

Abstract
Hybrid polar compounds (HPCs) have been synthesized that induce terminal differentiation and/or apoptosis in various transformed cells. We have previously reported on the development of the second-generation HPCs suberoylanilide hydroxamic acid (SAHA) and m-carboxycinnamic acid bishydroxamide (CBHA) that are 2,000-fold more potent inducers on a molar basis than the prototype HPC hexamethylene bisacetamide (HMBA). Herein we report that CBHA and SAHA inhibit histone deacetylase 1 (HDAC1) and histone deacetylase 3 (HDAC3) activity in vitro. Treatment of cells in culture with SAHA results in a marked hyperacetylation of histone H4, but culture with HMBA does not. Murine erythroleukemia cells developed for resistance to SAHA are cross-resistant to trichostatin A, a known deacetylase inhibitor and differentiation inducer, but are not cross-resistant to HMBA. These studies show that the second-generation HPCs, unlike HMBA, are potent inhibitors of HDAC activity. In this sense, HMBA and the second-generation HPCs appear to induce differentiation by different pathways.
Agents that induce malignant cells to generate differentiated progeny might prevent the progression of cancers (1). Hexamethylene bisacetamide (HMBA), a low molecular weight synthetic compound, induces terminal differentiation and apoptosis in transformed cells in culture (2–4). Although treatment with HMBA induced remissions in patients with myelodysplastic syndrome and acute myelogenous leukemia, it is not currently used as a clinical agent because of the high dosage required (millimolar blood levels) and toxic side effects (thrombocytopenia) (5).
We have reported (2) the synthesis of compounds structurally related to, but more potent than, HMBA. These compounds, along with HMBA, are called hybrid polar compounds (HPCs) because they have in common two polar groups separated by an apolar 5- to 6-carbon methylene chain. HPCs fall into two classes defined by the identities of their respective polar groups. Thus, suberoylanilide hydroxamic acid (SAHA) and m-carboxycinnamic acid bishydroxamide (CBHA) (Table 1, compounds 4 and 5, respectively) bear at least one hydroxamide in place of the amides found in HMBA and diethyl bis(pentamethylene-N,N-dimethylcarboxamide)malonate (EMBA) (Table 1, compounds 1 and 2, respectively). SAHA was synthesized to test the possibility that a hydrophobic phenyl group at one end of the molecule might enhance its activity. The structure of SAHA is related to that of trichostatin A (TSA; Table 1, compound 6) (6), a natural product isolated from Streptomyces hygroscopicus that was initially used as an antifungal antibiotic. Like the HPCs, TSA is a potent inducer of murine erythroleukemia (MEL) cell differentiation (7). TSA is also an inhibitor of histone deacetylase (HDAC) activity (8).
Table 1.
Differentiation activity of HPCs and TSA in MEL cells (DS19)
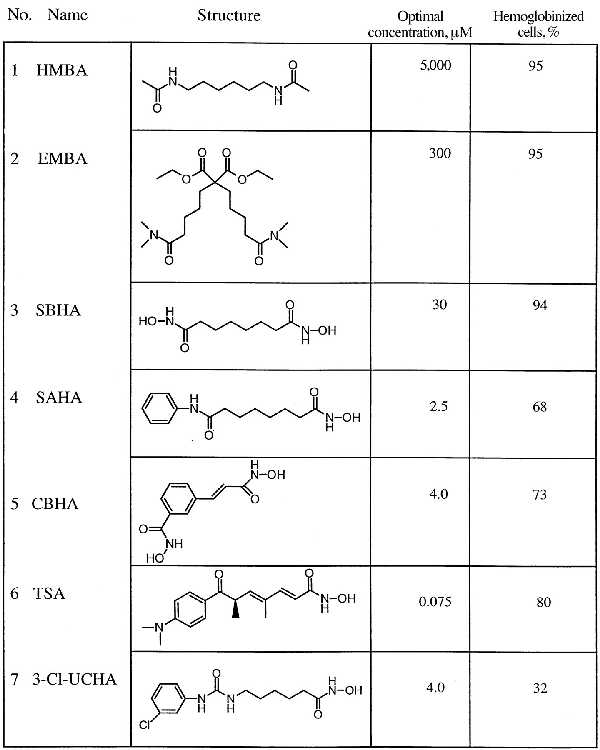
No receptors or binding proteins have been described for the HPCs, and the mechanism(s) by which they induce differentiation has not been elucidated. Indirect evidence suggests that HMBA and EMBA, on the one hand, and SAHA and CBHA, on the other, induce differentiation by different pathways (2). Thus, the MEL cell line variant R1, selected for resistance to HMBA, is sensitive to induction by SAHA and CBHA. In addition, the induction of differentiation by HMBA and EMBA is characterized by suppressed expression of c-myb, whereas SAHA and CBHA induce differentiation without suppression of c-myb expression.
