

Abstract
Peroxisomal disorders are lethal inherited diseases caused by either defects in peroxisome assembly or dysfunction of single or multiple enzymatic function(s). The peroxisomal matrix proteins are targeted to peroxisomes via the interaction of peroxisomal targeting signal sequences 1 and 2 (PTS1 or PTS2) with their respective cytosolic receptors. We have studied human skin fibroblast cell lines that have multiple peroxisomal dysfunctions with normal packaging of PTS1 and PTS2 signal-containing proteins but lack catalase in peroxisomes. To understand the defect in targeting of catalase to peroxisomes and the loss of multiple enzyme activities, we transfected the mutant cells with normal catalase modified to contain either PTS1 or PTS2 signal sequence. We demonstrate the integrity of these pathways by targeting catalase into peroxisomes via PTS1 or PTS2 pathways. Furthermore, restoration of peroxisomal functions by targeting catalase-SKL protein (a catalase fused to the PTS1 sequence) to peroxisomes indicates that loss of multiple functions may be due to their inactivation by H2O2 or other oxygen species in these catalase-negative peroxisomes. In addition to enzyme activities, targeting of catalase-SKL chimera to peroxisomes also corrected the in situ levels of fatty acids and plasmalogens in these mutant cell lines. In normal fibroblasts treated with aminotriazole to inhibit catalase, we found that peroxisomal functions were inhibited to the level found in mutant cells, an observation that supports the conclusion that multiple peroxisomal enzyme defects in these patients are caused by H2O2 toxicity in catalase-negative peroxisomes. Moreover, targeting of catalase to peroxisomes via PTS1 and PTS2 pathways in these mutant cell lines suggests that there is another pathway for catalase import into peroxisomes and that an abnormality in this pathway manifests as a peroxisomal disease.
Keywords: peroxisomal disease, peroxisomal targeting signal
Peroxisomes are becoming the focus of increased interest because they are involved in a number of important cellular metabolic processes, including the oxidation of fatty acids; biosynthesis of cholesterol, bile acids, and plasmalogen; and detoxification of H2O2 (1, 2). Abnormality in peroxisomal functions leads to progressive neurological disorders (1–3). The peroxisomal disorders identified, so far, are divided into two groups. One group has a specific functional defect due to an abnormality in its DNA sequence (e.g., X chromosome-linked adrenoleukodystrophy). The other group, with multiple enzyme defects, is again divided into two subgroups: one with intact peroxisomes (e.g., rhizomelic chondrodysplasia punctata) and others that lack peroxisomes (e.g., Zellweger syndrome).
Peroxisomal proteins, studied to date, are synthesized on free polysomes, and posttranslational targeting to peroxisomes is controlled by targeting signal sequences (4–6). Two types of targeting sequences have been characterized for peroxisomal matrix proteins, peroxisomal targeting signal sequences 1 and 2 (PTS1 or PTS2). PTS1 is the carboxyl-terminal tripeptide SKL or its variant (4–8) and PTS2 is first 11–16 amino acids of the protein sequence (9–14). Catalase, a major peroxisomal enzyme responsible for the metabolism of H2O2, is devoid of such conserved targeting signal sequence. However, recently it has shown that like the SKL sequence, the carboxyl-terminal sequence KANL of catalase is important and sufficient to target catalase into peroxisomes in humans and yeast (15).
Immunofluorescence studies for import of peroxisomal proteins and subsequent cloning of PTS1 receptor (PTS1R) and PTS2 receptor (PTS2R) sequence have identified selective import defects due to mutation in these receptors. Fibroblasts from patients with nALD are selectively deficient in the PTS1 pathway because of a mutation in PXR1, human PTS1 receptor (16). Yeast mutants such as Pas8 of P. pastoris (17) and pas10 of Saccharomyces cerevisiae (18) are also selectively deficient in the import of PTS1 proteins. On the other hand, the pas7 mutant of S. cerevisiae (19) and patients suffering from rhizomelic chondrodysplasia punctata are deficient only in the PTS2 receptor (PTS2R)-dependent import pathway (11, 20).
We investigated human skin fibroblasts (Rh and Sm cells) from patients with Zellweger-like clinical features and multiple peroxisomal enzymatic deficiencies but found that catalase was mislocalized to the cytosol. These cell lines have morphologically intact peroxisomes with normal level of acyl-CoA oxidase, a PTS1 protein, and 3-keto acyl-CoA thiolase, a PTS2 protein, but patients with this disorder appear to have multiple enzyme deficiencies (21). In this article, we report that the multiple peroxisomal enzyme defects in these patients is caused by the absence of catalase in peroxisomes because multiple peroxisomal functions were normalized when catalase was reintroduced into peroxisomes via PTS1 pathway. These observations suggest that in humans targeting of catalase to peroxisomes is mediated by a pathway that is independent of PTS1R or PTS2R. Moreover, these studies also show that the absence of catalase in peroxisomes results in the inactivation of multiple peroxisomal functions that gives a phenotype very similar to Zellweger syndrome in which cells lack peroxisomes.
MATERIALS AND METHODS
Fetal calf serum, trypsin, and tissue culture media were purchased from Life Technologies, and α-cyclodextrin was purchased from Sigma. [1-14C]Lignoceric acid was synthesized from n-tricosanoylbromide and K14CN as described (22). [2,3-3H]Phytanic acid was synthesized from [2,3-3H]dihydrophytol as described (23). Antibodies against catalase and chloramphenicol acetyltransferase (ChAT) were purchased from Biodesign International (Kennebunkport, ME) and 5 Prime–3 Prime, Inc., respectively.
Generation of Human Catalase Expression Constructs.
