does not lead to an energy surface with a qualitatively different set of traps. Here we illustrate this practical strategy for self-consistently optimizing energy functions by finding an optimal associative memory (AM) energy function with a simple encoding (5, 30, 31). AM energy functions are explicitly based on a database of known protein structures. They provide a very flexible way of using database information. They are simultaneously closely related to both neural networks that predict structures from sequence (32) and to the empirical (33–35) energy functions that use reduced descriptions of the protein. AM energy functions also can exploit knowledge of homology by preprocessing the sequence to increase the funnel-like nature of the landscapes. They are thus a very useful general framework for discussing a range of structure prediction schemes. The strategies we illustrate here can be used for more conventional energy functions as well.
The organization of this paper is as follows: By using a schematic view of the global density of states we quantify landscape features that control folding speed by approximating the characteristic temperatures (Tf, Tc and Tg.) Combining expressions for the folding and glass transition temperatures in terms of the energy parameters with additional relations for Tc and for the rigidity of the short-range structures, we describe the generic constrained self-consistent optimization strategy. We then briefly review the AM hamiltonian formulation and ways of including short-range interactions such as hydrogen bonding. We then apply the framework to obtain energy functions that lead to fast folding for a representative set of α-helical proteins. We describe briefly how the quality of the structure prediction changes with the various statistical landscape constraints. Finally, we identify the future prospects for using energy landscape ideas for improving practical protein structure prediction.
Constrained Self-Consistent Optimization Methodology
Fig. 1 illustrates schematically, in two different ways, some of the relevant statistics of a protein folding energy landscape. There are a large number of configurations at the top of the funnel that are nearly random coils, with few nonlocal contacts. These provide the bulk of the configurational entropy, Src = Nlnν where ν is the number of conformational states per monomer. As contacts are made the energy on the average decreases, but these contacts may be native-like and specific or indiscriminate and nonspecific. If even nonspecific contacts are sufficiently favorable energetically, a collapsed but fluid set of configurations becomes thermodynamically relevant and indeed may be a separate phase. In the lower half of the figure we sketch histograms of the energies of the configurations. The nonspecific collapse can be a first order or continuous transition depending on the backbone rigidity. This is reflected in the detailed shape of the entropy curves. Approximately one kB of entropy per residue would be lost because of collapse of the chain per residue. The average energy difference between a collapsed configuration and a random coil is δEc = 〈E〉rc − 〈E〉mg. Finally, the folded configurations are well separated from these disordered configurations by a stability gap from the molten globule, δEs = 〈E〉mg − Ef and by the amount δEs + δEc from the coils. For any energy function both δEs and δEc are easily computed once a set of disordered globule configurations has been generated. From a set of globule configurations, one also can compute the variance of energies or ruggedness ΔE2. The ruggedness along with the collapsed states entropy gives an estimate of the depth of the deepest misfolded trap by using spin glass theory.
Figure 1.

(A) Schematic diagrams of a protein’s energy landscape and energetic distribution of its density of states. The landscape is characterized by the protein’s energy and entropy. The top of the funnel depicts the large ensemble of random coil configurations. As the energy decreases so does the number of configurations until the lowest energy state is reached, which is the protein’s unique native structure. (B) The density of states histograms show the same picture but emphasize the relationships between the characteristic temperatures (Tf, Tc and Tg) and δEs, δEc, and ΔE. They depict two possible folding scenarios. (Left) An energy landscape in which the protein will first collapse upon cooling and then fold. (Right) A case that will lead to faster folding for the stability gap is large enough that the kinetically relevant ruggedness at the transition state will be smaller and folding will occur directly from a random coil. Because we want to optimize against even the worst folders, we will use the scenario at left for our optimization procedure.
