

Abstract
The extracellular matrix protein “elastin” is the major component of elastic fibers present in the arterial wall. Physiological degradation of elastic fibers, enhanced in vascular pathologies, leads to the presence of circulating elastin peptides (EP). EP have been demonstrated to influence cell migration and proliferation. EP also induce, at circulating pathophysiological concentrations (and not below), an endothelium- and NO- dependent vasorelaxation mediated by the 67-kDa subunit of the elastin-laminin receptor. Here, by using the techniques of patch-clamp, spectrofluorimetry and confocal microscopy, we demonstrate that circulating concentrations of EP activate low specificity calcium channels on human umbilical venous endothelial cells, resulting in increase in cytoplasmic and nuclear free calcium concentrations. This action is independent of phosphoinositide metabolism. Furthermore, these effects are inhibited by lactose, an antagonist of the elastin-laminin receptor, and by cytochalasin D, an actin microfilament depolymerizer. These observations suggest that EP-induced signal transduction is mediated by the elastin-laminin receptor via coupling of cytoskeletal actin microfilaments to membrane channels and to the nucleus. Because vascular remodeling and carcinogenesis are accompanied by extracellular matrix modifications involving elastin, the processes here described could play a role in the elastin-laminin receptor-mediated cellular migration, differentiation, proliferation, as in atherogenesis, and metastasis formation.
Elastic fibers in arteries, pulmonary alveolar septa, certain ligaments and skin are normally subjected to stretching. In vascular walls, elastic fibers organize into concentric sheets that endow the arteries with resiliency. In physiological conditions, elastin is synthesized only during the late stages of gestation and early infancy. Although elastin is a stable protein (1), a slow and regular elastin degradation mediated by specialized proteases—the elastases—occurs, contributing to the age-dependent increase in vessel stiffness. This process leads to the presence of elastin peptides (EP) in the circulation (10−6–10−2 mg⋅ml−1) (2, 3), increased in some vascular pathologies, as for instance arteriosclerosis (3, 4). EP influence cell migration (5) and proliferation (6) and, in adult rats, EP induce, at circulating concentrations (and not below), an endothelium-dependent vasodilation mediated by NO (7). EP act via binding to the 67-kDa subunit of the high affinity elastin-laminin receptor, present on the cell membranes of the vascular endothelial cells and on numerous other cell types, including arterial medial smooth muscle cells (8–11). Moreover, activation of the 67-kDa subunit of the elastin-laminin receptor also produces a variety of biological reactions, as for instance modifying cell migration (12), differentiation (13), proliferation (6), and enhancing metastatic potential of transformed cells (14, 15). The presence and density of the 67-kDa subunit on transformed cell membranes was claimed to be a marker of the cell metastatic potential (14).
Several vascular diseases are accompanied by extracellular matrix degradation including elastin (16–18). Therefore, we investigated the effect of EP, at circulating concentrations, on intracellular calcium signaling in endothelial cells. Our results show that binding of EP to the 67-kDa subunit of the elastin-laminin receptor induces the activation of calcium membrane channels resulting in an increase in both cytoplasmic- and nuclear-free calcium concentration ([Ca2+]), independent of phosphoinositide metabolism. Moreover, this action is mediated by the involvement of cytoskeletal actin microfilaments.
MATERIALS AND METHODS
Human Umbilical Venous Endothelial Cells (HUVEC).
HUVEC were obtained according to Jaffe (19, 20). Under a sterile hood the umbilical vein was cannulated and perfused for washing with a physiological buffer solution (Hepes-buffered saline) containing mM: 140 mM NaCl, 4 mM KCl, 7.6 mM d-glucose, 15 mM Hepes, plus 0.1 mg⋅ml−1 streptomycin, 100 units⋅ml−1 penicillin, and 0.1% phenol red (pH 7.4). The vein was then filled with Hepes-buffered saline containing 0.1% collagenase 1A and placed in a Hepes-buffered saline bath at 37°C for 10 min. Collagenase action was stopped and detached cells were obtained by perfusing the vein with culture medium followed by Hepes-buffered saline. The culture medium was medium 199 containing 22% human serum, 20 mM Hepes, 10 mM NaHCO3, 2 mM l-glutamine, 0.075 mg⋅ml−1 streptomycin, 75 units⋅ml−1 penicillin and 0.1% phenol red. The cell suspension was centrifuged at 200 × g for 10 min, the pellet was resuspended in culture medium (≈105 cells⋅ml−1) and placed in 0.25 mg⋅ml-1 fibronectin-coated dishes (21) at 37°C, 5% CO2, humid atmosphere. The culture medium was replaced after 2 hr and the cells were grown in the same conditions. The cells were used from first to fourth passage after primary culture.