To construct vectors for expression of full-length catalase (pFS1), normal catalase was subcloned into the KpnI–NotI site of pcDNA3 (Invitrogen). Plasmid pFS2 with SKL at carboxyl terminus of normal catalase was generated by PCR using primers 5′-TCAGAGTTTTAGCAGATTTGCCTTCTC-3′ followed by a stop codon and 5′-CGCACGCTATGGCTGACAG-3′ and cloned in pcDNA3. This spans positions −8 to +1,651 of the normal catalase transcript with additional nucleotides for SKL at the carboxyl terminus. For construction of the vector for expression of normal catalase with PTS2 signal (pFS3), a PTS2 fragment was generated by annealing 5′-AGCATGAATTCGGTCCGATGCACAGACTACAGgtagtgctgggacac-3′ and 5′-ATTCCGCGGACCGCAACGACCTGCCAGgtgtcccagcactac-3′ and a double-stranded fragment was obtained by a polymerizing reaction with the Klenow fragment of DNA polymerase I. The EcoRI/SacII-digested PTS2 fragment was then ligated to SacII/NotI-cleaved catalase from pZEM vector (Promega) and EcoRI/NotI-digested pcDNA3. The pFS4 vector construct of pcDNA3 expressing bacterial ChAT gene with an extra tetrapeptide, KANL, at the carboxyl terminus was generated by PCR using mutagenic primer 5′-CTTCTAGATTACAGATTTGCTTTCGCCCCGCCCTG-3′. The sequence and correct orientation were confirmed by DNA sequencing.
Transfection of the Cells.
Exponentially growing cells of 60–80% confluence were transfected with vector constructs by using Lipofectin as described by the manufacturer (Life Technologies). Briefly, 2 μg of plasmid DNA was diluted in 100 μl of serum-free medium per 35-mm dish. Lipid was prepared by diluting 10 μl of Lipofectin in 100 μl of serum-free medium, and then 0.8 ml of serum-free medium was added to the DNA–lipid complex. Cells were washed with serum-free medium immediately before transfection, overlaid with 1 ml of DNA–lipid complex to each dish, incubated for 6 h, and then replaced with fresh medium supplemented with 15% fetal calf serum. For transient expression experiments, the cells were used 24, 48, and 72 h after transfection. The stable transformants were selected by incubating the cells with G418 antibiotic (200 μg/ml) for 72 h after transfection and continuing the incubation in antibiotic at 50 μg/ml for 2 weeks. The transient and stable transformants were tested in the following biochemical and morphological studies.
Cell Culture and Studies of Intracellular Distribution of Catalase.
Normal human fibroblasts, mutant cells, and transfected cells cultured in DMEM supplemented with 15% fetal calf serum were harvested. Cytosolic and membrane bound catalase were distinguished by permeabilization of plasma membrane with digitonin. Catalase activity was measured as described (24).
Immunofluorescence Studies for Catalase and ChAT.
Immunofluorescence staining was performed essentially as described (21). Briefly, fibroblasts cultured on glass slides were fixed in freshly prepared 4% paraformaldehyde in 0.15 M potassium phosphate (pH 7.4) and permeabilized with 0.1% Triton X-100 in PBS. The cells were incubated for 2 h with rabbit anti-catalase antibodies or rabbit anti-ChAT antibodies and finally with fluorescein isothiocyanate-conjugated anti-rabbit IgG. The punctate appearance of peroxisomes was observed with a confocal microscope and images were recorded.
Enzyme Assays.
For fatty acid β-oxidation, lignoceric acid β- oxidation was measured as described (25). Briefly, the reaction was started by the addition of α-cyclodextrin-solubilized [1-14C]lignoceric acid (12 μM) to fibroblasts (50–100 μg of protein) suspended in Hanks’ balanced salt solution (HBSS) to a final volume of 0.25 ml. The reaction was stopped by the addition of 0.625 ml of 1 M KOH in methanol followed by incubation at 60°C in a shaking water bath for 1 h. The amount of radioactivity in the upper phase of Folch partition represents the rate of β-oxidation of [1-14C]lignoceric acid.
For fatty acid α-oxidation, phytanic acid α-oxidation of α-cyclodextrin-solubilized substrate was measured as described (26). Briefly, [2,3-3H]phytanic acid (3 μM) was added to the fibroblast suspended in 250 μl of HBSS buffer. The reaction was stopped by adding 0.625 ml of 1 M KOH in methanol and processed as for β-oxidation of [1-14C]lignoceric acid.
Dihydroxyacetone phosphate acyltransferase (DHAP-AT) activity was measured as described (27). Briefly, [32P]DHAP (0.1 mM) synthesized from dihydroxyacetone, MgCl2, and [γ-32P]ATP with glycerokinase was added to a reaction mixture containing 8 mM MgCl2, 8 mM NaF, 0.8 mg of fatty acid-poor BSA, 0.1 mM palmitoyl-CoA, 75 mM Tris⋅HCl (pH 7.5), and 5–50 μg of protein sample. The reaction was stopped by adding chloroform/methanol (2:1) and processed as described (27).
Quantitation of Plasmalogens and Fatty Acids.
Plasmalogens were measured as dimethylacetal (DMA) derivatives as described elsewhere (28). The relative amount of plasmalogens is reflected in the ratio of C16:0 DMA and C18:0 DMA to their corresponding fatty acid methyl esters. The measurement of very long chain fatty acids in fibroblasts was carried out by the direct trans-esterification method using gas chromatography (29).
RESULTS
Expression and Targeting of Catalase Chimeras (Catalase-PTS1 and PTS2-Catalase) into Catalase-Negative Peroxisomes of Mutant Cells.
Previous and present studies from our laboratory showed that in these cells both PTS1 and PTS2 signaling pathways were functional but catalase activity remained localized in the cytoplasm instead of peroxisomes (ref. 21, Fig. 1, and Table-1). These observations indicated that the lack of targeting of catalase to peroxisomes may be either due to a mutation in the catalase nucleotide sequence or an alteration in the import machinery for catalase. To test the first hypothesis, we sequenced the transcript for catalase in full in these cells. However, despite our extensive study, no mutation was found in the catalase transcript from these cells (data not shown). A recent study reported that KANL at the carboxyl-terminal end of human catalase is the targeting sequence for import of catalase into peroxisomes (15). To determine further that the abnormality in these cells was in the import of catalase, we constructed a vector (pFS4) to express ChAT fused to KANL (ChAT-KANL) and transfected mutant (Fig. 1A) and normal (Fig. 1B) cells. The subcellular distribution of ChAT-KANL was analyzed by immunocytochemical staining with anti-ChAT antibody. Interestingly, ChAT-KANL was targeted to peroxisomes in control cells, but we were unable to detect punctate fluorescence of peroxisomes in mutant cells, indicating that some factor required for catalase import was missing in the mutant cells.