The three energy statistics δEs, δEc, and ΔE2 lead to the three characteristic temperatures Tf, Tc, and Tg. Precise values of these temperatures depend on how entropy decreases with collapse and ordering but simple estimates can be made that are monotonically related to the more exact values. If the histogram is like the left of Fig. 1B, upon cooling the protein first collapses and then folds. With a larger energy gap direct folding from the random coils occurs as in the right of Fig. 1B. The second case will lead to faster folding because the kinetically relevant ruggedness at the transition state will be smaller. On the other hand, it is the first case that will apply for the worst folders of a training set so we will optimize by using the worst-case scenario that Tf ≤ Tc. This still guarantees good performance for good folders. In this scenario we can approximate the folding temperature by equating the free energy of the globule and the native structure giving Tf = δEs/Smg. The collapse temperature is approximated by the first-order transition formula, by using the known entropy loss of 1kB per residue, Tc = δEc/NkB. This not a bad estimate of where a nonspecific molten globule can form, even when that transition is not first order. When a nonspecific molten globule forms, there is a peak in heat capacity near Tc. Thirumalai defines a temperature Tχ as this peak. When Tχ ≅ Tc this is a useful definition but when collapse is specific, i.e., Tχ ≅ Tf, Tχ incompletely characterizes the phase diagram (just as we do not say the sublimation point of dry ice is its boiling point). Only in the collapsed phase is trapping kinetically serious. The characteristic temperature for traps is the glass transition Tg, which within the random energy approximation, is Tg = 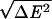 /
/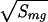 . Tg is always less than Tc (15, 18).
. Tg is always less than Tc (15, 18).
By using these formulas, unconstrained maximization of Tf/Tg is equivalent to maximizing the ratio δEs/ΔE. Optimization is simplest when considering parameters in the energy function that enter in a linear fashion, E = ∑γiξi. The γi’s are the strengths of the interactions terms whereas the ξi’s are the basic forms of the various interactions terms. In the present study, ξi will depend on hydrophobicity and proximity of two amino acids in a protein sequence, but there are many other possibilities. Varying the strength parameters γ leads to an optimization problem that can be explicity solved with linear algebra. The stability gap giving Tf can be written as δEs = Aγ, whereas the energetic variance giving Tg can be written ΔE2 = γ B γ. A and γ are vectors of dimensionality equal to the number of interaction types and B is a matrix given by
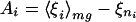 |
1 |
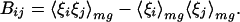 |
2 |
These averages depend on the frequencies at which any given interaction occurs in molten globule and native configurations. Maximizing the energy ratio Aγ/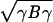 gives the solution that γ = B−1A up to a scalar multiple. (We note performing an average over training proteins gives rise to the harmonic mean expression used below in Eq. 4.) The collapse temperature is also a linear function of the energy parameters γ, Tc = A′γ where A′i = 〈ξi〉mg/N. If we want to control collapse it is reasonable to impose a constraint on Tc. This is a linear constraint that gives a new optimization functional [A − λ1A′]γ − λ*γBγ. The Lagrange multiplier λ1 can be chosen to maintain the ratio of Tf/Tc close to 1 while λ* sets the energy scale.
gives the solution that γ = B−1A up to a scalar multiple. (We note performing an average over training proteins gives rise to the harmonic mean expression used below in Eq. 4.) The collapse temperature is also a linear function of the energy parameters γ, Tc = A′γ where A′i = 〈ξi〉mg/N. If we want to control collapse it is reasonable to impose a constraint on Tc. This is a linear constraint that gives a new optimization functional [A − λ1A′]γ − λ*γBγ. The Lagrange multiplier λ1 can be chosen to maintain the ratio of Tf/Tc close to 1 while λ* sets the energy scale.
In estimating Tg the simple random energy approximation assumes the molten globule states have little or no native secondary or tertiary contacts (Q ≈ 0). Thus to find B we should subtract out the native contributions to Bij that only give heterogeneous but still native contacts. We can do this by redefining the variance in the molten globule distributions B′ij to reflect only the non-native fluctuations. In other words the “random” part of the energy of any molten globule configuration is defined by projecting out the contribution from its overlap QN with the native state, E′mg = Emg(1 − QN).
In molecular dynamics polymer chains move by locally overcoming barriers through backbone φ,ψ isomerizations. If there are local configurations with too low an energy, they will act as traps for individual segmental motions. One way to control the ruggedness of the short-range interactions or rigidity is to also ensure this local contribution to energetic fluctuations does not grow large. Imposing this constraint leads to another optimization functional [A − λ1A′]γ − λ2γB′sγ − λ*γB′γ. The fluctuation matrix B′s is determined by the local in sequence interactions. The new Lagrange multiplier λ2 then can be selected in each optimization iteration so that γB′sγ/γB′γ remains constant. Constrained optimization leads to the simple variational equation
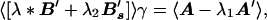 |
3 |
where 〈〉 indicates an average over a set of training proteins.