Patch-Clamp.
Single-channel currents were recorded at room temperature (22°C) from cell-attached patches on HUVEC membranes (holding potential = 20 mV) and analyzed by using standard procedures and instrumentation (20). Before recording, the cells were washed twice then bathed in a physiological salt solution (PSS) containing 118 mM NaCl, 5.6 mM KCl, 2.4 mM CaCl2, 1.2 mM MgCl2, 10 mM Hepes, and 11 mM D-glucose (pH 7.4). The patch pipette was filled with 90 mM Ba(CH3COO)2 and 10 mM Hepes (pH 7.4). Because Ba2+ was shown to be more permeant than Ca2+ through most of the Ca2+ channels (22), Ba2+ was chosen as the current carrier instead of Ca2+ to improve signal resolution. HUVEC membrane resting potential (Vm) was measured to −57 ± 4 mV (n = 35) for nondividing cells.
Suspended HUVEC Intracellular Free Calcium Concentration ([Ca2+]i).
Sub-confluent adhering HUVEC were washed twice with PSS and trypsinized. Trypsinization was stopped by addition of PSS containing 50% human serum. Cells were then centrifuged 5 min at 200 × g and resuspended in 6 ml PSS. Forty microliters of a 0.5 mM INDO1/AM solution in dimethyl sulfoxide were added to a 6ml cell suspension (final concentrations: 3.3 μM INDO1/AM, <1% dimethyl sulfoxide) before a 30-min incubation at 22°C in the dark. The cells were then pelleted by a 5-min 200 × g centrifugation and the cells were resuspended in 3 ml PSS (≈106 cells⋅ml−1). After a 10-min equilibration period (gentle stirring, 37°C), cells were photoexcited at 355 nm and the 90° fluorescence emission was detected at 405 nm and 475 nm in a dual-emission quartz cuvette-based spectrofluorimeter (Deltascan Model 4000, Photon Technology International, Princeton, NJ). The measurements were ended by adding 0.04% digitonin followed by 45 mM EGTA (maximal and minimal fluorescence ratio, respectively). Autofluorescence was subtracted from the measured values. Kd of INDO 1 for calcium was taken at 250 nM (23) and fluorescence ratios to [Ca2+]i conversions were assessed as described (23).
Adhering HUVEC Cytoplasmic and Nuclear [Ca2+].
Recordings on adherent HUVEC were performed by using a Zeiss LSM 410 confocal laser scanning microscope (Zeiss). Excitation: 488 nm, emission collection: 540 nm. The optical slices passed through the nuclei. The HUVEC were cultured on glass slides coated with 0.25 mg⋅ml−1 fibronectin. The cells (first to second passages) were washed twice with PSS, then 0.75 ml PSS was added followed by addition of 0.25 ml of a 2% BSA and 0.08% Pluronic F127 solution in PSS containing 5 μl of a 0.5 mM FLUO3/AM solution in dimethyl sulfoxide (final concentrations: 2.5 μM FLUO3/AM, <1% dimethyl sulfoxide). After a 20-min incubation in the dark, the cells were washed twice and bathed in 1.5 ml PSS 10 min before and during the measurements (22°C). F/Fo is the ratio of current fluorescence to initial fluorescence and is assumed to represent nuclear and cytoplasmic [Ca2+].
EPs and Chemicals.
Chemicals were obtained from Sigma. Elastin peptides were obtained by hydrolysis of highly purified bovine ligamentum nuchae elastin in 1 M KOH in 80% ethanol/20% H2O (vol/vol) (24). Average molecular mass of these peptides was 75 kDa.
Computer Analysis.
Kinetic analysis of the patch-clamp recordings were performed by using the biopatch software (Biologic, Claix, France). For suspended HUVEC [Ca2+]i measurements, fluorescence ratio to calcium level conversions were performed by using software provided by the spectrofluorimeter manufacturer. For fluorescence measurements performed by confocal microscopy, the time-series analysis was done with software written by the authors in C language. Briefly, the contours of the cells were drawn interactively on the computer screen using a view of the microscopic field in transmitted light. The fluorescence intensities were then measured for each cell throughout the time-series.
