

Abstract
UV irradiation induces apoptosis in U937 human leukemic cells that is accompanied by the activation of both the stress-activated protein kinase (SAPK) and p38 mitogen-activated protein kinase (MAPK) signal transduction pathways. The MAPK phosphatase, MKP-1, is capable of inactivating both SAPK and p38 MAPK in vivo. To determine whether MKP-1-mediated inhibition of SAPK and/or p38 MAPK activity provided cytoprotection against UV-induced apoptosis, a U937 cell line conditionally expressing MKP-1 from the human metallothionein IIa promoter was established. Conditional expression of MKP-1 was found to abolish UV-induced SAPK and p38 MAPK activity, and inhibit UV-induced apoptosis as judged by both morphological criteria and DNA fragmentation. MKP-1 was also found to inhibit other biochemical events associated with apoptosis, including activation of caspase-3 and the proteolytic cleavage of the caspase-3 substrate, poly(ADP ribose) polymerase. These findings demonstrate that MKP-1 acts at a site upstream of caspase activation within the apoptotic program. The cytoprotective properties of MKP-1 do not appear to be mediated by its ability to inhibit p38 MAPK because the p38 MAPK specific inhibitor SB203580 had no effect on UV-induced apoptosis in U937 cells. Furthermore, by titrating the level of MKP-1 expression it was found that MKP-1 inhibited UV-induced SAPK activity, DNA fragmentation, and caspase-3 activation in a similar dose-dependent manner. The dual-specificity phosphatase, PAC1, which does not inhibit UV-induced activation of SAPK, did not provide a similar cytoprotection against UV-induced apoptosis. These results are consistent with a model whereby MKP-1 provides cytoprotection against UV-induced apoptosis by inhibiting UV-induced SAPK activity.
Recently, much emphasis has been placed on the role of stress-activated protein kinase [SAPK/c-Jun N-terminal protein kinase (JNK)] and p38 mitogen-activated protein kinase (MAPK) in coupling cellular stress signals with the apoptotic machinery (1). Multiple apoptotic stress stimuli, including UV- and γ-irradiation, heat shock, protein synthesis inhibitors, DNA-damaging agents, and the proinflammatory cytokines tumor necrosis factor α and interleukin 1 (IL-1), are potent activators of the SAPKs (2–5). Although SAPK and p38 MAPK are coordinately regulated in response to many forms of apoptotic cellular stress, the role of these kinases in activating apoptosis remains unclear. Expression of a constitutively active mutant of MEK kinase-1 (MEKK1), an upstream activator of SAPK, has been shown to induce apoptosis in a variety of cell types (6–8). Furthermore, cells expressing dominant-inhibitory mutants within the SAPK pathway become highly resistant to stress-induced apoptosis (7–11). In this regard, expression of dominant-inhibitory mutants of MEKK1, SAPK/extracellular signal-regulated kinase (ERK) kinase 1, or SAPK is sufficient to block apoptosis in response to UV- and γ-irradiation (8–11). However, there are conflicting reports concerning the involvement of SAPK and p38 MAPK in mediating tumor necrosis factor α- or Fas-induced apoptosis (9, 12–16). Thus, distinct molecular mechanisms may mediate apoptosis in response to specific stress stimuli, some of which may involve activation of the SAPK signal transduction pathway.
The MAPKs—ERK, SAPK, and p38 MAPK—are activated by the reversible dual threonine and tyrosine phosphorylation of a conserved T-X-Y motif (3–5). The reversible nature of MAPK phosphorylation suggests that phosphatases play a key role in regulating MAPK activity. A growing family of dual specificity phosphatases, termed MAPK phosphatases (MKP), have been identified that inactivate MAPKs by dephosphorylation of these phosphothreonine and phosphotyrosine residues. The prototypic member of this family, MKP-1 (CL100/3CH134/erp), was originally identified as an ERK-specific phosphatase (17–20). However, it is now well established that MKP-1 is also capable of inactivating both SAPK and p38 MAPK (5, 21–28). Furthermore, we have shown that MKP-1 exhibits a higher specificity for p38 MAPK and SAPK than for ERK2 in U937 cells (28). Thus, substrate specificity of the MKP family members varies significantly. In this regard, the dual-specificity phosphatase, PAC1, which is closely related to MKP-1, is capable of inactivating ERK2 and p38 MAPK, while exhibiting very little activity toward SAPK (22, 29, 30).
In this study, we have exploited the ability of MKP-1 to inactivate SAPK and p38 MAPK as a unique means to examine whether inhibition of UV-induced activation of these stress kinases would provide cytoprotection against apoptosis. Inhibition of UV-induced SAPK and p38 MAPK activation by conditional expression of MKP-1 was found to inhibit UV-induced apoptosis, DNA fragmentation, activation of caspase-3, and cleavage of the death substrate poly(ADP-ribose) polymerase (PARP). The cytoprotective properties of MKP-1 do not appear to be mediated by p38 MAPK, as evidenced by the inability of a pharmacologic inhibitor of p38 MAPK to effect UV-induced apoptosis. Titration of the level of MKP-1 expression demonstrates that the dose-dependency of this cytoprotection correlates most closely with the inactivation of SAPK, and not p38 MAPK. Furthermore, conditional expression of the dual-specificity phosphatase, PAC1, which does not inhibit SAPK activity, does not elicit a similar protective response. In aggregate, these findings support a model whereby MKP-1 may play a physiological cytoprotective role by inhibiting activation of SAPK and blocking activation of the apoptotic machinery.