Proteins do not all have the same global energy landscape shapes, and therefore when the energy parameters γ are optimized by averaging over the set of training proteins, there will be a range of Tf/Tg values. To correct for the variation in these values, we scaled each training protein’s A and B matrices by a factor ω. The value of ω for a given proteins was chosen to equal its corresponding Tg/Tf so that the proteins with the lowest Tf/Tg values contribute the highest weight to the global γ values. The iterative optimization leads to equation
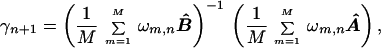 |
4 |
where B̂ = B′ + λ2B′s, Â = A−λ1A′, M is the total number of training proteins, and ωm,n is the Tg/Tf value for the corresponding training protein evaluated with the γn values. During simulated annealing, B̂, Â, and the ωm,n depend on the γn values, so Eq. 4 iterated until convergence. This procedure is similar to the harmonic mean average suggested by others (8). Averaging procedures are far from unique, but results are only weakly dependent on them.
The minima of the misfolded structures for each training protein are generated through molecular dynamics simulations using the γn values. Even misfolded structures are partially ordered and have a tendency to satisfy any especially large interaction energy terms. Thus iteratively maximizing the ratio of δE/ΔE where the new stability gap is defined as the difference between the energy of the native fold and the mean energy of the thermally generated misfolded structures, and the new standard deviation is over the energy distribution of these structures, increasing the discrimination between correct and the typically misfolded structure found by simulated annealing. Because the misfolded structures themselves depend on the energy function, the optimal γ values for each iteration are calculated with Eq. 4 in which the 〈ξi〉mg values in Eqs. 1 and 2 now denote the average frequency of occurrence in the current set of misfolded structures. To smooth convergence, successive over-relaxation was used. Each round of optimization combines the interaction parameters from previous optimizations, γn = (1 − ɛ)γn−1 + ɛγn′. Here we chose ɛ = 0.25.
For the first step of optimization, the molten globule states were generated by translating the target sequences along scaffolds of unrelated folds (5, 6). In the subsequent rounds the ensemble of misfolded structures were generated in constant temperature simulations and constrained to have Q < 0.4.
AM Potential and Backbone
The AM potential introduced earlier by the Illinois group (5, 31) is based on the correlations between a target protein’s sequence and the sequence-structure patterns in a set μ of memory proteins. The interaction terms are obtained by averages over the memory set. One may either average without using knowledge of precisely which pair of residues in the target structure may correspond with a given pair in a memory protein, or a correspondence can be set up by using a memory preliminary alignment procedure. Here we associate pairs by the mean-field alignment of the target sequence to the structural scaffold provided by the memory proteins using an energy-based threading algorithm developed earlier (7). The threading procedure places insertions and deletions so that residues i and j of the the target align to residues i′ and j′ in the memory protein. The energy parameters γ encode similarity (defined by a few amino acid properties Pi) between residues pair i and j of the target protein and the corresponding pair in the memory protein. Between nonadjacent residues only four types of interactions are considered, i.e., between Cα − Cα, Cα − Cβ, Cβ − Cα, and Cβ − Cβ). The AM potential encoding these patterns and interactions is given by
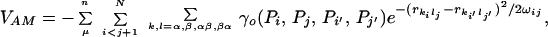 |
5 |
where the structural similarity between the target and memory protein is measured by a gaussian function. rkilj is the distance between the k and l atoms of residues i and j in the target protein; rki′lj′ is the distance between k and l atoms of residues i′ and j′ in the memory protein μ; ωij is a tolerance specifying how close pairing distances should match and is equal to (j − i)0.3, allowing a more generous mismatch for residues distant in sequence. We consider here as in the previous study a simple binary hydrophobicity scale with three proximity classifications: short range, where j − i < 5; intermediate or tertiary range where j − i > 4 and ri′j′ < 8.0Å; and long range where j − i > 4 and ri′j′ > 8.0Å. We also consider an energy function (EF-15) with a maximal cut-off distance, j − i > 4 and 8.0Å < ri′j′ < 15.0Å.