RESULTS
Single-channel currents were recorded from cell-attached patches on HUVEC. EP induced cyclic inward currents (Fig. 1 A and B), through activation of 16 pS channels (Fig. 1C). These results suggest that EP activate calcium channels because: (i) Ba2+ was the only permeant charge carrier in the patch pipette, (ii) Ba2+ had an inwardly directed driving force at a holding potential of 20 mV, (iii) single channel inward current decreased when membrane potential increased and, (iv) similar 16 pS calcium channels were previously described on endothelial cells (25). However, because the reversal membrane potential was different from that of specific calcium channels, it could be hypothesized that the channels observed here are of low specificity. These channels exhibited cyclic EP-induced activity bursts, including long-lasting opening periods, separated by low-activity periods. Statistical analysis of patch-clamp recordings indicated that the unitary current has an amplitude of ≈1.2pA (Fig. 2). Kinetic analysis of the recordings indicated that, unlike high activity periods (Fig. 2 B, D, and F), low activity periods (Fig. 2 A, C, and E) do not exhibit multiunitary open states. Moreover, low activity periods exhibited only short decay constants (open and closed times) whereas high activity periods exhibited both short and long decay constants (open and closed times). These results suggest that EP enhance inward calcium currents both by increasing the number of simultaneously activated channels and by lengthening the opening periods.
Figure 1.
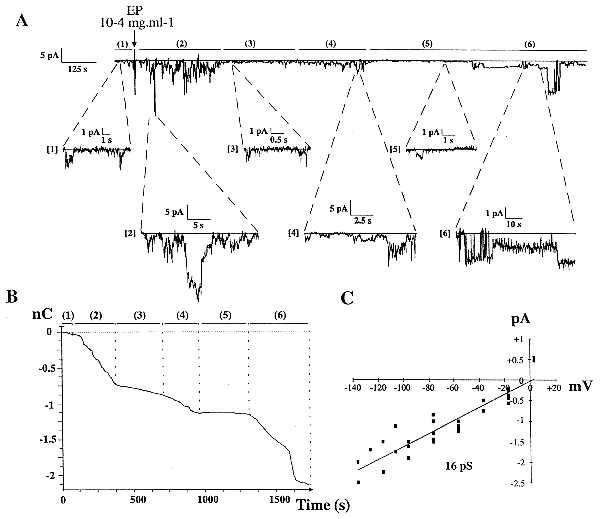
EP-induced single channel inward currents recorded from cell-attached membrane patch on HUVEC, seen as downward deflections from baseline. (A) EP-induced inward currents (filtering 1 kHz, sampling 3 kHz). This recording is typical of 20 similar experiments performed on 20 different cells from 20 different culture dishes (Upper). EP (final concentration 10−4 mg⋅ml−1) induced cyclic bursts of inward current [periods (2), (4), and (6)] separated by low activity periods [periods (3) and (5)]. Below, magnified parts of the recorded trace are shown: 1 min before, then 1, 6, 14, 19, and 25 min after EP addition. (B) Integration of inward current recording presented in Fig. 1A. High activity periods [periods (2), (4), and (6)] exhibit charge inward 3- to 30-fold higher than those measured in low activity periods [periods (1), (3), and (5)]. (C) Current-voltage relationship, obtained by linear regression from pooled data of five patches, exhibiting 16 ± 1 pS channels with reversal membrane potential in the range of 1.5 mV.
Figure 2.