Figure 1.
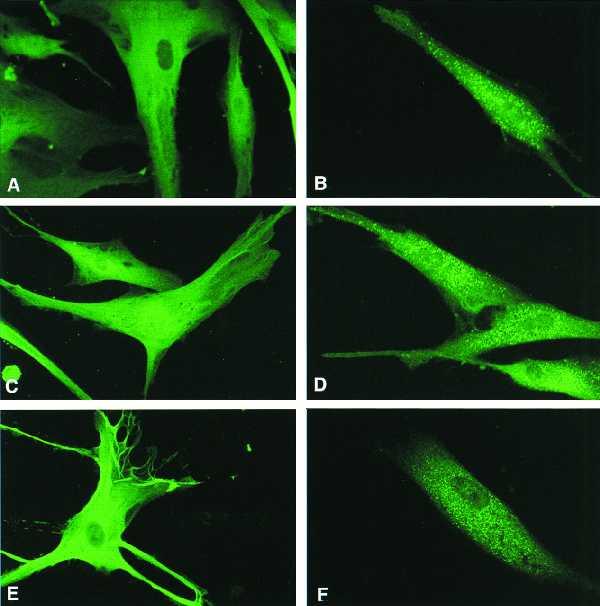
Indirect immunofluorescence (with ChAT antibody) of mutant cells (A) and normal cells (B) transfected with a pFS4 construct expressing ChAT-KANL. Catalase antibody was used to detect intracellular catalase in C–F. (C and E) Mutant cells transfected with only vector and pFS1 expressing normal catalase, respectively. (D and F) Mutant cells transfected with pFS2, which expresses catalase-SKL chimera (catalase with PTS-1 at the carboxyl terminus), and pFS3, which expresses normal catalase with PTS2 signal peptide at the amino terminus, respectively.
To decipher further that the abnormality is in the targeting of catalase into peroxisomes, we constructed vectors that express normal catalase (pFS1), chimeric catalase containing PTS1 (pFS2), or PTS2 (pFS3). The mutant cells, transfected with pFS1, pFS2, and pFS3 expressing normal catalase, catalase-SKL, and PTS2-catalase, respectively, were analyzed by immunocytochemistry for catalase-containing peroxisomes. The staining with anti-catalase antibody detected a punctate pattern of peroxisomes in pFS2- (Fig. 1D) and pFS3- (Fig. 1F)-transfected cells, whereas no catalase-containing peroxisomes were detected in pFS1-transfected cells (Fig. 1E) even though catalase activity in these cells increased by 3-fold (Fig. 2A). These studies and the cytosolic distribution of ChAT-KANL clearly demonstrate that the cytoplasmic localization of catalase is due to a defect in the import machinery of catalase into peroxisomes.
Figure 2.
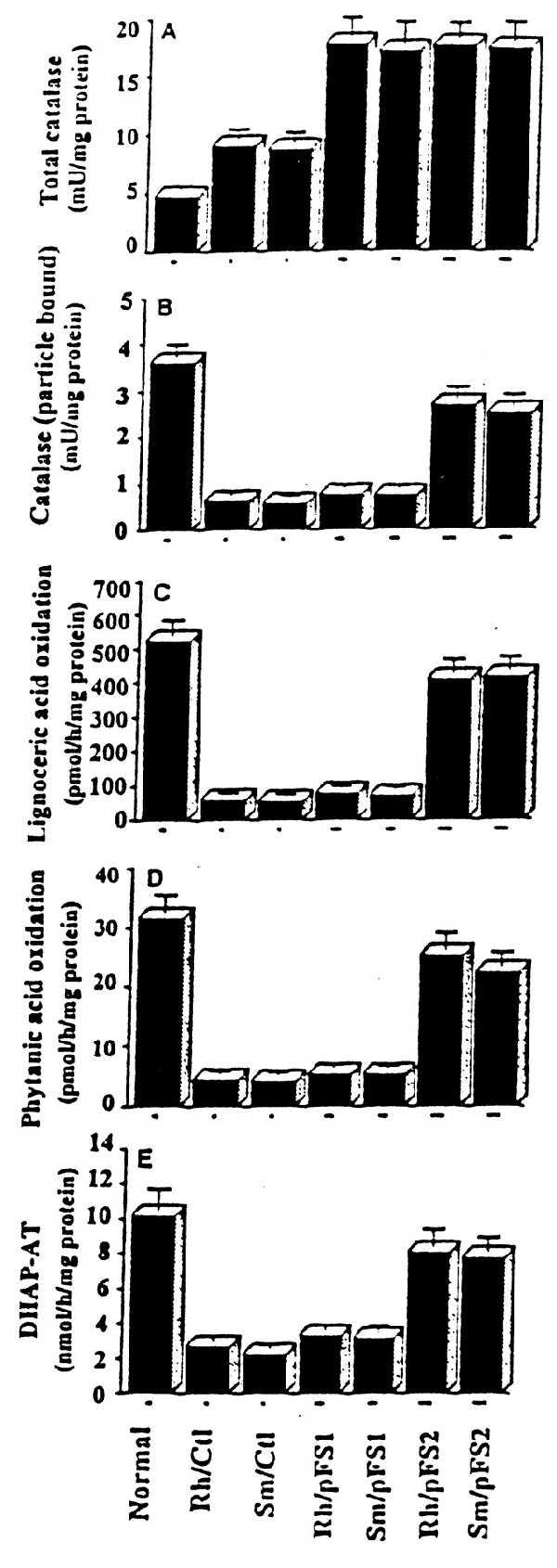
Normalization of peroxisomal functions upon relocalization of catalase into peroxisomes via PTS1 import pathway. Rh and Sm mutant cells were transfected with pFS1 and pFS2 and enzyme activities were measured in cells stably expressing normal catalase (Rh/pFS1 and Sm/pFS1) and catalase-SKL chimera (Rh/pFS2 and Sm/pFS2). Total catalase (A), particulate-bound catalase (B), activities of lignoceric acid oxidation (C), phytanic acid oxidation (D), and DHAP-AT (E) were measured. Rh/Ctl and Sm/Ctl were nontransfected control mutant cells.