MATERIALS AND METHODS
Plasmids, Reagents, and Production of Stable Cell Lines.
The human metallothionein IIa (hMTIIa)-MKP1(myc) expression vector has been described (28). hMTIIa-PAC1 was prepared by subcloning a 1.5-kb EcoRI fragment from pMT2T-PAC1 (a gift from K. Kelly, ref. 22) into pcDNA3. The PAC1 insert was then subcloned into the KpnI/XhoI site of pMEP4 (Invitrogen). U937 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated bovine calf serum (31). Stable cell lines were established as described (32). UV irradiation was performed by using a UV Stratalinker 1800 (Stratagene). Glutathione S-transferase (GST)-cJun(5–89) has been described (27). GST-ATF2(1–109) was provided by M. Green (University of Massachusetts, Worcester). The anti-Myc epitope mAb 9E10 was provided by N. Tonks and H. Sun (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). The p38 MAPK (C-20), PAC1 (N-19), and PARP (C-19) antibodies were purchased from Santa Cruz Biotechnology. The ERK2 (107) antibody was obtained from Zymed. Ac-YVAD-AMC, Ac-DEVD-AMC, and the p38 MAPK inhibitor, SB203580, were purchased from Alexis Biochemicals (San Diego).
Protein Kinase Assays, Western Blot Analysis, and Immunoprecipitation.
Whole-cell extracts were prepared as described (28). SAPK activity was measured by the solid-phase kinase assay by using GST-cJun(5–89) as substrate (2). p38 MAPK activity was determined by immunocomplex kinase assay by using anti-p38 MAPK immunoprecipitates and GST-ATF2(1–109) as substrate (5, 28). Inducible MKP-1(myc) protein expression from the hMTIIa promoter was analyzed exactly as described (28). For Western blot analysis, whole-cell extracts were resolved by SDS/PAGE and electroblotted onto nitrocellulose. PARP and PAC1 expression were determined by Western blot analysis as described (27).
Analysis of Apoptosis.
For DNA fragmentation studies, cells (0.5–1 × 106) were harvested and lysed as described (33). DNA was resolved in 2% agarose gels containing ethidium bromide in TAE (40 mM Tris-acetate and 1.0 mM EDTA, pH 8.0) and was visualized by UV illumination. DNA fragmentation was quantified by fluorescence-activated cell sorter analysis (FACS) of cells stained with propidium iodide (PI). Cells (≈5 × 105) were incubated for 30 min at 23°C in stain solution (0.3% saponin/25 μg/ml PI/0.1 mM EDTA/125 units/ml RNase A). Cells were then analyzed for cell cycle distribution by FACS analysis and cells appearing as a sub-G1 peak were scored as apoptotic. For analysis of caspase activity, cells were lysed for 10 min on ice in caspase lysis buffer (50 mM Tris, pH 7.4/1 mM EDTA/10 mM EGTA/10 μM digitonin). Soluble protein (50 μg) was incubated for 30 min at 37°C in 100 μl of caspase lysis buffer containing 20 μM Ac-DEVD-AMC (caspase-3) or Ac-YVAD-AMC (IL1-β-converting enzyme) and fluorescence monitored on a PerSeptive Biosystems Cytofluor Series 4000 multiwell plate reader with an excitation wavelength of 360 nm and an emission wavelength of 460 nm. Substrate autoflourescence was subtracted from each value, and data are presented as fold activation over lysates from untreated cells.
RESULTS
Conditional Expression of MKP-1 Abolishes UV-Induced SAPK and p38 MAPK Activity.
We have recently shown that conditional expression of the MAPK phosphatase, MKP-1, preferentially inhibits PMA-induced p38 MAPK and SAPK activity in U937 cells (28). We thus sought to determine whether conditional expression of MKP-1 inhibited UV-induced SAPK and p38 MAPK activity in U937 cells. Conditional expression of MKP-1 was accomplished using the heavy metal-inducible hMTIIa promoter (28). U937 cells were transfected with either a hMTIIa vector or hMTIIa-MKP1 and stable cell lines derived by selection in the presence of hygromycin. Very little MKP-1 protein expression was detected in uninduced hMTIIa-MKP-1 cells, whereas treatment for 4 h with 1 μM CdSO4 induced an approximate 50-fold increase in MKP-1 protein expression (Fig. 1A). To determine the effect of conditional expression of MKP-1 on UV-induced SAPK and p38 MAPK activity, U937 cells stably expressing hMTIIa-MKP-1 were exposed to UV radiation (200 J/m2) and kinase activities determined after various periods of time. UV radiation induced a rapid activation of both SAPK and p38 MAPK (Fig. 1B, −Cd++). Activation of both SAPK and p38 MAPK was transient with maximal activation occurring 30 min postirradiation. UV radiation induced a more potent activation of SAPK (10- to 20-fold) than p38 MAPK (2- to 5-fold). Consistent with previous studies, UV radiation was found to have no effect on ERK2 phosphorylation in U937 cells (ref. 28, data not shown). Conditional expression of MKP-1 by pretreatment with CdSO4 abolished UV-induced SAPK and p38 MAPK activity (Fig. 1B, +Cd++).