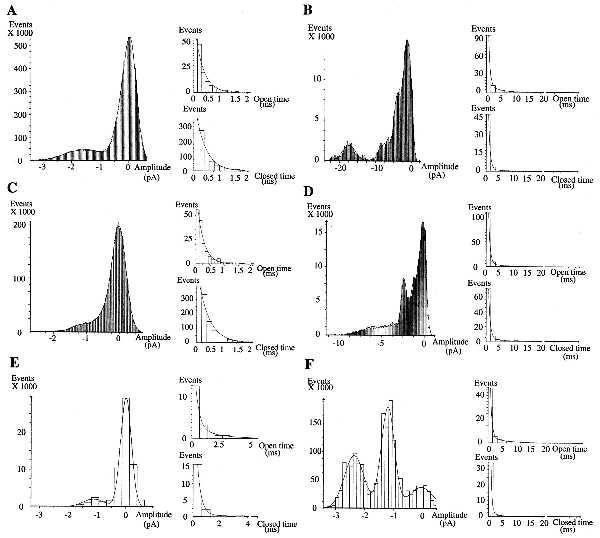
Amplitude and kinetic analyses of the patch-clamp recordings. A–F correspond respectively to the extracts [1] to [6] presented in Fig. 1A downward. Probability density histograms (Left) exhibited unitary current amplitude of ≈1.2 pA. Kinetic analysis of low activity periods (A, C, and E) exhibit only short lasting decay constants (Right), except for (E), and no presence of multiunitary open states (Left). Unitary open state decay constants (Right, Upper) are 0.31 ms (A), 0.24 ms (C), 0.32 ms (E, short) and 1.9 ms (E, long). Corresponding closed state decay constants (Right, Lower) are: 0.37 ms, 0.40 ms, 0.27 ms (short), and 3.4 ms (long), respectively. By contrast, high activity periods (B, D, and F) exhibit the presence of numerous multiunitary open states (Left) and of both short and long decay constants (Right). Short and long open state decay constants are (Upper): 0.53 ms/4 ms (B), 0.57 ms/5.4 ms (D), and 0.58 ms/6 ms (F), respectively, whereas corresponding closed state decay constants (Lower) are: 0.58 ms/7 ms (B), 0.60 ms/4.7 ms (D), and 0.44 ms/3.6 ms (F).
To study the intracellular effects of membrane calcium channels, we measured [Ca2+]i in suspended HUVEC incubated with INDO1/AM, this fluorescent dye being poorly present in the nucleus and, unlike FURA2, diffusely distributed in the endothelial cell cytosol (26). As compared with the control (Fig. 3A), EP induced a long-lasting several-fold increase in [Ca2+]i whose amplitude (average value: ≈2-fold) depended on the experiment (Fig. 3B). Evidence for suspended HUVEC functionality is provided by their standard response to bradykinin (27) (Fig. 3C). Moreover, after EP stimulation, addition of nickel, a calcium channel blocker (28), induced a return of [Ca2+]i to its basal value (Fig. 3D). To confirm the implication of membrane calcium channels in this EP-induced [Ca2+]i increase, we performed measurements on cells treated with thapsigargin, an endoplasmic reticulum calcium ATPase blocker (29) that induces depletion of the endoplasmic reticulum calcium and blocks its restoration. Thapsigargin had no effect on EP-induced [Ca2+]i increase in the presence of 2.4 mM extracellular calcium (Fig. 3E), whereas a decrease in extracellular calcium concentration ([Ca2+]o = 10−7 M) strongly inhibited EP effect (Fig. 3F).
Figure 3.
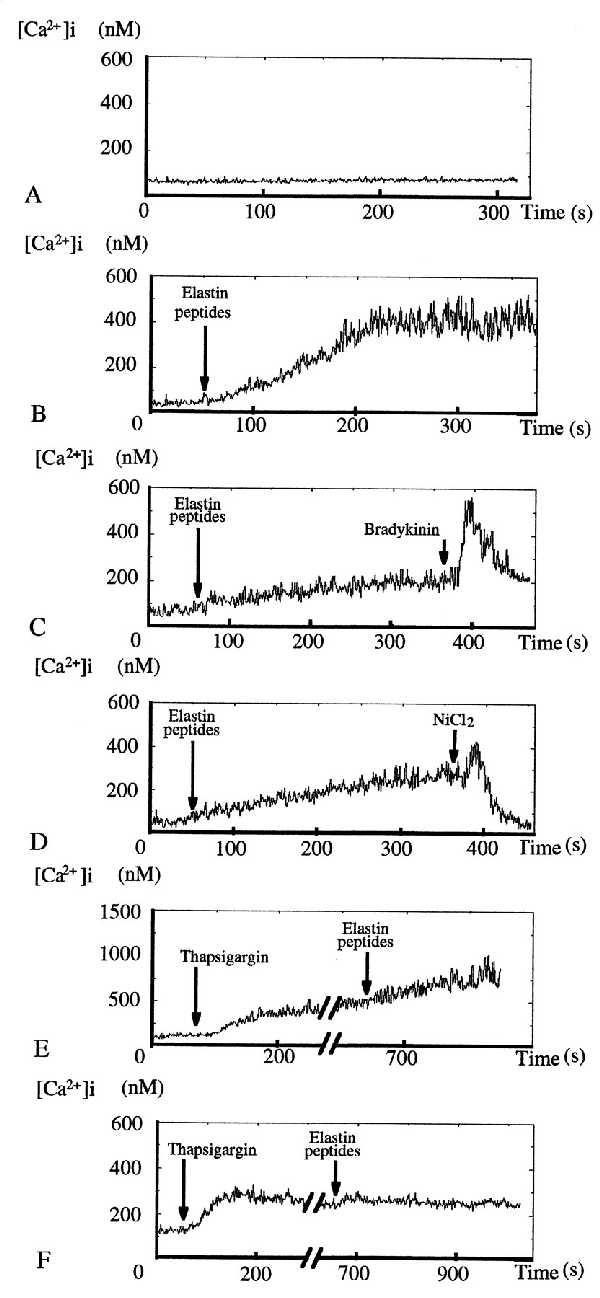