In addition to the side-chain sequence-dependent interaction, simulated annealing requires specifying a backbone potential that maintains chain connectivity and encourages correct peptide stereochemistry. The backbone potential is similar to Friedrichs et al. (31) with an additional hydrogen-bonding term Vhb and a more realistic Ramachandran potential (Vrama) to describe the allowed torsional angles of the backbone. The total potential used in the molecular dynamics simulations is
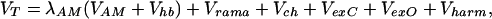 |
6 |
where Vch is a chirality potential that biases L-amino acid chirality; VexC and VexO are excluded volume potentials to prevent nonbonded carbon and oxygen atoms from coming within 3 Å and 4 Å of each other, respectively. Vharm is a sum of three quadratic potentials that are used along with a series of SHAKE constraints shown in Fig. 2 to provide backbone rigidity and assure the planarity of the peptide bond. The sequence-dependent potentials VAM and the hydrogen bond strength are simultaneously optimized. Both potentials are rescaled at the beginning of any annealing run so that the total AM energy of a target protein is approximately 4N. The scale factors for the other backbone terms have been empirically chosen (unpublished data).
Figure 2.

A schematic diagram of the constraints and the Vharm and Vrama potentials on a protein’s backbone. The blue dashed bonds indicate the pairs of atoms whose distances are kept fixed (shaken). Harmonic potentials are applied to the atom pairs connected by the red wavy lines. The potentials are used to maintain the rigidity and planarity of the peptide backbone.
Results
The energy parameters γ were optimized by using a set of known, well-resolved structures from the α-helical folding class. The 10 proteins in the training set represent five of the six general folds characterized by Orengo et al. (36): α metal rich, orthogonal, EF-hand, α up-down, and globin. After aligning the 10 training proteins to a representative set of 34 proteins from the topology subgroup of the mostly α class of proteins (36), the 20 lowest energy alignments for each training protein were chosen as its memory set. A training protein has, at most, two structural analogs included in its memory set. If a training protein had more than two structural analogs, the two lowest energy structures with the lowest sequence identities were selected. The structural analogs of the training proteins also were aligned by using a modified Needleman-Wunsch (P-NW) alignment algorithm (7). The alignments between the two different methods were compared, and the ones with the highest Q-scores were used.
At each iteration, we can characterize the discrimination by the value of Tf/Tg = δEs/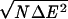 for each training protein. This averaged ratio is given in Fig. 3 as a function of the iteration step. The mean energy and variance for the molten globule ensembles were evaluated from a set of 100 misfolded structures (Q < 0.4 when compared with the native structure, and a radius of gyration ranging from 80% to 130% of the native structure) generated during the molecular dynamics simulation with the nth iterate energy function.
for each training protein. This averaged ratio is given in Fig. 3 as a function of the iteration step. The mean energy and variance for the molten globule ensembles were evaluated from a set of 100 misfolded structures (Q < 0.4 when compared with the native structure, and a radius of gyration ranging from 80% to 130% of the native structure) generated during the molecular dynamics simulation with the nth iterate energy function.
Figure 3.

Comparison of Tf/Tg (•) and Tf/Tc (▪) ratios averaged over the 10 training proteins. The colors represent different iterations of the EF γ values used to calculate the ratios. All of the ratios evaluated with the γ values obtained from the GSW optimization procedure (5) are shown in black. We calculated the average ratios with respect to each set of molten globule ensembles. The −1.0 mark represents the molten globule ensemble generated by translation, the 0.0 mark represents the ensembles generated with the 0th iterate γ values, etc. The plot reveals that the second iterate γ values produce the highest Tf/Tg on average and that the Tf/Tc ratio is being conserved through each round of optimization. For the second iterate optimization λ1 = −0.1 and λ2 = 78.0.
We see the second iterate γ values give the largest discrimination with respect to any of the molten globule ensembles. The average Tf/Tc values shown in the graph verify the constraint on the collapse transition imposed in Eq. 3. Similar results are seen for the EF-15 γ values (data not shown). Self-consistent optimization without the constraint gives collapse temperatures ranging from much greater than Tf to values much smaller than Tχ. By maintaining this ratio close to one the folding and collapse transitions nearly coincide, which conveniently meshes with an efficient annealing schedule.
Our previous work (GSW, ref. 5) indicated that kinetic traps are best avoided when the energy is distributed evenly between the short-range, tertiary, and long-range interactions. The ratio between the average short range and tertiary interactions in the native structures by using the GSW optimization was 0.83. When unconstrained, optimization leads to a dramatically larger part of the energy fluctuations coming from the local in sequence term. The formation of local secondary structure overshadows the effect of any specific tertiary interaction leading to very inefficient folding at temperatures sufficient to stabilize the tertiary fold. When the Lagrange multiplier for short-range ruggedness is introduced, the short- to long-range ruggedness ratio using these γ values is equal to 1.03.