Figure 1.
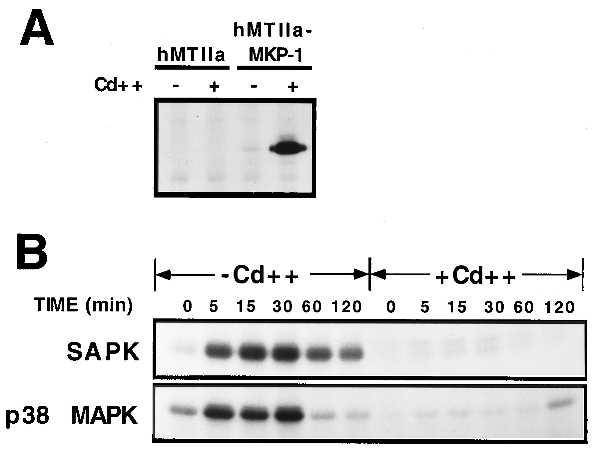
Conditional expression of MKP-1 inhibits UV-induced SAPK and p38 MAPK activity. (A) U937 cells were transfected with either hMTIIa vector or hMTIIa-MKP-1(myc) and stable cell lines derived by selection in the presence of hygromycin B. Cells were metabolically labeled for 4 h with [35S]methionine in the presence or absence of 1 μM CdSO4 (Cd++). MKP-1(myc) protein was immunoprecipitated from whole-cell extracts with the anti-Myc antibody 9E10, resolved by SDS/PAGE, and visualized by fluorography. (B) U937 cells expressing hMTIIa-MKP-1 were pretreated in the absence or presence of 1 μM CdSO4 for 4 h before exposure to UV irradiation (200 J/m2). Cells were incubated for the indicated times and SAPK and p38 MAPK activity determined as described in Materials and Methods.
Conditional Expression of MKP-1 Inhibits UV-Induced Apoptosis.
Because conditional expression of MKP-1 abolished UV-induced SAPK and p38 MAPK activity, which have been implicated in UV-induced apoptosis, the ability of MKP-1 to protect U937 cells against UV-induced apoptosis was examined. Exposure of hMTIIa-MKP-1 U937 cells to UV radiation induced a time-dependent increase in internucleosomal DNA fragmentation (Fig. 2A, −Cd++), which is characteristic of apoptotic cell death (34). Preinduction of MKP-1 protein expression with CdSO4 abolished UV-induced DNA fragmentation in the hMTIIa-MKP-1 U937 cell line (Fig. 2A, +Cd++). DNA fragmentation was quantified by FACS analysis of cells stained with PI. Cells were analyzed for cell cycle distribution by FACS analysis and cells appearing as a sub-G1 peak were scored as apoptotic. UV radiation was found to induce 30–40% DNA fragmentation after 8 h and conditional expression of MKP-1 reduced this by 80% (Fig. 2B). Therefore, conditional expression of MKP-1 provided significant protection against UV-induced DNA fragmentation and apoptosis.
Figure 2.
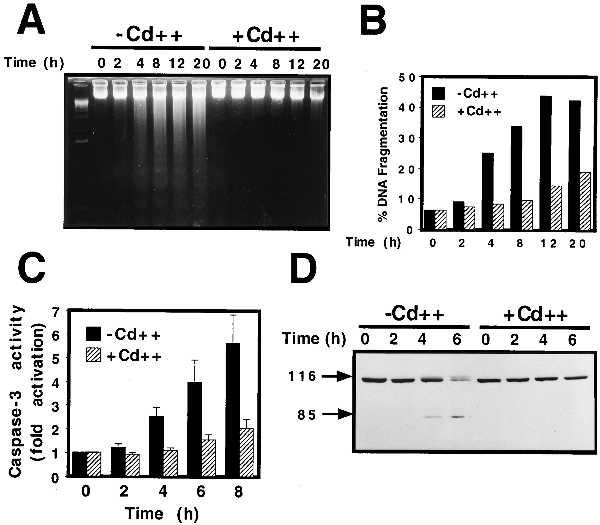
Conditional expression of MKP-1 inhibits UV-induced apoptosis. (A) U937 cells expressing hMTIIa-MKP-1 were pretreated for 4 h with 1 μM CdSO4 (Cd++) as indicated before exposure to UV radiation (200 J/m2). At the indicated times, chromosomal DNA was isolated, resolved on a 2% agarose gel and visualized by ethidium bromide staining. (B) DNA fragmentation was quantified by cell cycle FACS analysis on aliquots of cells treated as above and stained with PI. Cells appearing as a sub-G1 peak were scored as apoptotic. (C) Cells were treated as above and caspase-3 activity was measured by cleavage of the Ac-DEVD-AMC fluorogenic substrate as described in Materials and Methods. Data are presented as fold activation over untreated cells and are means +/− SEM of three experiments performed in triplicate and assayed in duplicate. (D) Cells were treated as above and PARP expression and cleavage was determined by Western blot analysis.