Herein we report that SAHA and CBHA, like TSA, inhibit HDAC activity in vitro and cause accumulation of hyperacetylated histone H4 in cultured cells, whereas HMBA and EMBA do not. Moreover, MEL cells selected for resistance to the differentiation-inducing activity of SAHA are cross-resistant to TSA but not to HMBA. These results suggest that two pathways, one of which involves inhibition of HDAC and one of which does not, can trigger terminal differentiation of malignant cells.
MATERIALS AND METHODS
Cell Culture.
MEL DS19/Sc9 cells, derived from 745A cells, were maintained in α-MEM containing 10% fetal calf serum (FCS). Cell cultures were initiated with cells in logarithmic growth phase at a cell density of 1 to 2 × 105 cells per ml. Cells density, benzidine reactivity, and commitment to terminal differentiation were determined as described (9). HMBA was obtained from Sigma and the second-generation compounds suberic bishydroxamic acid (SBHA), SAHA, and CBHA were synthesized as described (2). The HPC 6-(3-chlorophenylureido)caproic hydroxamic acid (3-Cl-UCHA) was synthesized as described (10). SAHA-resistant MEL cells were selected by growth in 5 μM SAHA by following a standard mutagenesis procedure (11). For mutagenesis, DS19 cells were cultured with methyl methanesulfonate (100 μg/ml) for 30 min. Jurkat cells were cultured in Opti-MEM (GIBCO/BRL) supplemented with 2% FCS (HyClone), penicillin (50 units/ml), streptomycin (50 μg/ml), and 2 mM glutamine at 37°C in a humidified 95% air/5% CO2 atmosphere. T24 bladder carcinoma cells were obtained from the American Type Culture Collection and maintained in α-MEM containing 10% FCS. Human myeloma ARP-1 cells were obtained from J. Hardin (Arkansas Cancer Research Center, Little Rock, AR) and maintained in RPMI 1640 medium containing 10% FCS as described (12).
Isolation of Histones and Acid Urea/Triton X-100 (AUT) Gel Electrophoresis.
Histones were extracted from cells after culture with HPCs or TSA (Wako Biochemicals, Richmond, VA) as indicated and as described by Yoshida et al. (8). AUT gel electrophoresis was used for detection of acetylated histone molecules. Histones (20 μg of total protein) were electrophoresed at 170 V for 24 h at 4°C as described by Panyim and Chalkley (13) and modified Yoshida et al. (8). Gels were stained with Coomassie brilliant blue R-250, dried, and photographed.
Immunoprecipitation–HDAC Assays.