Action of EP on suspended HUVEC [Ca2+]i. (A) Time-control tracing (n = 2). (B) Addition of 10−2 mg⋅ml−1 EP induced a slow but strong increase in [Ca2+]i, starting from a steady low [Ca2+]i before reaching a new high level steady state after 200 s (n = 10). [Ca2+]i increase amplitudes varied with the experiment and were from 1.5-fold up to 7-fold. (C) Addition of 1 μM bradykinin induced an immediate strong but short-lasting [Ca2+]i rise (n = 5). (D) Addition of 1 mM NiCl2 totally abolished EP-induced [Ca2+]i increase (n = 4). (E) Ten minute incubation in PSS ([Ca2+]o = 2.4 mM) with 1 μM thapsigargin had no effect on EP-induced [Ca2+]i increase pattern (n = 6). (F) EP became unable to induce a [Ca2+]i increase when the same experiments (n = 5) were performed in low calcium PSS ([Ca2+]o ≈ 10−7 M).
FLUO3 fluorescence is slightly different in the HUVEC nucleus (homogeneous spatial distribution) as compared with the cytosol (inhomogeneous spatial distribution) (30), requiring the use of fluorescence ratios for intercompartment comparisons. Confocal microscopy of adhering HUVEC loaded with FLUO3 showed that EP strongly enhance both cytoplasmic and nuclear [Ca2+] (Fig. 4A). Nevertheless, the EP-induced [Ca2+] increase was higher in the cytoplasm than in the nucleus (Fig. 4 B and C). Moreover, there was no synchronous response of the different cells in the same field of observation and some cells did not show any response even several minutes after EP stimulation (Fig. 4 B and C). Finally, the confocal microscopy experiments showed that the EP-induced increase in cytoplasmic and nuclear [Ca2+] was abolished by lactose, an antagonist of the elastin-laminin receptor (8), as well as by cytochalasin D, an inhibitor of actin microfilament polymerization and depolymerization (31) (Fig. 4 D and E). The same inhibitory effects of lactose and cytochalasin D were obtained on suspended HUVEC (data not shown).
Figure 4.
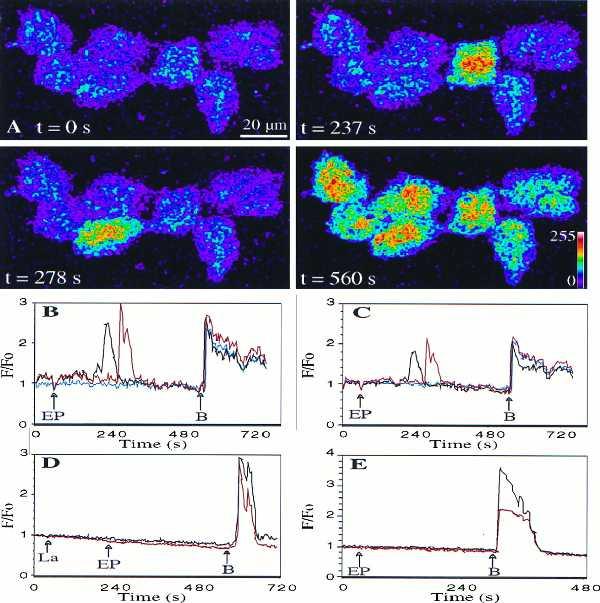
(A) Images of adherent HUVEC response to 10−3 mg⋅ml−1 EP (scale: fluorescence intensity code). From a basal level (t = 0 s), fluorescence increased transiently, 180 s after EP addition, in a first cell (t = 237 s), then in a second cell (t = 278 s), before all the cells respond simultaneously to 1 μM bradykinin (t = 560 s) [see also (B) and (C)]. Fluorescence increase of the first EP-responding cells started between 20 s and 180 s after stimulation (four experiments, n = 20). In this field, EP-induced [Ca2+] transient increases were higher in the cytoplasm (B) than in the nucleus (C) of two selected cells (black and red), whereas a third cell (blue) did not respond. Preincubation for 2–3 min with 10−4 M lactose (La) (four experiments, 20 cells studied) (D) or for 45 min with 1 μM cytochalasin D (four experiments, 20 cells studied) (E) abolished 10−3 mg⋅ml−1 EP-induced but not 1 μM bradykinin-induced nuclear (red) and cytoplasmic (black) [Ca2+] increases (response of one cell presented in each case).