Apoptosis is characterized biochemically by the activation of a family of IL1-β-converting enzyme-like cysteine proteases termed caspases (35, 36). Caspase-3, a central executioner of most forms of apoptosis (35, 36), is activated in U937 cells induced to undergo apoptosis in response to various stress-inducing agents (37, 38). To determine whether MKP-1-mediated inhibition of UV-induced DNA fragmentation was accompanied by an inhibition of UV-induced caspase-3 activity, lysates from UV-irradiated cells were analyzed for caspase-3 activity using the fluorogenic caspase-3 substrate, Ac-DEVD-AMC (39). In uninduced hMTIIa-MKP-1 U937 cells, UV radiation induced a time-dependent increase in caspase-3 activity (Fig. 2C, −Cd++). Conditional expression of MKP-1 nearly abolished UV-induced caspase-3 activity for up to 8 h postirradiation (Fig. 2C, +Cd++). No activation of IL1-β-converting enzyme-like caspases was observed in response to UV radiation as determined utilizing the fluorogenic substrate Ac-YVAD-AMC (data not shown), suggesting that IL-β-converting enzyme-like proteases do not play a role in UV-induced apoptotic cell death in U937 cells.
A variety of caspase-3 substrates have been identified that may play a role in mediating apoptosis, including PARP (35, 36). Caspase-3 cleaves PARP at a consensus DEVD motif producing an 85-kDa fragment (36). To determine whether MKP-1 inhibited caspase-3-mediated cleavage of PARP in vivo, cell lysates from UV irradiated cells were examined for PARP cleavage by Western blot analysis. UV radiation induced the cleavage of PARP from a 116-kDa holo-enzyme to its characteristic 85-kDa fragment in uninduced hMTIIa-MKP-1 cells (Fig. 2D, −Cd++). Preinduction of MKP-1 protein expression, however, abolished the appearance of this PARP fragment (Fig. 2D, +Cd++). Conditional expression of MKP-1 also inhibited UV-induced cleavage of PKCδ (data not shown), which is also a caspase-3 substrate in U937 cells induced to undergo apoptosis (33). Although heavy metals, such as Zn2+ and Cd2+, have been shown to inhibit both Ca2+-dependent endonuclease and caspase-3 activity (40, 41), CdSO4 pretreatment had little effect on UV-induced DNA fragmentation, caspase-3 activation or PARP cleavage in a U937 cell line stably expressing the hMTIIa vector alone (data not shown). Thus, the protective effects observed upon treatment of hMTIIa-MKP-1 U937 cells with CdSO4 are caused by the induction of MKP-1 protein expression and not by an indirect effect of Cd2+ on the apoptotic machinery. These studies suggest that conditional expression of MKP-1 serves a cytoprotective function in U937 cells by inhibiting UV-induced activation of the apoptotic pathway at a site upstream of caspase-3.
MKP-1-Mediated Protection Against UV-Induced Apoptosis Correlates with the Inhibition of SAPK Activity.
We have previously demonstrated that the relative level of MKP-1 expression from the hMTIIa promoter is exquisitely sensitive to the concentration of CdSO4 used for induction (28). By titrating the level of MKP-1 expression from the hMTIIa promoter, we have been able to demonstrate a substrate specificity for MKP-1 of p38 MAPK ≫ SAPK > ERK2 (28). This approach was exploited to determine whether the efficacy by which MKP-1 inhibits UV-induced SAPK correlated with the inhibition of UV-induced apoptosis. hMTIIa-MKP-1 U937 cells were pretreated with increasing concentrations of CdSO4 for 4 h, exposed to UV radiation and SAPK activity, caspase-3 activity and DNA fragmentation analyzed. Titration of the level of MKP-1 protein expression before exposure to UV radiation resulted in a dose-dependent inhibition of UV-induced SAPK activity measured 30 min postirradiation (Fig. 3A). The concentration of CdSO4 required to inhibit 50% of UV-induced SAPK activity was ≈30 nM (Fig. 3 A and B). We have previously shown that the IC50 for inhibition of p38 MAPK activity is 10-fold lower than this (≈3 nM) (28). UV-induced caspase-3 activity measured 8 h postirradiation was inhibited with an almost identical dose-dependency to that observed for SAPK (≈30 nM) (Fig. 3B). Furthermore, a similar dose-dependent inhibition of UV-induced DNA fragmentation was observed (Fig. 3C). When quantitated by FACS analysis of PI-stained cells, a slightly higher concentration of CdSO4 was required for half-maximal inhibition of DNA fragmentation (Fig. 3C, % frag). This may indicate that a threshold level of caspase-3 activity is required for the commitment to apoptosis and DNA fragmentation. In aggregate, these results demonstrate that the cytoprotective effects of MKP-1 correlate in a dose-dependent manner with the inhibition of UV-induced SAPK activity.