Jurkat cells (5 × 107 cells) were washed twice with ice-cold PBS and resuspended in 1 ml of lysis buffer (50 mM Tris⋅HCl, pH 7.5/120 mM NaCl/5 mM EDTA/0.5% Nonidet P-40) in the presence of protease inhibitors (Complete, Mini protease inhibitor mixture tablets, Boehringer Mannheim). The lysate was incubated for 1 h on ice and cleared by centrifugation at 12,000 × g for 10 min at 4°C. Supernatants were precleared with 30 μl of a 50% protein G-Sepharose slurry for 1 h at 4°C. Beads were pelleted by centrifugation and supernatants were incubated for 1 h at 4°C with 10 μg of IgG fraction from anti-HDAC1 or HDAC3 polyclonal antisera (preincubated 2 h at room temperature with either the homologous or heterologous immunizing peptide). Both antisera were raised in rabbits against the carboxyl-terminal peptide of HDAC1 and HDAC3 by using synthetic peptides coupled to keyhole limpet hemocyanin (14). As a control we used 10 μl of the corresponding preimmune serum. Thirty microliters of a 50% protein G-Sepharose slurry was added for 1 h at 4°C. Immune complexes were pelleted by centrifugation and washed three times with 1 ml of lysis buffer. Beads were resuspended in 200 μl of HDAC buffer (20 mM Tris⋅HCl, pH 8.0/150 mM NaCl/10% glycerol), and a HDAC assay was performed as described with an 3H-acetylated peptide corresponding to amino acids 1–24 of histone H4 (15). Released [3H]acetic acid was quantified by scintillation counting. For inhibitions studies, the immunoprecipitated complexes were preincubated with the different drugs for 30 min at 4°C.
RESULTS
Effect of Second-Generation HPCs on Accumulation of Acetylated Histone H4 in MEL Cells.
The structural similarity between SAHA and TSA lead us to examine the effect of SAHA and the prototype HPC HMBA on HDAC activity in MEL cells by determining the acetylation status of histone H4. MEL cells were cultured for 6, 24, and 48 h with HMBA (5 mM) or SAHA (2.5 μM) at concentrations that are optimal for inducing differentiation (2). Histones were isolated and subjected to AUT gel electrophoresis (Fig. 1A). HMBA had no detectable effect on the acetylation level of histone H4 at 6, 24, or 48 h, whereas after 6 h of culture with SAHA, there was an appreciable accumulation of acetylated histone H4. The pattern of accumulation of acetylated forms of histone H4 did not change with continued culture with SAHA for 24 and 48 h. The degree of histone H4 hyperacetylation during culture with SAHA was similar to that observed with TSA (Fig. 1A).
Figure 1.
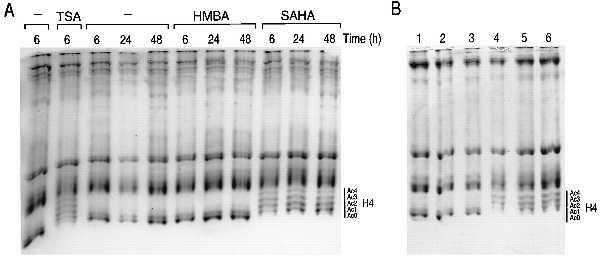
Effect of HPCs on histone acetylation in MEL cells. (A) MEL cells were cultured with TSA (75 nM), HMBA (5 mM), and SAHA (2.5 μM) for the indicated times. Histones were extracted from the treated cells and were analyzed on an AUT gel. (B) MEL cells were cultured for 6 h with no addition (lane 1), 5 mM HMBA (lane 2), 0.3 mM EMBA (lane 3), 30 μM SBHA (lane 4), 2.5 μM SAHA (lane 5), and 4.0 μM CBHA (lane 6). Histones were isolated and analyzed on an AUT gel. The degree of histone acetylation of histone H4 is indicated as follows: Ac0, nonacetylated H4; Ac1, monoacetylated H4; Ac2, diacetylated H4; Ac3, triacetylated H4; Ac4, tetraacetylated H4.
To further explore the structural requirements for accumulation of acetylated H4, we determined the effect of several other compounds structurally related to HMBA that we have previously synthesized (Table 1 and Fig. 1B). The bisamide EMBA, which is a 10-fold more active inducer of differentiation on a molar basis than HMBA, has four exposed polar groups balanced by two apolar groups. As we observed for HMBA, EMBA does not cause acetylated histone H4 accumulation (Fig. 1B, lane 3). SBHA (Table 1, compound 3) was synthesized with the expectation that a hydroxamic acid could bind to a cellular target more strongly than the amide of HMBA and was found to be 100 times as potent as HMBA in inducing differentiation of MEL cells (16). SBHA caused the accumulation of acetylated H4 (Fig. 1B, lane 4), suggesting that hydroxamic acid groups contribute to increased potency and to inhibition of HDAC activity. CBHA (Table 1, compound 5) was synthesized to determine the effect of increasing the rigidity of the flexible hexamethylene spacer between the two polar groups by interposing a phenyl ring (2). This modification resulted in a further 20-fold increase in differentiation activity on a molar basis compared with SBHA. CBHA, like SAHA and SBHA, caused accumulation of acetylated histone H4 (Fig. 1B, lane 6).