Eight proteins were annealed by molecular dynamics using the EF-15(γn = 3) and EF(γn = 2) energy parameters. Basically the annealing protocols of Friedrichs et al. (31) were used. Each run takes approximately 8–20 hr on a SGI INDY work station. As shown in Table 1, the predictions were highly structurally similar to the correct structures for 6 of 8 targets using the EF-15 (γn = 3) values and 7 of 8 for the EF (γn = 2) values. The rms deviation from the x-ray structure is considerably improved over the GSW optimized γ values (5). In fact, we may say the algorithm achieves some “creativity” for two structures, the bovine calcium-binding domain (3icb), the results of which are shown in Fig. 4, and Escherichia coli the gamma delta resolvase (DNA binding domain) (1res). In these cases either the Q-score or the rms of the structures obtained from simulated annealing are better than any example used to construct the energy function.
Table 1.
Results of simulated annealing for eight targets
| Protein | Best memory
|
Predicted structure EF-15
|
Predicted structure EF
|
|||||
|---|---|---|---|---|---|---|---|---|
| NRES | Q | %I | rms*† | Q | rms* | Q | rms* | |
| 1r69 | 63 | 0.89 | 52.4 | 0.85 | 0.73 | 2.17 | 0.68 | 6.08 |
| 1utg | 70 | 0.90 | 55.7 | 0.78 | 0.31 | 8.30 | 0.53 | 6.58 |
| 3icb | 75 | 0.52 | 28.0 | 3.28 | 0.57 | 3.51 | 0.60 | 3.38 |
| 5pal | 109 | 0.89 | 44.4 | 0.85 | 0.62 | 3.69 | 0.77 | 2.33 |
| 1ccr | 112 | 0.87 | 57.4 | 1.39 | 0.79 | 1.96 | 0.80 | 2.4 |
| 1res | 43 | 0.48 | 16.3 | 13.02 | 0.33 | 6.84 | 0.42 | 7.77 |
| 1hyp | 75 | 0.43 | 22.7 | 5.46 | 0.32 | 8.14 | 0.29 | 12.13 |
| 5cyt(R) | 103 | 0.75 | 34.3 | 1.56 | 0.32 | 9.99 | 0.44 | 5.94 |
The Protein Data Bank designation of the protein is listed in column 1. The next four columns list the number of residues (NRES), the degree of sequence identity of the target with the most homologous structure in the memory set as well as the Q-score and rms deviation. Columns 6 and 7 give the Q-score and rms deviation for predicted structures using the EF-15 (γn=3) values with respect to the correct structure, and the last two columns given the results for using EF (γn=2 values).
Based on Cα atoms.
Based on alignment to homolog.
Figure 4.

(A) Comparison of the predicted structure (red), the most similar memory scaffold (blue), and the native structure of 3icb (green). (B) rms deviation (based on Cα atoms only) between the native structure of 3icb and the predicted structure (red) and the rms between the best memory scaffold and the native structure (blue). Both representations demonstrate the “creativity” achieved by prediction with this algorithm. The rms deviation plot clearly shows the areas in which our predicted structure more accurately represents the native state. The circled values indicate the insertions in the alignment to the memory scaffold.
The predicted structures of rabbit uteroglobin (1utg), 3icb, rice embryo cytochrome c (1ccr), and cytochrome c (5pcyR) indicates that although they essentially reproduce the native structures, the core elements tend to be slightly overcollapsed (see Fig. 5). 1utg, a very open structure, is the most overcollapsed. Some part of the overcollapse is probably because of the assignment of equal radii for all Cβ atoms and from imposing an overidealized backbone potential. In general, including spatially distant correlations leads to more accurate predictions of more opened structures such as 1utg. In the case of 1r69 and 1ccr the end helices have the highest rms deviation. The poorest result is for the small hydrophobic soybean seed protein (1hyp), which has four disulfide bonds.
Figure 5.

Superposition of the predicted (blue) and native (yellow) structures of (A) uteroglobin (1utg), (B) gamma delta resolvase (1res), (C) rice embryo cytochrome c (1ccr), and (D) cytochrome c (5cyt(R)). All of the predicted structures show the correct topology for the native structure although each predicted structure tends to be slightly overcollapsed. The 1utg predicted structure has the largest deviation from its native structure caused by overcollapse. 1res is one of the two predicted structures that exhibit “creativity,” i.e., the rms of the predicted structure is higher than the rms of the best scaffold in its memory set.