Figure 3.
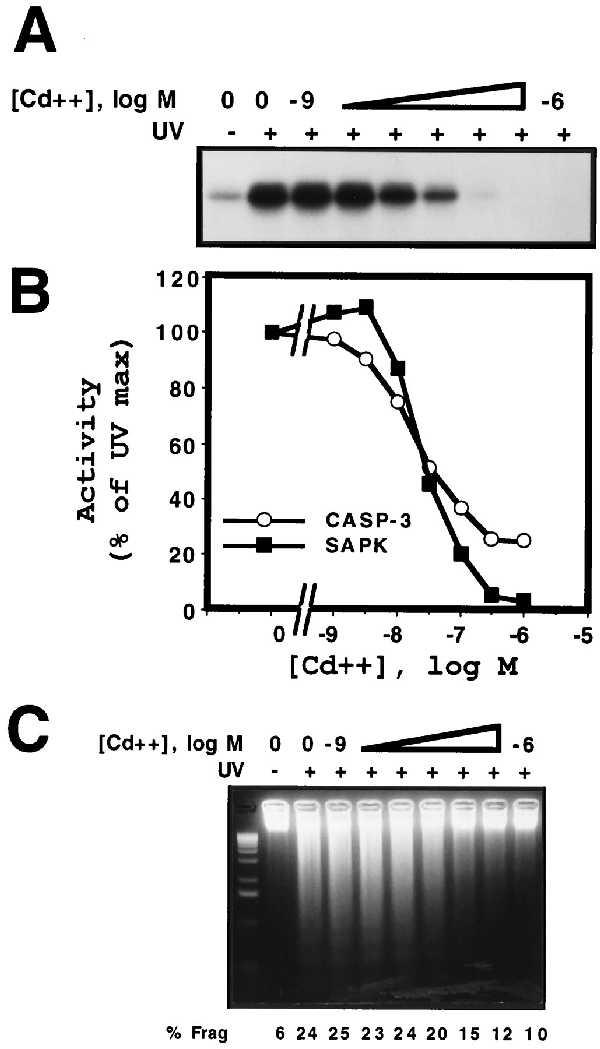
MKP-1 cytoprotection against UV-induced apoptosis correlates with inhibition of SAPK activity. (A) U937 cells expressing hMTIIa-MKP-1 were pretreated with the indicated concentrations of CdSO4 for 4 h before exposure to UV radiation (200 J/m2) and after 30 min whole-cell lysates were prepared. SAPK activity was determined as described (28). (B) Cells were treated as above and caspase-3 activity (○) was determined 8 h postirradiation and is plotted together with SAPK activity (▪) as determined in A above. (C) Cells were treated as described above and DNA fragmentation was analyzed 8 h postirradiation as described in Fig. 2. DNA fragmentation was also quantified by FACS analysis and is reported under each lane (% frag).
UV-Induced Apoptosis in U937 Cells Is Not Mediated by p38 MAPK.
Although the cytoprotective effects of MKP-1 correlate in a dose-dependent manner with the inhibition of UV-induced SAPK activity, we directly examined whether p38 MAPK may also play a role in mediating UV-induced apoptosis by using the highly selective p38 MAPK inhibitor, SB203580 (42, 43). Parental U937 cells were pretreated with 10 μM SB203580 for 1 h before exposure to UV radiation as indicated (Fig. 4). This treatment protocol has been shown to result in the functional inhibition of p38 MAPK activity in U937 cells (44). Pretreatment with SB203580 was found to have no effect on either UV-induced caspase-3 activity (Fig. 4A) or DNA fragmentation (Fig. 4B). Furthermore, the p38 MAPK inhibitor had no effect on UV-induced cleavage of the death substrate, PARP (data not shown). These findings suggest that p38 MAPK activation does not play an integral role in UV-induced apoptotic signaling in U937 cells. Thus, although MKP-1 abolished UV-induced activation of both SAPK and p38 MAPK, it is unlikely that the cytoprotective effects of MKP-1 are mediated by inhibition of UV-induced p38 MAPK activity.
Figure 4.
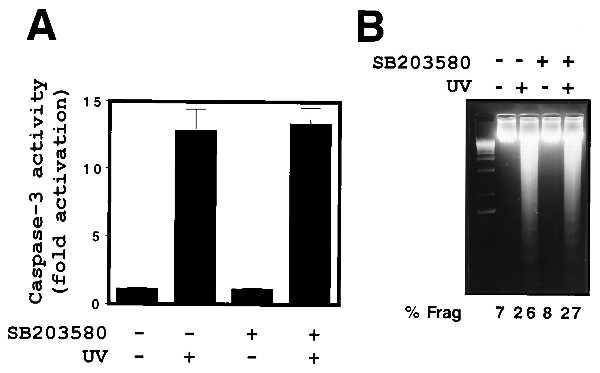
Inhibition of p38 MAPK activity has no effect on UV-induced apoptosis. Parental U937 cells were pretreated with the p38 MAPK inhibitor SB203580 (10 μM) for 1 h before exposure to UV radiation (200 J/m2) as indicated. (A) Caspase-3 activity and (B) DNA fragmentation were analyzed 8 h postirradiation as described (33). UV-induced DNA fragmentation was also quantified by FACS analysis and is shown under each lane (% frag).