Distribution of Catalase and the Catalase-SKL Chimera Expressed in Mutant Cells.
The mutant cells transfected with pFS1 and pFS2 expressing catalase and catalase-SKL chimeric protein, respectively, were tested for the expression of catalase activity. In transient and stable transfection, the particulate-bound catalase activity increased linearly in pFS2-transfected cells (Table 1), but there was no such tendency in cells transfected with pFS1, which expresses normal catalase protein, even though total catalase activity increased (Fig. 2 A and B). Specific activity of particulate-bound catalase in stable transformants expressing the chimeric catalase-SKL was also increased but not in cells transfected with pFS1 (Fig. 2B).
Table 1.
Peroxisomal function of transiently transfected mutant cells
| Cell type | Peroxisomal function | Without transfection | After transfection
|
||
|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | |||
| Rh (pFS2) | Catalase (particulate bound) | 7.85 ± 1.48 | 14.90 ± 3.95 | 18.90 ± 2.54 | 27.46 ± 3.72 |
| Lignoceric acid oxidation | 52.80 ± 3.39 | 72.40 ± 2.12 | 115.30 ± 9.89 | 137.26 ± 3.16 | |
| DHAP-AT | 1.02 ± 0.07 | 1.15 ± 0.06 | 1.58 ± 0.09 | 2.56 ± 0.08 | |
| Sm (pFS2) | Catalase (particulate bound) | 6.60 ± 1.73 | 16.80 ± 8.48 | 21.00 ± 3.67 | 28.82 ± 4.85 |
| Lignoceric acid oxidation | 46.70 ± 4.24 | 72.05 ± 2.47 | 111.55 ± 11.52 | 136.10 ± 4.94 | |
| DHAP-AT | 0.89 ± 0.05 | 1.42 ± 0.06 | 1.52 ± 0.07 | 2.32 ± 0.12 | |
Rh and Sm mutant cells were transfected with pFS2 expressing the catalase-SKL chimera. The cells were also transfected simultaneously with vector alone and pFS1 construct that expresses normal catalase. No improvement was noticed with only vector and pFS1-transfected cells (data not shown). Activities of particulate-bound catalase, lignoceric acid oxidation, and DHAP-AT are expressed in milliunits/mg, pmol per h per mg, and nmol per h per mg of protein, respectively. These values in normal cells were 77.59 ± 5.82, 513 ± 22.6, and 10.03 ± 0.14, respectively.
Catalase-SKL Chimera in Stable Transformants Restored Peroxisomal Activities.
To test the hypothesis that the observed multiple enzyme abnormalities (e.g., β- and α-oxidation of fatty acids and synthesis of plasmalogens) were due to the inactivation of enzymes by excessive H2O2 accumulation, we tested peroxisomal functions in transient and stable transformants.
Interestingly, the peroxisomal activities increased as the catalase targeted to peroxisomes increased in transient transfection with pFS2 (Table 1). Next, we examined the recovery of functions in stable transformants. Oxidation of lignoceric acid is severely affected in Rh and Sm cells (Fig. 2C) and transfection of these cells with pFS2 restored this enzyme activity in both Rh and Sm cells but not in pFS1-transfected cells expressing normal catalase (Fig. 2C). Phytanic acid is α oxidized in peroxisomes (30) and was reduced to 10–15% in these mutant cells compared with normal activity, indicating a partially active peroxisomes (21). The function of phytanic acid oxidation was also restored to 80% of the normal values by introduction of catalase-SKL into peroxisomes but not in cells transfected with pFS1 expressing normal catalase (Fig. 2D).
The early steps of plasmalogen biosynthesis occur in peroxisomes (1, 27). Activity of DHAP-AT, the first enzyme in the plasmalogen biosynthesis pathway, was reduced severely in these cells (Fig. 2E and ref. 21). This activity was also restored to 80% of the normal values in cells stably expressing catalase-SKL chimera but not in cells transfected with pFS1 (Table 1 and Fig. 1E).
Normalization of in Situ Levels of VLC Fatty Acids and Plasmalogens After Targeting of Catalase into Catalase-Negative Peroxisomes.
Next we tested whether the normalization of enzymatic activities would also correct the in situ levels of metabolites (e.g., VLC fatty acids and plasmalogens) after targeting catalase to catalase-negative peroxisomes. As shown in Fig. 3, the steady-state level of VLC fatty acids (C26:0 and C24:0) and their ratios (C26:0/C22:0 and C24:0/C22:0) were normalized in cells transfected with pFS2 but not in cells transfected with pFS1. Steady-state level of plasmalogens was also normalized in mutant cells transfected with pFS2 but not with pFS1 (Fig. 4). Thus, the normalization of peroxisomal enzymatic activities and normalization of steady-state level of metabolites of peroxisomes after targeting catalase to catalase-negative peroxisomes support the conclusion that abnormalities in peroxisomal functions in these mutant cell lines may be due to inactivation of these enzymes by excessive accumulation of H2O2, produced by oxidases present in catalase-negative peroxisomes.
Figure 3.