Characterization of SAHA-Resistant MEL Cells.
MEL cells resistant to SAHA were developed by mutagenizing cells with methyl methanesulfonate. SAHA-resistant cells (SAHA R-4 and SAHA R-6) were selected in 5 μM SAHA and maintained thereafter in the absence of SAHA. These cells were then tested for their ability to commit to terminal erythroid differentiation (17). DS19, SAHA R-4, and SAHA R-6 were cultured with 2.5 μM SAHA for 48 h in suspension culture. The cells were then washed with SAHA-free medium and plated in methylcellulose in the absence of SAHA. The abilities to proliferate into colonies and to express hemoglobin were determined after a 5-day culture period (Fig. 2A). The SAHA-sensitive DS19 cells are committed to differentiate by SAHA, forming small colonies (less than 32 cells) that accumulate hemoglobin. SAHA R-4 and SAHA R-6 cells formed large benzidine-negative colonies (>32 cells) after exposure to SAHA, indicating that the cells are resistant to induction of differentiation by SAHA in terms of both accumulation of hemoglobin and cessation of cell division (Fig. 2A). We also examined whether the SAHA-resistant DS19 cells were cross-resistant to TSA and to HMBA by performing commitment assays (Fig. 2A). The SAHA-resistant clones were resistant to induction of terminal differentiation by SAHA and TSA but were not cross-resistant to HMBA.
Figure 2.
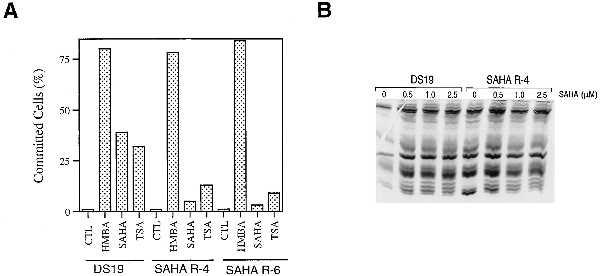
Characterization of SAHA-resistant cell clones, SAHA R-4 and SAHA R-6. (A) Effect of HPCs and TSA on commitment to terminal differentiation. Cells were cultured with no addition (CTL), 5 mM HMBA, 3 μM SAHA, or TSA (12.5 ng/ml), as indicated, and commitment assays performed after 48 h of culture. After a 5-day growth period in methylcellulose, cells were scored as committed to terminal differentiation if they formed small colonies (<32 cells) and were expressing hemoglobin as determined by benzidine staining. (B) Effect of SAHA on histone acetylation in DS19 and SAHA R-4 cells. Cells were cultured in 0, 0.5, 1.0, and 2.5 μM SAHA for 2 h. Histones were isolated and analyzed on an AUT gel as indicated.
Resistance to SAHA could result from an inability to inhibit the HDAC. We tested this possibility by culturing DS19 and SAHA R-4 cells with 0.5, 1.0, and 2.5 μM SAHA for 6 h and examined the acetylation state of the histones as described above (Fig. 2B). Both the DS19 and SAHA R-4 cells showed a similar pattern of acetylated H4 in response to culture with SAHA. These results suggest that the resistance of the SAHA R-4 cells to SAHA is distal to the inhibition of the deacetylase. These results also demonstrate that the resistance is not caused by a failure of SAHA to enter the cell.
Effect of HPCs on Histone Acetylation Levels in T24 and ARP-1 Cells.