Conditional Expression of PAC1 Does Not Provide Cytoprotection Against UV-Induced Apoptosis.
To determine whether cytoprotection against UV-induced apoptosis was a general characteristic of all dual-specificity phosphatases or was dependent on substrate specificity toward SAPK, similar studies were performed with the dual-specificity phosphatase, PAC1 (29, 30). PAC1 has been shown to inactivate both ERK2 and p38 MAPK, but not SAPK, in transient transfection studies (22, 30). The ability of PAC1 to inhibit UV-induced apoptosis was examined by establishing an inducible hMTIIa-PAC1 U937 cell line. Treatment of the hMTIIa-PAC1 U937 cell line with CdSO4 for 4 h resulted in a potent induction of PAC1 protein expression (Fig. 5A). In a manner identical to MKP-1 (28), conditional expression of PAC1 inhibited PMA-induced ERK2 phosphorylation (Fig. 5B Upper). However, unlike MKP-1, conditional expression of PAC1 had no effect on UV-induced SAPK activity (Fig. 5B Lower). This substrate specificity for PAC1 is consistent with previous transient transfection studies (22) and demonstrates that although conditional expression of PAC1 was functionally active, it exhibited no activity toward UV-induced SAPK activity in U937 cells. We have proposed that the anti-apoptotic effects of MKP-1 are mediated by the inhibition of UV-induced SAPK activity. We therefore examined the effect of conditionally expressing PAC1 on UV-induced DNA fragmentation. Conditional expression of PAC1 had no effect on UV-induced DNA fragmentation (Fig. 5C). These findings indicate that, unlike MKP-1, the dual-specificity phosphatase PAC1 does not provide cytoprotection against UV-induced U937 cell apoptosis.
Figure 5.
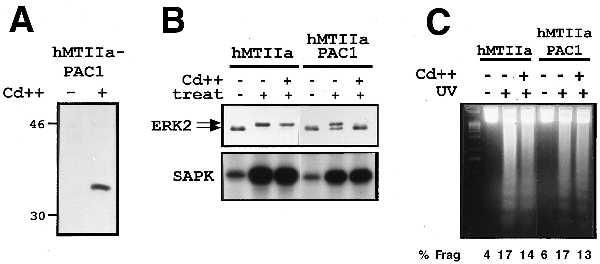
Conditional expression of PAC1 does not provide cytoprotection against UV-induced apoptosis. (A) U937 cells were transfected with hMTIIa-PAC1 and a stable cell line derived by selection in the presence of hygromycin B. Cells were treated for 4 h with 1 μM CdSO4 and PAC1 expression determined by Western blot analysis. (B) Cells were pretreated with 1 μM CdSO4 and then treated with either PMA (200 nM) (Upper) or UV radiation (200 J/m2) (Lower). After 30 min, cell extracts were analyzed for ERK2 phosphorylation (Upper), or SAPK activity (Lower) as described in Materials and Methods. (C) Cells were pretreated with 1 μM CdSO4 for 4 h and exposed to UV irradiation (200 J/m2) as indicated. DNA fragmentation was analyzed 8 h postirradiation as described (33).
DISCUSSION
Our results demonstrate that conditional expression of MKP-1 is capable of providing cytoprotection against UV-induced apoptosis. Furthermore, MKP-1 was found to act at a site upstream of caspase-3 activation, which is thought to be the commitment step in the execution phase of the apoptotic pathway (36). This indicates that a member of the dual-specificity phosphatase family plays a role in mediating cell survival by attenuating signaling through the SAPK pathway(s). Whereas both p38 MAPK and SAPK have been implicated in mediating stress-induced apoptosis (1, 7), UV irradiation is a more potent activator of SAPK than p38 MAPK activity in U937 cells. A similar preferential activation of SAPK by UV irradiation has been reported in Jurkat T cells (8), 293T cells (8) and a panel of small cell lung cancer cell lines (11). Furthermore, our inability to block UV-induced apoptosis with the p38 MAPK inhibitor, SB203580, and the dose-dependent correlation between MKP-1-mediated inhibition of SAPK activity and apoptosis, suggest that the molecular mechanism mediating the cytoprotective effects of MKP-1 involves the inhibition of SAPK activity, and not p38 MAPK activity. Given the broad substrate specificity of MKP-1, however, it is possible that the cytoprotection provided by MKP-1 results from the inhibition of both SAPK and p38 MAPK. However, such a mechanism is not supported by previous studies, in which selective inhibition of SAPK activation using dominant-inhibitory mutants of SAPK or its upstream activators, SEK1 or MEKK1, is sufficient to block UV-induced apoptosis in a variety of cell types (8–11). Thus, while we have previously demonstrated that MKP-1 exhibits a substrate specificity of p38 MAPK>SAPK, the anti-apoptotic properties of MKP-1 with regard to UV-induced apoptosis in U937 cells correlate with inhibition of SAPK activity. Our findings do not rule out the possibility that additional, perhaps more relevant, MKP-1 substrates may play a role in mediating the cytoprotective effects of MKP-1.