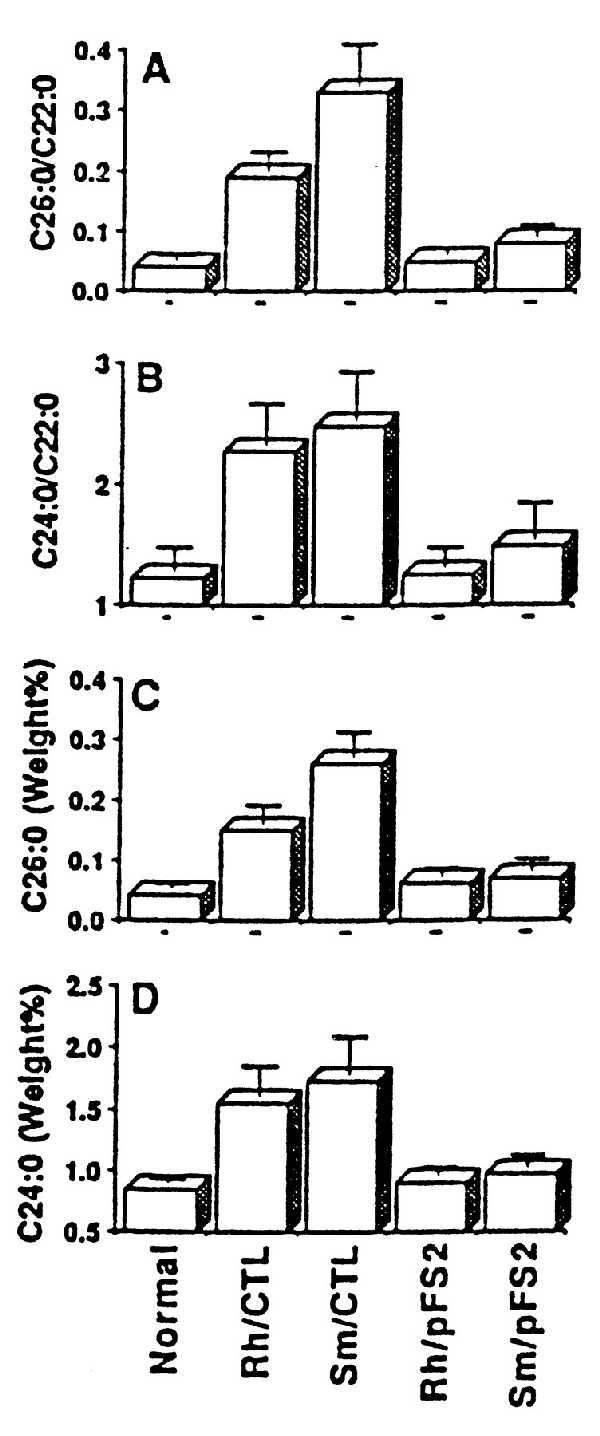
Normalization of fatty acid levels in cells (Rh and Sm) stably expressing catalase-SKL chimera with pFS2. (A and B) Ratio of C26:0/C22:0 and C24:0/C22:0, respectively. (C and D) Weight percent of C26:0 and C24:0, respectively. Rh/Ctl and Sm/Ctl were nontransfected mutant cells.
Figure 4.
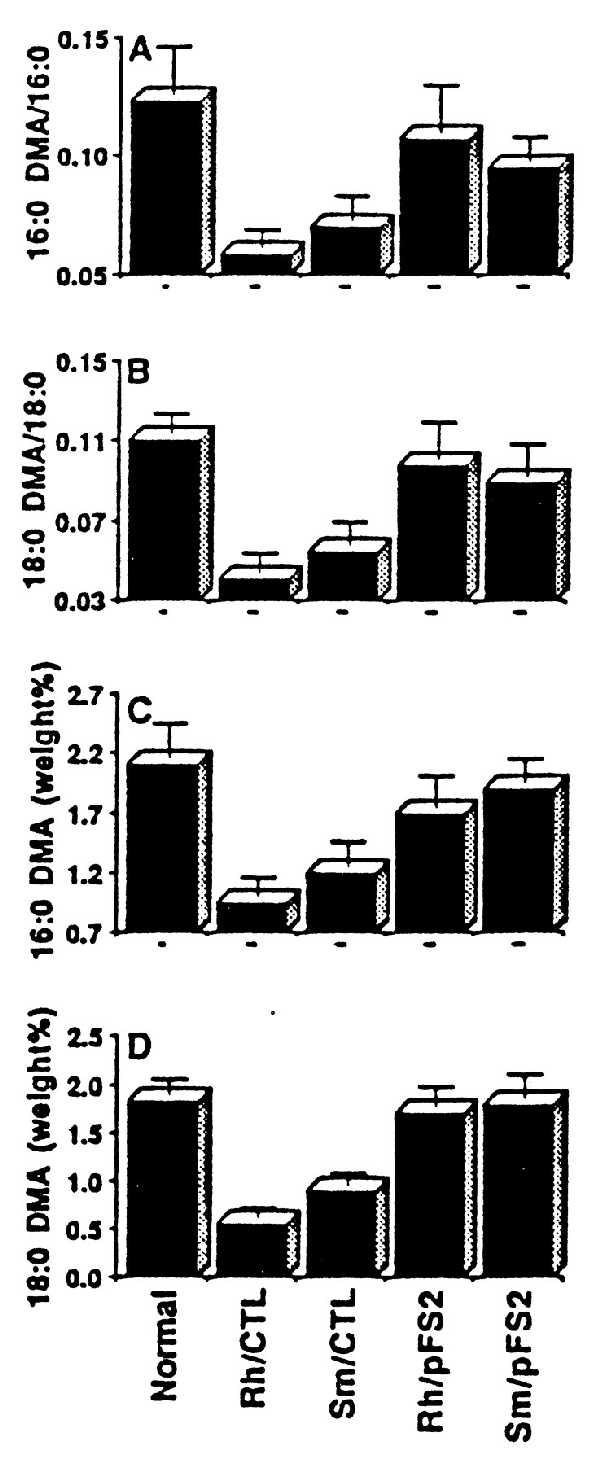
Normalization of plasmalogen levels in cells (Rh and Sm) stably expressing catalase-SKL chimera with pFS2. (A and B) Ratio of C16:0DMA/C16:0 and C18:0DMA/C18:0, respectively. (C and D) DMA weight percent of C16:0 and C18:0, respectively. Rh/Ctl and Sm/Ctl were nontransfected mutant cells.
H2O2 Inhibits Peroxisomal Activities in Normal Fibroblast Cells.
To support our hypothesis, we investigated the effect of endogenously produced H2O2 on peroxisomal functions by inhibition of catalase with aminotriazole (ATZ) (42) in cultured control skin fibroblasts. Normal fibroblast cells were treated with various concentrations of ATZ for 24 h and their peroxisomal activities were measured. All three major peroxisomal activities were reduced severely in parallel with inactivation of catalase (Fig. 5A). In a parallel study, we treated normal cells with a constant concentration of ATZ for various periods. The enzymatic activities were found to be reduced drastically within the first 24 h of treatment (Fig. 5B). To investigate whether the loss of fatty acid oxidation in ATZ-treated cells was due to any impairment of substrate/cofactor transport, we examined the oxidation of palmitic acid, a mitochondrial function, and oxidation of lignoceric acid, a peroxisomal function, in intact and homogenized cells. ATZ inhibited the β-oxidation of lignoceric acid but not of palmitic acid, indicating that the loss of peroxisomal fatty acid oxidation in ATZ-treated cells is not due to alteration in fatty acids/cofactors transport properties of cells. These studies further support the conclusion that excessive H2O2 may be responsible for the observed loss of peroxisomal functions in mutant cells that lack catalase in peroxisomes.