In addition to MEL cells, HPCs induce differentiation of the human bladder carcinoma cell line T24 (3) and induce apoptosis of the human myeloma cell line ARP-1 (4). We determined the effect of HMBA, the second-generation HPC 3-Cl-UCHA (Table 1, compound 7), and TSA on the accumulation of acetylated histone H4 in T24 and ARP-1 cells. 3-Cl-UCHA is the most potent HPC in T24 and ARP-1 cells. T24 and ARP-1 cells were cultured for 6 h in the presence of the indicated concentration of HPC, histones were isolated, and AUT gel electrophoresis was performed (Fig. 3). Consistent with the evidence from MEL cells, HMBA did not cause accumulation of hyperacetylated histone H4, whereas 3-Cl-UCHA and TSA did.
Figure 3.
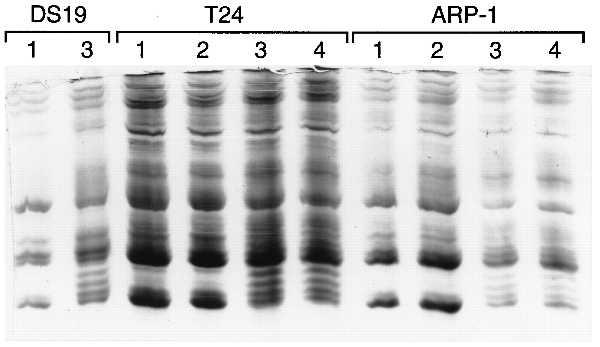
Effect of HPCs on histone acetylation in T24 and ARP-1 cells. MEL cells were cultured for 6 h with no addition (lane 1) or 4 μM 3-Cl-UCHA (lane 3). T24 cells were cultured for 6 h with no addition (lane 1), 10 mM HMBA (lane 2), 3 μM 3-Cl-UCHA (lane 3), or TSA (500 ng/ml; lane 4). ARP-1 cells were cultured for 6 h with no addition (lane 1), 5 mM HMBA (lane 2), 1 μM 3-Cl-UCHA (lane 3), or TSA (20 ng/ml; lane 4). The histones were extracted from the treated cells and then analyzed on an AUT gel.
Effect of HPCs on HDAC Activity.
An increase in acetylated histone H4 can reflect either activation of histone acetyltransferase(s) or inhibition of HDAC(s). Because of the structural homology between SAHA and TSA, we examined the effect of the HPCs on the enzymatic activity of two recently identified human HDACs, HDAC1 (15) and HDAC3 (14, 18). HDAC1 and HDAC3 were immunoprecipitated from Jurkat cells by using specific antisera directed against each protein. The specificity of the antisera was demonstrated by preincubating the antiserum with the peptide used for immunization. Immunoprecipitation with the anti-HDAC1 antiserum was blocked by the HDAC1 peptide but not by the HDAC3 peptide (Fig. 4A). The opposite was observed with the anti-HDAC3 antiserum (Fig. 4A). Immunoprecipitates obtained with the anti-HDAC1 and anti-HDAC3 antisera were incubated with a synthetic peptide corresponding to the amino-terminal part of histone H4. This peptide was acetylated in vitro with [3H]acetate and we consider the release of [3H]acetate to be a measure of HDAC activity. Both activities were inhibited by TSA (Fig. 4A). Both HDAC1 and HDAC3 activities were inhibited by SBHA, SAHA, and CBHA at submicromolar concentrations (ID50 values for HDAC1 are 0.25, 0.01, and 0.01 μM, respectively, and for HDAC3 are 0.30, 0.02, and 0.07 μM, respectively; Fig. 4B). The concentration of HPC required to inhibit HDAC1 and HDAC3 correlated with the concentration of HPC required to induce differentiation. SBHA is 10-fold less potent in inducing differentiation than SAHA and CBHA (Table 1) and approximately 10-fold less potent in inhibiting HDAC1 and HDAC3 than SAHA and CBHA. Neither HMBA nor EMBA inhibited either HDAC1 or HDAC3 activity, at concentrations sufficient to induce differentiation, in agreement with the absence of histone modification in vivo in response to these compounds.
Figure 4.