Unlike MKP-1, conditional expression of PAC1 did not provide cytoprotection against UV-induced apoptosis. This finding demonstrates that the survival advantage provided by MKP-1 is not a universal characteristic of all dual-specificity phosphatases. Various members of the MKP family of dual-specificity phosphatases have been shown to exhibit distinct substrate specificities for ERK, SAPK, and p38 MAPK (22). Consistent with previous reports (22, 30), our studies demonstrate that conditional expression of PAC1 abolishes phorbol 12-myristate 13-acetate-induced activation of ERK2, while having no activity toward UV-induced SAPK activity in U937 cells. The differential substrate specificities of PAC1 and MKP-1 toward SAPK, provides further evidence that the molecular mechanism mediating the anti-apoptotic effects of MKP-1 involves the specific inhibition of UV-induced SAPK activity. It remains to be determined whether other dual-specificity phosphatases that are capable of inactivating SAPK, such as MKP-2 (22, 45) or M3/6 (46), may play a similar cytoprotective role.
Although we have demonstrated that ectopic expression of MKP-1 is capable of inhibiting UV-induced apoptosis, it remains to be established whether MKP-1 plays a physiological cytoprotective role by attenuating signaling through the SAPK pathway. MKP-1 is an immediate early gene product that is potently induced in response to both mitogenic and stress stimuli (28, 47–52). We and others have shown that MKP-1 expression is mediated predominantly by ERK activation (23, 28, 52). Xia et al. (7) have proposed that the coordinate regulation of both growth factor-induced ERK and stress-induced SAPK or p38 MAPK activity ultimately determines whether a cell undergoes apoptosis. Thus, ERK-mediated induction of MKP-1 may play an important role in attenuating signaling through the SAPK pathway and promoting cell survival.
Recent evidence suggests that the duration of SAPK activation may also play a key role in determining cell fate. Whereas mitogenic stimulation of Jurkat T cells leads to a transient activation of SAPK, apoptotic stimuli lead to the persistent activation of SAPK (8). Furthermore, inhibition of tyrosine phosphatase activity in Jurkat T cells by cotreatment with sodium orthovanadate prolongs SAPK activation in response to mitogenic stimuli and induces apoptosis, rather than proliferation (8). Thus, not only does the duration of SAPK activity appear to be an important determinant directing either proliferation or apoptosis, this effect may be regulated by the coordinate activity of a dual-specificity phosphatase (8).
Recent studies on prostate carcinogenesis have also demonstrated a correlation between MKP-1 expression and apoptosis in vivo (53). MKP-1 was found to be overexpressed in the preinvasive stage of prostate cancer (53). Consistent with the studies described here, MKP-1 expression was found to be inversely related to SAPK activity and apoptosis (53). Furthermore, treatment by androgen ablation caused a reduction in MKP-1 expression and an increase in SAPK activity, which correlated with the induction of apoptosis, whereas a subpopulation that did not undergo androgen ablation-induced apoptosis maintained high levels of MKP-1 expression (53). These findings support a role for MKP-1 in regulating apoptosis in vivo. In aggregate, these findings along with the results presented here suggest that MKP-1 may play a physiological role in enhancing cell survival by attenuating signaling through the SAPK pathway.
Acknowledgments
We thank N. Tonks for the MKP-1(myc) plasmid and 9E10 antibody, K. Kelly for the pMT2T-PAC1 plasmid, M. Green for the GST-ATF2(1–109), J. Cohen for use of the Cytofluor fluorimeter, and C. Lamb for help with the caspase assays. This work was supported by American Cancer Society Grant IRG-5–37 (to C.C.F.), University of Colorado Cancer Center Wines for Life Seed Grant (to C.C.F.), and National Institutes of Health Grants DK44741 and CA42533 (to A.S.K.)