Figure 5.
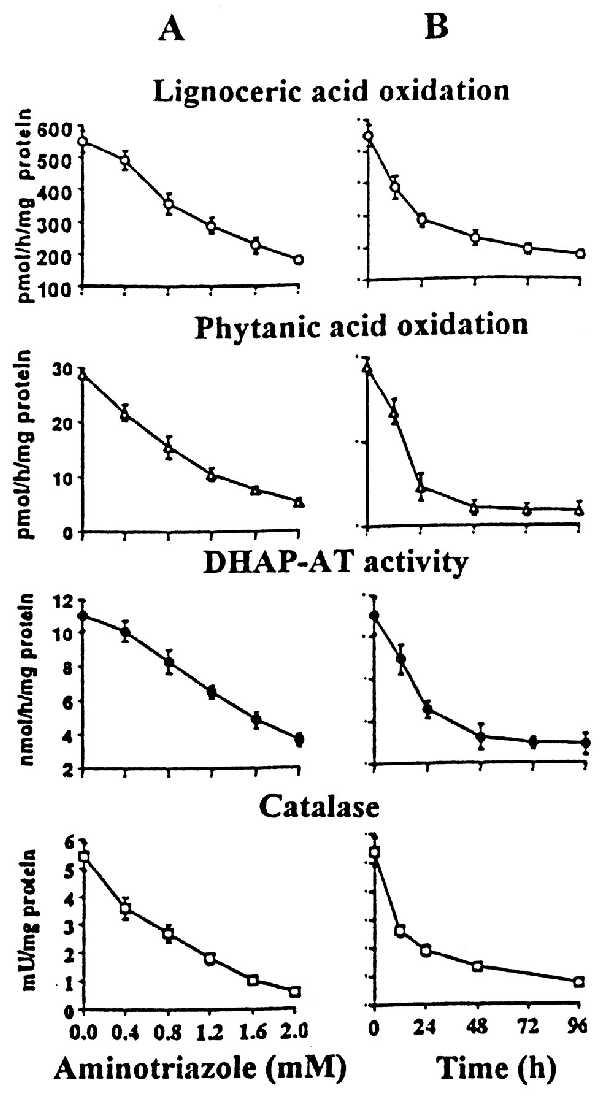
Inhibition of peroxisomal functions by treatment of normal cells with ATZ. Normal cells were incubated with ATZ from 0.4 to 2.0 mM (A) and with 1 mM ATZ for various times (B). Cells were harvested and peroxisomal functions (activities of lignoceric acid oxidation, phytanic acid oxidation, plasmalogen biosynthesis, and catalase) were measured.
DISCUSSION
Fundamental peroxisomal functions, such as α- and β-oxidation of fatty acids and plasmalogen biosynthesis are defective in disorders of peroxisomal biogenesis (1–3), in pseudo-Zellweger syndrome (31), in Zellweger-like syndrome (32), and in pseudo-infantile refsum disease (33). Recently, we reported patients with clinical features very similar to disorders of peroxisomal biogenesis (21, 34). The biochemical features of these patients differ from those of pseudo-Zellweger syndrome (31), Zellweger-like syndrome (32), and pseudo-infantile refsum disease (33) reported previously. Peroxisomes from skin fibroblasts of these mutant cells were morphologically intact (refs. 21 and 34 and Fig. 1). The PTS1 and PTS2 pathways of protein import were also functional (Fig. 1). Thus, the studies reported in this article clearly demonstrate that multiple peroxisomal enzymatic deficiencies in these patients were due to abnormality in targeting of catalase into peroxisomes. These conclusions are based on the following observations. (i) Targeting of catalase via PTS1 pathway into peroxisomes corrected multiple enzymatic defects. (ii) Inhibition of catalase in normal fibroblast reduced enzymatic activity to levels that were similar to those found in the mutant cells.
Peroxisomal proteins are synthesized on free polysomes and posttranslationally targeted to existing peroxisomes via peroxisomal targeting signals. Catalase is a major peroxisomal enzyme. The peroxisomal targeting signal for catalase differs from classical PTS1 consensus sequence. The PTS1 signal has an absolute requirement of a basic residue, lysine, at position −2 from the carboxyl terminus in mammals (4–6). Whereas in catalase asparagine is present at this position (15). The PTS for catalase is KANL and the lysine at position −4 from the carboxyl terminus is required for targeting of catalase to peroxisomes, but when the arginine at position −2 was replaced with different amino acids, there was little or no effect on targeting of catalase into peroxisomes (15). The targeting of PTS1-containing proteins is mediated by a cytoplasmic receptor (PTS1R). This receptor binds PTS1-signal-containing proteins in the cytosol and delivers them to peroxisomal membranes via its interaction with the cytoplasmic SH3-domain-containing peroxisomal membrane protein Pex13p (35–37). In addition to the abnormality in targeting of PTS1 proteins, the targeting of PTS2-signal-containing proteins and catalase into peroxisomes was defective in the Pex13 mutant. These observations suggest that Pex13p may be the common translocation machinery for catalase and other proteins with PTS1 or PTS2 signals into peroxisomes (35–37). Studies described in this article clearly show that PTS1R or PTS2R is not involved in the targeting of catalase into peroxisomes. This conclusion is based on the following observations. (i) The PTS1 and PTS2 pathway of protein import into peroxisomes are normal in these mutant cells; however, catalase with an intact KANL sequence is not translocated into peroxisomes (Figs. 1A and 2A). (ii) ChAT fused to KANL at the carboxyl terminus was targeted to peroxisomes in normal cells but not in mutant cells (Fig. 1 A and B). (iii) Overexpression of normal catalase could not increase the particulate-bound catalase in mutant cells (Fig. 2 A and B), whereas the catalase chimera with the PTS1 signal catalase-SKL was efficiently targeted into peroxisomes (Fig. 1D) with an increase in particulate-bound catalase activity (Fig. 2B).