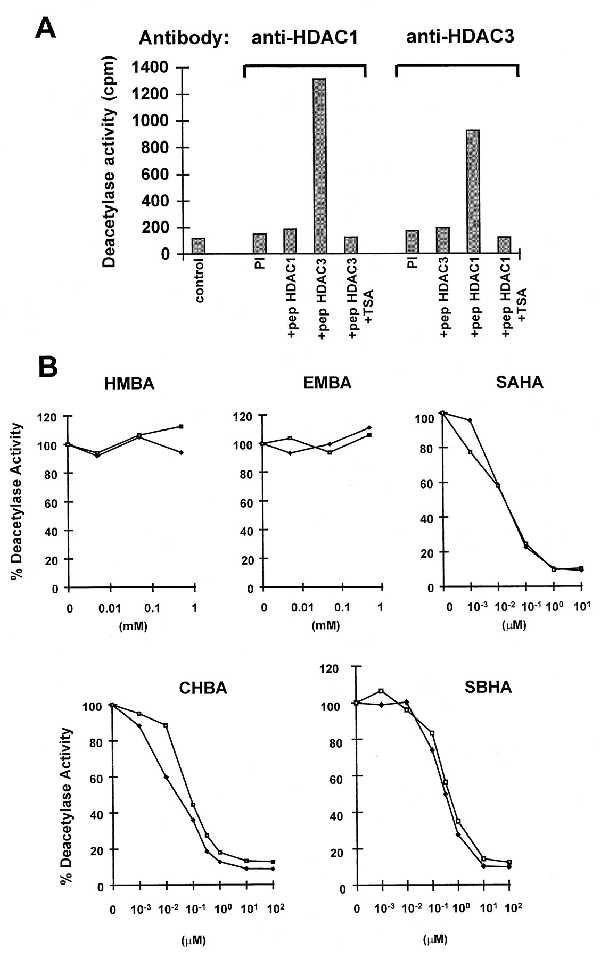
Inhibition of HDAC1 and HDAC3 by HPCs. (A) Cellular extracts from Jurkat cells (5 × 107 cells) were subjected to immunoprecipitation with a polyclonal antiserum against HDAC1 and HDAC3 in the presence of a 100-fold excess of HDAC3 peptide (residues 413–428) or HDAC1 peptide (residues 467–482). As an additional control, the corresponding preimmune serum was used. Immunoprecipitated complexes were tested for HDAC activity by measuring the release of [3H]acetate from an acetylated amino-terminal H4 peptide (in cpm), in the absence or presence of 400 nM TSA. (B) Cellular extracts from Jurkat cells were subjected to immunoprecipitation with polyclonal antiserum against HDAC1 (♦) or HDAC3 (□) and tested for HDAC activity in the absence or the presence of increasing concentration of the following inhibitors: HMBA, 0.005, 0.05, and 0.5 mM; EMBA, 0.005, 0.05, and 0.5 mM; SAHA, 0.001, 0.01, 0.1, 1, and 10 μM; CBHA or SBHA, 0.001, 0.01, 0.1, 0.33, 1, 10, and 100 μM.
DISCUSSION
We report the finding of potent HDAC inhibitors that are inducers of transformed cell differentiation and apoptosis. We have screened several hundred HPCs for induction of differentiation by using the MEL cell model system and have identified more than 150 active inducers of differentiation, about 75 of which are hydroxamides. Of the inducers tested, those which contain at least one hydroxamic acid moiety are inhibitors of HDAC activity in vitro and cause the accumulation of acetylated histone H4 in cells in culture. The HPCs, like TSA, appear to be general inhibitors of HDAC acetylase activity because they inhibit both HDAC1 and HDAC3 activity with the same potency. n-Butyrate is another inhibitor of histone deacetylation in cells in culture and is also a weak inducer of MEL cell differentiation (19, 20). These studies collectively suggest that a relationship exists between hyperacetylation of histones and induction of differentiation by HPCs with a hydroxamic acid moiety.