DISCUSSION
EP were shown to influence many properties of both normal and transformed cells. To investigate a possible pathway for intracellular signaling in the vascular system, and because EP have been shown to be active on the vascular tone at circulating concentrations (and not below) (7), we studied the effects of circulating concentrations of EP on [Ca2+]i of human endothelial cells. Using patch-clamp technology, spectrofluorimetry and confocal microscopy, we found that EP increased both cytoplasmic and nuclear [Ca2+] by activating calcium channels in the cell membrane.
EP induced cyclic inward current, through activation of 16 pS low specificity channels, which resulted in a 2-fold increase in [Ca2+]i, resembling the nontransient [Ca2+]i rises induced on neurons by N-cadherin or by laminin (32), or on endothelial cells by caffeine (33). This [Ca2+]i increase is independent of intracellular calcium stores and, following EP-addition to HUVEC, no increase in intracellular IP3 could be detected by using a standard method (34) (data not shown). These results suggest that EP increase [Ca2+]i in HUVEC by enhancing calcium influx via calcium channel opening rather than via mobilization of intracellular calcium stores, unlike their effect on leukocytes where EP signal transduction was shown to involve both calcium influx and IP3 pathways (34). Confocal microscopy of adhering HUVEC loaded with FLUO3 showed that EP strongly enhanced both cytoplasmic and nuclear [Ca2+], although the delay and amplitude of the response varied from cell to cell in the same field.
The elastin-laminin receptor, present on endothelial cells (9), is linked to cytoskeletal actin microfilaments (9) and contains a lectin domain that, when occupied by lactose, inhibits elastin binding (8). Because, as for suspended HUVEC [Ca2+]i, lactose or cytochalasin D specifically abolished EP-induced increases in cytoplasmic and nuclear [Ca2+] of adhering HUVEC, it is suggested that EP act via the elastin-laminin receptor, and that actin microfilaments are involved in EP signal transduction.
Two general conclusions can be drawn from these results. First, the elastin-laminin receptor-mediated transduction of EP signals involves cytoskeletal actin microfilaments, resulting in membrane calcium channel activation and [Ca2+]i increase. This EP-induced [Ca2+]i rise could play a role in the previously observed NO-mediated vasodilatory action of EP (7), because a similar amplitude increase in [Ca2+]i in endothelial cells leads to activation of NO synthase and NO production (35, 36). Moreover, in certain pathophysiological situations, such as atherosclerosis, circulating EP concentration is chronically elevated (3). The EP-triggered intracellular mechanisms could induce a sustained overproduction of NO by the endothelial cells, leading to a short term vasorelaxation counterbalancing the hypertensive and vasoconstrictive effects of atherosclerosis, but also possibly contributing to a long term enhancement of the previously demonstrated NO/superoxide free radical/peroxinitrite-mediated damage to the vessel wall (37, 38). Secondly, elastin-laminin receptor activation by EP induced an actin microfilament-mediated rise in both cytoplasmic and nuclear [Ca2+]. Similar cytoskeletal as well as cytosolic and nuclear [Ca2+] changes have been linked to modifications of gene expression in other systems (39, 40). It is unlikely that the elastin-peptide-induced [Ca2+] increase is due to an indirect NO effect on membrane channels or on other intracellular pathways because NO tends to lower, not to enhance, the induced [Ca2+]i increases in endothelial cells (41).