Conclusion
The results of this paper show that self-consistent optimization augmented by constraints on short-range interactions and collapse temperature can lead to efficient structure prediction algorithms by using simulated annealing. Although the present constrained self-consistent optimization strategy is effective, we have not optimized the magnitude of the constraints on Tc and short-range ruggedness. This requires detailed exploration of the dependence of folding speed on these parameters and is a nonlinear problem. There is no barrier to carrying out such studies. Searching through a few constraints is much more convenient than a complete search through the entire energy parameter space. Likewise information about how specific structure patterns influence folding speed can be incorporated into a structure prediction strategy. Because some interactions are more important in the folding transition state than others (37–39) and their specific contributions can be inferred from theory (40), these interactions can be emphasized in the folding speed optimization strategy. Perhaps the most pressing future development is the application of the decoding strategies using a finer division of the amino acids into classes than that used here based on hydrophobicity. All of these advances should help achieve even greater efficiency and accuracy in structure prediction.
Acknowledgments
We thank Corey Harden for supplying the hydrogen bond potential and José Nelson Onuchic and Klaus Schulten for reading the manuscript. Computations were carried out in part at the National Center for Supercomputing Applications in Urbana, IL. This work was supported by National Institutes of Health Grant No. 1R01 GM44557.
ABBREVIATION
- AM
associative memory
Appendix
The 10 training proteins (shown in bold) and their corresponding memory proteins were selected from the Protein Data Bank (PDB) (41). Their PDB codes are: 1R69: 3CRO(L), 1LMB(3), 1ROP, 1UTG, 1SCT(A), 1ABK, 1MBA, 451C, 1LE4, 1CC5, 149L, 1GDJ, 256B(A), 2SPC(A), 2HHB(A), 2TMV(P), 1COL(A), 1YCC, 2CCY(A), 3ICB; 1UTG: 1CCD, 1CC5, 1HBH, 4CPV, 1ALA, 1ABK, 1ROP, 3ICB, 1LE4, 2WRP(R), 2SPC(A), 2CCY(A), 149L, 1GDJ, 1MBC, 256B(A), 1POD, 1R69, 2SAS, 4FIS; 3ICB: 4CPV, 5TNC, 2SAS, 1ABK, 1ALA, 1MBC, 149L, 1COL(A), 256B(A), 1MBA, 2TMV(P), 1GDJ, 1HBH(A), 1UTG, 1LE4, 451C, 2SPC(A), 1ITH, 2WRP(R), 2CCY(A); 256B(A): 2CCY(A), 1LE4, 1MBC, 1UTG, 1HBH(A), 1GDJ, 2SAS, 2HHB(A), 2SPC(A), 2WRP(R), 1MBA, 149L, 1CC5, 1ALA, 1ROP(A), 2TMV(P), 4CPV, 1POD, 1COL(A), 1SCT(A); 5PAL: 1RRO, 4CPV, 149L, 3ICB, 1MBC, 2SAS, 256B(A), 1MBA, 1ABK, 1ALA, 1GDJ, 2TMV(P), 451C, 2HHB(A), 1POD, 1LE4, 1HBH(A), 1R69, 1UTG, 2SCP(A); 1CCR: 1YCC, 3C2C, 3SPD, 1MBA, 1CC5, 1MBC, 1ALA, 1GDJ, 149L, 1ITH, 2SCP(A), 451C, 1SCT(A), 2WRP(R), 1HDD(A), 2SAS, 2HHB(A), 2SPC(A), 3ICB, 1LE4; 2MHR: 2HMZ(A), 1GDJ, 1POD, 1ALA, 2HHB(A), 1ABK, 1LE4, 1MBC, 1MBA, 3SDP, 2TMV(P), 1SCT(A), 4CPV, 2SPC(A), 149L, 1PRC(L), 1COL(A), 1HBH(A), 3ICB, 2SAS; 1MOH: 1MBA, 1MBC, 1POD, 4CPV, 1LE4, 1YCC, 1ABK, 2SAS, 2CCY(A), 2SCP(A), 256B(A), 1ALA, 1CC5, 2MHZ(A), 2TMV(P), 3ICB, 451C, 1LMB(3), 1PRC(L), 1COL(A); 2MYE: 1MBA, 1GDJ, 1ABK, 1LE4, 149L, 2SAS, 1COL(A), 2SCP(A), 1ALA, 2TMV(P), 1POD, 256B(A), 1YCC, 2WRP(R), 1PRC(L), 2CCY(A), 2SPC(A), 451C, 1CC5, 2MHZ(A); 1GDJ: 1MBC, 1HBH(A), 149L, 2CCY(A), 1ALA, 1ABK, 4CPV, 1POD, 1LE4, 256B(A), 2SAS, 2TMV(P), 2SCP(A), 1COL(A), 1YCC, 1PRC(L), 1CC5, 2MHZ(A), 1LMB(3), 2SPC(A).