Acetylation of core histones is a reversible modification associated with transcriptionally active chromatin (21). Chromatin fractions enriched in actively transcribed genes are also enriched in the more highly acetylated isoforms of the core histones (22). Histone acetylation appears to serve as a marker of the potential for transcription of genes expressed in differentiated cells rather than as the trigger for transcription itself. The highly acetylated chromatin fraction in chicken erythrocytes is enriched for the β-globin gene but not for the unexpressed ovalbumin gene (23). But the β-globin gene is also enriched in the acetylated fraction of chromatin prepared from the erythrocytes of 5-day embryos, at a time when the β-globin gene is not yet expressed (24). The accumulation of hyperacetylated histone after inhibition of HDAC by TSA in mammalian cells results in the transcriptional regulation of a small fraction of cellular genes (25). Thus, these studies suggest that histone acetylation acts permissively to set the stage for transcription of selected genes to occur when the appropriate transcription factors are present.
The finding that the second-generation HPCs and TSA are both inducers of transformed cell differentiation and inhibitors of HDAC activity suggests that transformed cells have in some manner altered their regulation of histone acetylation. Altered regulation of histone acetyltransferases has been associated with leukemogenesis (26, 27) and the Rubinstein–Taybi syndrome (RTS), a developmental disorder in which patients have a propensity to develop malignancies (28). The CREB-binding protein gene (CBP), encoding a histone acetyltransferase, has been found fused to the MLL gene in therapy-related acute myeloid or lymphoid leukemias and myelodysplasia (26) and to the MOZ gene in acute myeloid leukemia (27). CBP is the gene responsible for RTS. Microdeletions, translocations, inversions, and point mutations in the CBP gene have been identified in patients with RTS (28).
HMBA-induced MEL cell differentiation most likely does not involve inhibition of HDAC activity. This is most clearly implied by (i) the lack of inhibition of HDAC at concentrations of HMBA effective for inducing differentiation, (ii) lack of the accumulation of acetylated H4 in cells cultured with HMBA, (iii) the finding that cells resistant to SAHA still are induced by HMBA, and (iv) the finding that potency for differentiation can be increased 10-fold (i.e., EMBA) without any increase in inhibition of HDAC activity. It is possible that HMBA alters histone acetylation more discretely, undetected by the current assays. Alternatively, HMBA may initiate some other modification of chromatin structure and function, allowing the expression of differentiation-specific gene sets.
These experiments show that the second-generation HPCs are potent inhibitors of HDAC activity. These findings further support the model that altered regulation of histone acetylation plays a role in the development of cancer and that modulation of HDAC activity might lead to new therapeutic opportunities against cancer.
Acknowledgments
We thank Lang Ngo and Gisela Venta-Perez for expert technical assistance. These investigations were supported, in part, by Grant CA-0874823 from the National Cancer Institute and grants from the Japan Foundation for the Promotion of Cancer Research Fund, the DeWitt Wallace Fund for Memorial Sloan–Kettering Cancer Center, the Westbranch Leukemia Research Fund, and the Picower Institute for Medical Research.