There is increasing evidence that extracellular matrix components regulate gene expression through interactions with transmembrane receptors. The signaling pathways from receptor to responsive elements in the nucleus are progressively elucidated. Laminin is able to regulate gene expression via a transduction pathway including sequentially: membrane receptors, cytoskeleton, and nuclear matrix (42). Our finding of an EP-induced increase in nuclear and cytoplasmic [Ca2+] mediated by the elastin-laminin receptor suggests that circulating degradation products of extracellular matrix could play a role in altering gene expression or influencing cell motility and/or differentiation.
Acknowledgments
We thank F. Cand, C. Lanthelme, and Drs. A. Queyroy, G. Lallement, and G. Blanchet for their help.
ABBREVIATIONS
- EP
elastin peptides
- [Ca2+]
free calcium concentration
- [Ca2+]i
intracellular free calcium concentration
- HUVEC
human umbilical venous endothelial cells
- PSS
physiological salt solution
References
- 1.Robert, L. & Chadwick, D. J., eds. (1995) Ciba Found. Symp. 192.
- 2.Kucich U, Christner P, Lippmann M, Fein A, Goldberg A, Kimbel P, Weinbaum G, Rosenbloom J. Am Rev Respir Dis. 1983;127:S28–S30. doi: 10.1164/arrd.1983.127.2P2.S28. [DOI] [PubMed] [Google Scholar]
- 3.Fülöp T, Jr, Wei S M, Robert L, Jacob M P. Clin Physiol Biochem. 1990;8:273–282. [PubMed] [Google Scholar]
- 4.Robert L, Hornebeck W, editors. Elastin and elastases. Boca Raton, FL: CRC; 1989. [Google Scholar]
- 5.Senior R M, Griffin G L, Mecham R P. J Clin Invest. 1980;66:859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghuysen-Itard A F, Robert L, Jacob M P. C R Acad Sci Ser III. 1992;315:473–478. [PubMed] [Google Scholar]
- 7.Faury G, Ristori M T, Verdetti J, Jacob M P, Robert L. J Vasc Res. 1995;32:112–119. doi: 10.1159/000159084. [DOI] [PubMed] [Google Scholar]
- 8.Hinek A, Wrenn D S, Mecham R P, Barondes S H. Science. 1988;239:1539–1541. doi: 10.1126/science.2832941. [DOI] [PubMed] [Google Scholar]
- 9.Yannariello-Brown J, Wewer U, Liotta L, Madri J A. J Cell Biol. 1988;106:1773–1786. doi: 10.1083/jcb.106.5.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robert L, Jacob M P, Fulop T, Timar J, Hornebeck W. Pathol Biol. 1989;37(6):736–741. [PubMed] [Google Scholar]
- 11.Mecham R P. FASEB J. 1991;5:2538–2546. doi: 10.1096/fasebj.5.11.1651264. [DOI] [PubMed] [Google Scholar]
- 12.Hinek A, Boyle J, Rabinovitch M. Exp Cell Res. 1992;203:344–353. doi: 10.1016/0014-4827(92)90008-v. [DOI] [PubMed] [Google Scholar]
- 13.Grant D S, Tashiro K I, Segui–Real B, Yamada Y, Martin G R, Kleinman H K. Cell. 1989;58:933–943. doi: 10.1016/0092-8674(89)90945-8. [DOI] [PubMed] [Google Scholar]
- 14.Martignone S, Ménard S, Bufalino R, Cascinelli N, Pellegrini R, Tagliabue E, Andreola S, Rilke F, Colnaghi M I. J Natl Cancer Inst. 1993;85(5):398–402. doi: 10.1093/jnci/85.5.398. [DOI] [PubMed] [Google Scholar]
- 15.Timar J, Lapis K, Fulop T, Varga Z S, Tixier J M, Robert L, Hornebeck W. J Cancer Res Clin Oncol. 1991;117:232–238. doi: 10.1007/BF01625430. [DOI] [PubMed] [Google Scholar]
- 16.Hornebeck W, Adnet J J, Robert L. Exp Gerontol. 1978;13:293–298. doi: 10.1016/0531-5565(78)90037-2. [DOI] [PubMed] [Google Scholar]
- 17.Robert L, Robert A M. In: Frontiers of Matrix Biology. Robert A M, Robert L, editors. Vol. 8. Basel: Karger; 1980. pp. 130–173. [Google Scholar]