References
- 1.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 4.Onuchic J N, Luthey-Schulten Z A, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein R A, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein R, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koretke K K, Luthey-Schulten Z, Wolynes P G. Protein Sci. 1996;5:1043–1059. doi: 10.1002/pro.5560050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirny L A, Shakhnovich E I. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 9.Liwo A, Pincus M R, Wawak R J, Rackovsky S, Oldziej S, Scheraga H A. J Comp Chem. 1997;18:874–887. [Google Scholar]
- 10.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 13.Wolynes P G. In: Search and Recognition: Spin Glass Engineering as an Approach to Protein Structure Prediction. Peliti L, editor. New York: Plenum; 1991. pp. 15–37. [Google Scholar]
- 14.Gutin A, Abkevich V, Shakhnovich E I. Phys Rev Lett. 1996;77:5433–5436. doi: 10.1103/PhysRevLett.77.5433. [DOI] [PubMed] [Google Scholar]
- 15.Wolynes P G. Proc Natl Acad Sci USA. 1997;94:6170–6175. doi: 10.1073/pnas.94.12.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shakhnovich E I, Gutin A M. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hao M, Scheraga H. Proc Natl Acad Sci USA. 1996;93:4984–4989. doi: 10.1073/pnas.93.10.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryngelson J D, Wolynes P G. Biopolymers. 1990;30:177–188. [Google Scholar]
- 19.Socci, N. D., Onuchic, J. N. & Wolynes, P. G. (1998) Proteins Struct. Funct. Genet., in press.
- 20.Thirumalai D. J Physique I. 1995;5:1457–1467. [Google Scholar]
- 21.Klimov D K, Thirumalai D. Phys Rev Lett. 1996;76:4070–4073. doi: 10.1103/PhysRevLett.76.4070. [DOI] [PubMed] [Google Scholar]
- 22.Govindarajan S, Goldstein R A. Biopolymers. 1995;36:43–51. [Google Scholar]
- 23.Abkevich V I, Gutin A M, Shakhnovich E I. J Mol Biol. 1995;252:460–471. doi: 10.1006/jmbi.1995.0511. [DOI] [PubMed] [Google Scholar]
- 24.Saven J G, Wang J, Wolynes P G. J Chem Phys. 1994;101:11037–11043. [Google Scholar]
- 25.Wang J, Saven J G, Wolynes P G. J Chem Phys. 1996;105:11276–11284. [Google Scholar]
- 26.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 27.Plotkin S S, Wang J, Wolynes P G. Phys Rev E. 1996;53:6271–6296. doi: 10.1103/physreve.53.6271. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Plotkin S S, Wolynes P G. J Physique I. 1997;7:395–421. [Google Scholar]
- 29.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 30.Friedrichs M S, Wolynes P G. Science. 1989;246:371–373. doi: 10.1126/science.246.4928.371. [DOI] [PubMed] [Google Scholar]
- 31.Friedrichs M S, Goldstein R A, Wolynes P G. J Mol Biol. 1991;222:1013–1034. doi: 10.1016/0022-2836(91)90591-s. [DOI] [PubMed] [Google Scholar]
- 32.Bohr H G, Wolynes P G. Phys Rev A. 1992;46:5242–5248. doi: 10.1103/physreva.46.5242. [DOI] [PubMed] [Google Scholar]
- 33.Miyazawa S, Jernigan R L. Macromolecules. 1985;218:534–552. [Google Scholar]
- 34.Casari G, Sippl M J. J Mol Biol. 1992;224:725–732. doi: 10.1016/0022-2836(92)90556-y. [DOI] [PubMed] [Google Scholar]
- 35.Miyazawa S, Jernigan R L. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 36.Flores T P, Orengo C A, Moss D S, Thornton J M. Protein Sci. 1993;2:1811–1826. doi: 10.1002/pro.5560021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 38.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 39.Onuchic J N, Socci N D, Luthey-Schulten Z, Wolynes P G. Folding Design. 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 40.Shoemaker B A, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]