ABBREVIATIONS
- MEL
murine erythroleukemia
- HMBA
hexamethylenebisacetamide
- HPC
hybrid polar compound
- SAHA
suberoylanilide hydroxamic acid
- CBHA
m-carboxycinnamic acid bishydroxamide
- 3-Cl-UCHA
6-(3-chlorophenylureido)caproic hydroxamic acid
- EMBA
diethyl bis(pentamethylene-N,N-dimethylcarboxamide)malonate
- SBHA
suberic bishydroxamic acid
- TSA
trichostatin A
- HDAC
histone deacetylase
- AUT
acid urea/Triton X-100
References
- 1.Marks P A, Richon V M, Rifkind R A. Int J Hematol. 1996;63:1–17. doi: 10.1016/0925-5710(95)00428-9. [DOI] [PubMed] [Google Scholar]
- 2.Richon V M, Webb Y, Merger R, Sheppard T, Jursic B, Ngo L, Civoli F, Breslow R, Rifkind R A, Marks P A. Proc Natl Acad Sci USA. 1996;93:5705–5708. doi: 10.1073/pnas.93.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richon V M, Russo P, Venta-Perez G, Cordon-Cardo C, Rifkind R A, Marks P A. Cancer Res. 1997;57:2789–2798. [PubMed] [Google Scholar]
- 4.Siegel D S, Zhang X, Feinman R, Teitz T, Zelenetz A, Richon V M, Rifkind R A, Marks P A, Michaeli J. Proc Natl Acad Sci USA. 1998;95:162–166. doi: 10.1073/pnas.95.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreeff M, Stone R, Michaeli J, Young C W, Tong W, Sogoloff H, Ervin T, Kufe D, Rifkind R A, Marks P A. Blood. 1992;80:2604–2609. [PubMed] [Google Scholar]
- 6.Yoshida M, Horinouchi S, Beppu T. BioEssays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida M, Nomura S, Beppu T. Cancer Res. 1987;47:3688–3691. [PubMed] [Google Scholar]
- 8.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 9.Chen Z, Banks J, Rifkind R A, Marks P A. Proc Natl Acad Sci USA. 1982;79:471–475. doi: 10.1073/pnas.79.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb Y. Ph.D. thesis. New York: Columbia University; 1997. [Google Scholar]
- 11.Klungland A, Laake K, Hoff E, Seeberg E. Carcinogenesis. 1995;16:1281–1285. doi: 10.1093/carcin/16.6.1281. [DOI] [PubMed] [Google Scholar]
- 12.Hardin J, MacLeod S, Grigorieva I, Chang R, Barlogie B, Xiao H, Epstein J. Blood. 1994;84:3063–3070. [PubMed] [Google Scholar]
- 13.Panyim S, Chalkley R. Arch Biochem Biophys. 1969;130:337–346. doi: 10.1016/0003-9861(69)90042-3. [DOI] [PubMed] [Google Scholar]
- 14.Emiliani, S., Fischle, W., Van Lint, C., Al-Abed, Y., Spector, D. & Verdin, E. (1998) Proc. Natl. Acad. Sci. USA 95, in press. [DOI] [PMC free article] [PubMed]
- 15.Taunton J, Hassig C A, Schreiber S L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 16.Breslow R, Jursic B, Yan Z F, Friedman E, Leng L, Ngo L, Rifkind R A, Marks P A. Proc Natl Acad Sci USA. 1991;88:5542–5546. doi: 10.1073/pnas.88.13.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marks P A, Rifkind R A. Annu Rev Biochem. 1978;47:419–448. doi: 10.1146/annurev.bi.47.070178.002223. [DOI] [PubMed] [Google Scholar]
- 18.Yang W M, Yao Y L, Sun J M, Davie J R, Seto E. J Biol Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 19.Leder A, Leder P. Cell. 1975;5:319–322. doi: 10.1016/0092-8674(75)90107-5. [DOI] [PubMed] [Google Scholar]
- 20.Riggs M G, Whittaker R G, Neumann J R, Ingram V M. Nature (London) 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 21.Grunstein M. Nature (London) 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 22.Turner B M. Cell. 1993;75:5–8. [PubMed] [Google Scholar]
- 23.Hebbes T R, Clayton A L, Thorne A W, Crane-Robinson C. EMBO J. 1994;13:1823–1830. doi: 10.1002/j.1460-2075.1994.tb06451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebbes T R, Thorne A W, Clayton A L, Crane-Robinson C. Nucleic Acids Res. 1992;20:1017–1022. doi: 10.1093/nar/20.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Lint C, Emiliani S, Verdin E. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 26.Sobulo O M, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, Housman D, Doggett N A, Rowley J D, Zeleznik-Le N J. Proc Natl Acad Sci USA. 1997;94:8732–8737. doi: 10.1073/pnas.94.16.8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borrow J, Stanton V P, Jr, Andresen J M, Becher R, Behm F G, Chaganti R S, Civin C I, Disteche C M, Dube I, Frischauf A-M, et al. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 28.Petrij F, Giles R H, Dauwerse H G, Saris J J, Hennekam R C, Masumo M, Tommerup N, vanOmmen G J, Goodman R H, Peters D J M, Breuning M H. Nature (London) 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]