- 18.Botney M D, Mecham R P. In: Textbook of Respiratory Medicine. 2nd ed. Murray J, Nadel J, editors. Vol. 1. Philadelphia: Saunders; 1994. pp. 498–508. [Google Scholar]
- 19.Jaffe E A, Nachman R L, Becker C G, Mimick C R. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Queyroy A, Verdetti J. Biochim Biophys Acta. 1992;1108:159–168. doi: 10.1016/0005-2736(92)90021-d. [DOI] [PubMed] [Google Scholar]
- 21.Yamada K M, Older K. Nature (London) 1978;275:179–184. doi: 10.1038/275179a0. [DOI] [PubMed] [Google Scholar]
- 22.Hammond C, Tritsch D. Neurobiologie cellulaire. Paris: Doin; 1990. [Google Scholar]
- 23.Grynkiewicz G, Poenie M, Tsien R Y. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 24.Jacob M P, Hornebeck W. In: Frontiers of Matrix Biology. Robert L, Moczar M, Moczar E, editors. Vol. 10. Basel: Karger; 1985. pp. 92–123. [Google Scholar]
- 25.Revest P A, Abbott N J. Trends Pharmacol Sci. 1992;13(11):404–407. doi: 10.1016/0165-6147(92)90124-o. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg S F, Bilezikian J P, Al-Awqati Q. Am J Physiol. 1987;253:C744–C747. doi: 10.1152/ajpcell.1987.253.5.C744. [DOI] [PubMed] [Google Scholar]
- 27.Wang R, Sauvé R, De Champlain J. J Hypertens. 1995;13:993–1001. [PubMed] [Google Scholar]
- 28.Narahashi T, Herman M D. Methods Enzymol. 1992;207:620–643. doi: 10.1016/0076-6879(92)07045-p. [DOI] [PubMed] [Google Scholar]
- 29.Thastrup O, Dawson A P, Scharff O, Foder B, Cullen P J, Drobak B K, Bjerrum P J, Christensen S B, Hanley M R. Agents Actions. 1989;27(1–2):17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda M, Ariyoshi H, Kambayashi J, Fujitani K, Shinoki N, Sakon M, Kawasaki T, Monden M. J Cell Biochem. 1996;63:23–36. doi: 10.1002/(sici)1097-4644(199610)63:1<23::aid-jcb2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 31.Cooper J A. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bixby J L, Grunwald G B, Bookman R J. J Cell Biol. 1994;127:1461–1475. doi: 10.1083/jcb.127.5.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corda S, Spurgeon H A, Lakatta E G, Capogrossi M C, Ziegelstein R C. Circ Res. 1995;77:927–935. doi: 10.1161/01.res.77.5.927. [DOI] [PubMed] [Google Scholar]
- 34.Varga Z, Jacob M P, Robert L, Fülöp T., Jr FEBS Lett. 1989;258(1):5–8. doi: 10.1016/0014-5793(89)81602-3. [DOI] [PubMed] [Google Scholar]
- 35.Lüscher T F, Vanhoutte P M. The Endothelium: Modulator of Cardiovascular Function. Boca Raton, FL: CRC; 1990. pp. 23–53. [Google Scholar]
- 36.Korenaga R, Ando J, Ohtsuka A, Sakuma I, Yang W, Toyo-oka T, Kamiya A. Cell Struct Funct. 1993;18:95–104. doi: 10.1247/csf.18.95. [DOI] [PubMed] [Google Scholar]
- 37.Robert L. Atherosclerosis (Berlin) 1996;123:169–179. doi: 10.1016/0021-9150(96)05804-2. [DOI] [PubMed] [Google Scholar]
- 38.White C R, Brock T A, Chang L Y, Crapo J, Briscoe P, Ku D, Bradley W A, Gianturco S H, Gore J, Freeman B A, Tarpey M. Proc Natl Acad Sci USA. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Ze’ev A. Crit Rev Eukaryotic Gene Expression. 1992;2:265–281. [PubMed] [Google Scholar]
- 40.Nicotera P, Zhivotovsky B, Orrenius S. Cell Calcium. 1994;16:279–288. doi: 10.1016/0143-4160(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 41.Shin W S, Sasaki T, Kato M, Hara K, Seko A, Yang W D, Shimamoto N, Sugimoto T, Toyo-oka T. J Biol Chem. 1992;267:20377–20382. [PubMed] [Google Scholar]
- 42.Boudreau N, Myers C, Bissell M J. Trends Cell Biol. 1995;5:1–4. doi: 10.1016/s0962-8924(00)88924-2. [DOI] [PubMed] [Google Scholar]