Peroxisomes were named because of their association with the production and degradation of H2O2 (38, 39). Peroxisomes are estimated to consume between 5% and 20% of the total cellular oxygen in liver (39, 40). More than 90% of the oxygen consumed by mitochondria is converted to H2O and rest to O2−, whereas the oxygen consumed by peroxisomes is quantitatively converted to H2O2 and possibly a small amount is converted to O2− (40). These reactive oxygen species (ROS) are the normal byproducts of cellular metabolism and are accordingly kept in check by cellular defenses provided by antioxidant enzymes (1, 40). These antioxidant enzymes provide protection to the cell against ROS (O2− and H2O2) by detoxifying them at the site (compartments, organelle, and membrane) at which they are generated because ROS with the exception of H2O2 are not expected to diffuse away from the site of generation because of their highly reactive nature and short half-life. Lack of or deficiency in the detoxification of ROS could result in the oxidative injury, which includes oxidative modification of proteins, lipids, and nucleic acids. The possible role of catalase in protection of peroxisomal enzymes is also supported by loss of peroxisomal functions and catalase activity as a result of ischemia–reperfusion injury (41) and inactivation of its function when purified peroxisomes were treated with exogenous H2O2 (42). These observations indicate that excessive H2O2, produced by oxidases, by itself or OH⋅, a reaction product of H2O2 and O2−, inactivate other enzymes in the catalase-negative peroxisomes in these mutant cells. H2O2 is also known to inactivate superoxide dismutase (43, 44). Possibly, this inactivation also may increase O2−, which may react with H2O2 to generate highly reactive OH⋅ in peroxisomes, thus leading to inactivation of peroxisomal functions. The H2O2-induced toxicity in cells with catalase-negative peroxisomes may not induce significant toxicity in the cytoplasm because even if some of the excessive H2O2 diffuses out of peroxisomes, it may be detoxified by GPX and by the mistargeted catalase in the cytoplasm in these mutant cells. The redox in peroxisomes is maintained by antioxidant enzymes that are present in peroxisomes, and an alteration in the enzyme system that produces or degrades ROS can result in change in redox in peroxisomes (40, 44). Thus, we propose that the lack of catalase in peroxisomes results in an alteration in peroxisomal redox that results in inactivation of multiple enzyme activities in peroxisomes, because these enzymatic deficiencies are corrected by targeting of catalase to peroxisomes in these mutant cells.
In summary, experiments described in this study provide evidence for a mechanism to transport catalase into human perxoisomes. The catalase import pathway is independent of PTS1R and PTS2R pathways and impairment in this pathway delocalizes catalase to the cytosol. In the absence of particulate-bound catalase, the accumulated H2O2 suppressed the activities of many peroxisomal matrix proteins leading to a peroxisomal disease with severe neurological malfunctioning. This is the first description of such a disease.
Acknowledgments
We thank Mrs. Swarupa Pahan for technical help and Dr. Avtar K. Singh for reviewing the manuscript and valuable advice. This work was supported by grants from the National Institutes of Health (NS-22576 and NS-34741).
ABBREVIATIONS
- PTS
peroxisomal targeting signal
- PTS1R and PTS2R
PTS1 and PTS2 receptors, respectively
- Chat
chloramphenicol acetyltransferase
- DHAP-AT
dihydroxyacetone phosphate acyltransferase
- DMA
dimethylacetal
- ATZ
aminotriazole
- ROS
reactive oxygen species
References
- 1.Singh I. Mol Cell Biochem. 1997;167:1–29. doi: 10.1023/a:1006883229684. [DOI] [PubMed] [Google Scholar]
- 2.Moser H W, Moser A B. Ann NY Acad Sci. 1996;804:427–441. doi: 10.1111/j.1749-6632.1996.tb18634.x. [DOI] [PubMed] [Google Scholar]
- 3.Brown F R, Voight R, Singh A K, Singh I. Am J Dis Child. 1993;147:617–626. doi: 10.1001/archpedi.1993.02160300023015. [DOI] [PubMed] [Google Scholar]
- 4.Subramani S. Curr Opin Cell Biol. 1996;8:513–518. doi: 10.1016/s0955-0674(96)80029-9. [DOI] [PubMed] [Google Scholar]
- 5.Lazarow P B. J Neuropathol Exp Neurol. 1995;54:720–725. doi: 10.1097/00005072-199509000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Erdmann R, Veenhuis M, Kanau W-H. Trends Cell Biol. 1997;7:400–407. doi: 10.1016/S0962-8924(97)01126-4. [DOI] [PubMed] [Google Scholar]
- 7.Gould S J, Keller G-A, Hosken N, Wilkinson J, Subramani S. J Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller G A, Krisans S, Gould S J, Sommer J M, Wang C C, Schliebs W, Kunau W, Brody S, Subramani S. J Cell Biol. 1991;114:893–904. doi: 10.1083/jcb.114.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramani S. Nat Genet. 1997;15:331–333. doi: 10.1038/ng0497-331. [DOI] [PubMed] [Google Scholar]
- 10.Osumi T, Sukamoto T, Hata S, Yokota S, Miura S, Fuziki Y, Hijikata M, Miyazawa S, Hashimoto T. Biochem Biophys Res Commun. 1991;181:947–954. doi: 10.1016/0006-291x(91)92028-i. [DOI] [PubMed] [Google Scholar]
- 11.Braverman N, Steel G, Obie C, Moser A, Moser H, Gould S J, Valle D. Nature Genetics. 1997;15:369–375. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- 12.Erdmann R. Yeast. 1994;10:935–944. doi: 10.1002/yea.320100708. [DOI] [PubMed] [Google Scholar]
- 13.Glover J R, Andrews D W, Rachubinski A. Proc Natl Aca Sci USA. 1994;91:10541–10545. doi: 10.1073/pnas.91.22.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazawa S, Osumi T, Hashimoto T, Ohro K, Miura S, Fuziki Y. Mol Cell Biol. 1989;9:83–91. doi: 10.1128/mcb.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purdue P E, Lazarow P B. J Cell Biol. 1996;134:849–862. doi: 10.1083/jcb.134.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dodt G, Braverman N, Wong C, Moser A, Moser H W, Watkins P, Valle D, Gould S J. Nat Genet. 1995;9:115–125. doi: 10.1038/ng0295-115. [DOI] [PubMed] [Google Scholar]
- 17.McCollum D, Monosov E, Subramani S. J Cell Biol. 1993;121:761–774. doi: 10.1083/jcb.121.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Leij I, Franse M M, Elgersma Y, Distel B, Tabak H F. Proc Natl Acad Sci USA. 1993;90:11782–11786. doi: 10.1073/pnas.90.24.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marzioch M, Erdmann R, Veenhuis M, Kunau W H. EMBO J. 1994;13:4908–4918. doi: 10.1002/j.1460-2075.1994.tb06818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motley A, Lumb M J, Oatey P B, Jennings P R, De Zoysa P A, Wanders R J A, Tabak H F, Danpure C J. J Cell Biol. 1995;131:95–109. doi: 10.1083/jcb.131.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh I, Voigt R G, Sheikh F G, Kremser K, Brown F R., III Biochem Mol Med. 1997;61:198–207. doi: 10.1006/bmme.1997.2593. [DOI] [PubMed] [Google Scholar]
- 22.Hoshi M, Kishimoto Y. J Biol Chem. 1973;248:4123–4130. [PubMed] [Google Scholar]
- 23.Zenger-Hain J, Craft D A, Rizzo W B. In: New Development of Fatty Acid Oxidation. Coates P M, Tanaka K, editors. New York: Wiley–Liss; 1992. pp. 399–407. [Google Scholar]
- 24.Wanders R J, Kos M, Roest B, Meijer A J, Schrakamp G, Heymans H S, Tegelaers W H, van den Bosch H, Schutgens R B, Tager J M. Biochem Biophys Res Commun. 1984;123:1054–1061. doi: 10.1016/s0006-291x(84)80240-5. [DOI] [PubMed] [Google Scholar]
- 25.Lazo O, Contreras M, Hashmi M, Stanley W, Irazu C, Singh I. Proc Natl Acad Sci USA. 1988;85:7647–7651. doi: 10.1073/pnas.85.20.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh I, Pahan K, Dhaunsi G S, Lazo O, Ozand P. J Biol Chem. 1993;268:9972–9979. [PubMed] [Google Scholar]
- 27.Junes K M, Hajra A K. Clin Chem. 1994;40:946–947. [PubMed] [Google Scholar]
- 28.Dacremont, G. & Vincent, G. (1995) J. Inher. Metab. Dis. 18, Suppl. 1, 84–89. [DOI] [PubMed]
- 29.Dacremont, G., Cocquyt, G. & Vincent, G. (1995) J. Inherited Metab. Dis. 18, Suppl. 1, 76–83. [DOI] [PubMed]
- 30.Singh I, Pahan K, Singh A K, Barbosa E. J Lipid Res. 1993;34:1755–1764. [PubMed] [Google Scholar]
- 31.Clayton P T, Lake B D, Hjelm M, Stephenson J, B, Besley G, T, Wanders R J, Schram A, W, Tager J M, Schutgens R B, Lawson A M. J Inherited Metab Dis. 1988;11:165–168. doi: 10.1007/BF01804226. [DOI] [PubMed] [Google Scholar]
- 32.Mandel H, Berant M, Aizin A, Gershony R, Hemmli S, Schutgens R B H, Wanders R J A. J Inherited Metab Dis. 1992;15:381–384. doi: 10.1007/BF02435982. [DOI] [PubMed] [Google Scholar]
- 33.Aubourg P, Kremser K, Roland M, O, Rocchiccioli F, Singh I. Pediatr Res. 1993;34:270–276. doi: 10.1203/00006450-199309000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Burdette D E, Kremser K, Fink J K, Pahan K, Stanley W, Singh I. Neurology. 1996;46:829–831. doi: 10.1212/wnl.46.3.829. [DOI] [PubMed] [Google Scholar]
- 35.Erdmann R, Blobel G. J Cell Biol. 1996;135:111–121. doi: 10.1083/jcb.135.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gould S J, Kalish J E, Morrell J C, Bjorkman J, Urquhart A J, Crane D I. J Cell Biol. 1996;135:85–95. doi: 10.1083/jcb.135.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elgersma Y, Kwast L, Klein A, Voorn-Brouwer T, van den Berg M, Metzig B, America T, Tabak H F, Distel B. J Cell Biol. 1996;135:97–109. doi: 10.1083/jcb.135.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhodin J. Ph.D. thesis. Godvil, Stockholm: Aktiebolaget; 1954. [Google Scholar]
- 39.de Duve C, Baudhuin P. Physiol Rev. 1966;46:323–357. doi: 10.1152/physrev.1966.46.2.323. [DOI] [PubMed] [Google Scholar]
- 40.Singh I. Ann NY Acad Sci. 1996;804:612–627. doi: 10.1111/j.1749-6632.1996.tb18648.x. [DOI] [PubMed] [Google Scholar]
- 41.Singh A K, Dhaunsi G S, Asayama K, Orak J K, Rajagopalan P R, Singh I. Arch Biochem Biophys. 1994;315:331–338. doi: 10.1006/abbi.1994.1508. [DOI] [PubMed] [Google Scholar]
- 42.Gulati S, Ainol L, Orak J, Singh A K, Singh I. Biochim Biophys Acta. 1993;1182:291–298. doi: 10.1016/0925-4439(93)90071-8. [DOI] [PubMed] [Google Scholar]
- 43.Dhaunsi G S, Gulati S, Singh A K, Orak J K, Asayama K, Singh I. J Biol Chem. 1992;267:6870–6873. [PubMed] [Google Scholar]
- 44.Dhaunsi G, S, Singh I, Orak J, K, Singh A K. Carcinogenesis. 1994;15:1923–1930. doi: 10.1093/carcin/15.9.1923. [DOI] [PubMed] [Google Scholar]