ABBREVIATIONS
- JNK
c-Jun N-terminal protein kinase
- SAPK
stress-activated protein kinase (JNK)
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- hMTIIa
human metallothionein IIa
- MKP-1
MAPK phosphatase-1
- PARP
poly(ADP ribose) polymerase
- IL
interleukin
- GST
glutathione S-transferase
- FACS
fluorescence-activated cell sorter
- PI
propodium iodide
References
- 1.Kyriakis J M, Avruch J. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 2.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 3.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. Nature (London) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 4.Derijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 5.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 6.Johnson N L, Gardner A M, Diener K M, Lange-Carter C A, Gleavy J, Jarpe M B, Minden A, Karin M, Zon L I, Johnson G L. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 7.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y-R, Wang X, Templeton D, Davis R J, Tan T-H. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 9.Verheij M, Bose R, Lin X H, Yao B, Jarvis W D, Grant S, Birrer M J, Szabo E, Zon L I, Kyriakis J M, et al. Nature (London) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 10.Zanke B W, Boudreau K, Rubie E, Winnett E, Tibbles L A, Zon L, Kyriakis J, Liu F-F, Woodgett J R. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]
- 11.Butterfield L, Storey B, Maas L, Heasley L E. J Biol Chem. 1997;272:10110–10116. doi: 10.1074/jbc.272.15.10110. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z-G, Hsu H, Boeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 13.Natoli G, Costanzo A, Ianni A, Templeton D J, Woodgett J R, Balsano C, Levrero M. Science. 1997;275:200–302. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- 14.Lenczowski J M, Dominguez L, Eder A M, King L B, Zacharichuk C M, Ashwell J D. Mol Cell Biol. 1997;17:170–181. doi: 10.1128/mcb.17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juo P, Kuo C J, Reynolds S E, Konz R F, Raingeaud J, Davis R J, Biemann H-P, Blenis J. Mol Cell Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goillot E, Raingeaud J, Ranger A, Tepper R I, Davis R J, Harlow E, Sanchez I. Proc Natl Acad Sci USA. 1997;94:3302–3307. doi: 10.1073/pnas.94.7.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charles C H, Sun H, Lau L F, Tonks N K. Proc Natl Acad Sci USA. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noguchi T, Metz R, Chen L, Mattei M-G, Carrasco D, Bravo R. Mol Cell Biol. 1993;13:5195–5205. doi: 10.1128/mcb.13.9.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H, Charles C H, Lau L F, Tonks N K. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 20.Sun H, Tonks N K, Bar-Sagi D. Science. 1994;266:285–288. doi: 10.1126/science.7939666. [DOI] [PubMed] [Google Scholar]
- 21.Milne D M, Campbell L E, Campbell D G, Meek D W. J Biol Chem. 1995;270:5511–5518. doi: 10.1074/jbc.270.10.5511. [DOI] [PubMed] [Google Scholar]
- 22.Chu Y, Solski P A, Khosravi-Far R, Der C J, Kelly K. J Biol Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- 23.Brondello J-M, Brunet A, Pouyssegur J, McKenzie F R. J Biol Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- 24.Hirsch D D, Stork P J S. J Biol Chem. 1997;272:4568–4575. doi: 10.1074/jbc.272.7.4568. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S, Barrett T, Whitmarsh A J, Cavanagh J, Sluss H K, Derijard B, Davis R J. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Gorospe M, Yang C, Holbrook N J. J Biol Chem. 1995;270:8377–8380. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- 27.Franklin C C, Kraft A S. Oncogene. 1995;11:2365–2374. [PubMed] [Google Scholar]
- 28.Franklin C C, Kraft A S. J Biol Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 29.Rohan P J, Davis P, Moskaluk C A, Kearns M, Drutzsch H, Siebenlist U, Kelly K. Science. 1993;259:1763–1766. doi: 10.1126/science.7681221. [DOI] [PubMed] [Google Scholar]
- 30.Ward Y, Gupta S, Jensen P, Wartmann M, Davis R J, Kelly K. Nature (London) 1994;367:651–654. doi: 10.1038/367651a0. [DOI] [PubMed] [Google Scholar]
- 31.Franklin C C, Unlap T, Adler V, Kraft A S. Cell Growth Differ. 1993;4:377–385. [PubMed] [Google Scholar]
- 32.Franklin C C, Sanchez V, Wagner F, Woodgett J R, Kraft A S. Proc Natl Acad Sci USA. 1992;89:7247–7251. doi: 10.1073/pnas.89.15.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong W W, Kamen R, Weichselbaum R, et al. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin S J. Trends Cell Biol. 1993;3:141–144. doi: 10.1016/0962-8924(93)90128-n. [DOI] [PubMed] [Google Scholar]
- 35.Fraser A, Evan G. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 36.Chinnaiyan A M, Dixit V M. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- 37.Seimiya H, Mashima T, Toho M, Tsuruo T. J Biol Chem. 1997;272:4631–4636. doi: 10.1074/jbc.272.7.4631. [DOI] [PubMed] [Google Scholar]
- 38.Naito M, Nagashima K, Mashima T, Tsuruo T. Blood. 1997;89:2060–2066. [PubMed] [Google Scholar]
- 39.Margolin N, Raybuck S A, Wilson K P, Chen W, Fox T, Gu Y, Livingston D J. J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 40.Lohmann R D, Beyersmann D. Biochem Biophys Res Commun. 1993;190:1097–1103. doi: 10.1006/bbrc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 41.Perry D K, Smyth M J, Stennicke H R, Salvesen G S, Duriez P, Poirier G G, Hannun Y A. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 42.Lee J C, Laydon J T, McDonnell P C, Gallagher T F, Kumar S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, et al. Nature (London) 1994;372:739–745. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 43.Cuenda A, Rouse J, Doza Y N, Meier R, Cohen P, Gallagher T F, Young P R, Lee J C. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 44.Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee J C, Haegeman G, Cohen P, Fiers W. EMBO J. 1996;15:1914–1923. [PMC free article] [PubMed] [Google Scholar]
- 45.King A G, Ozanne B W, Smythe C, Ashworth A. Oncogene. 1995;11:2553–2563. [PubMed] [Google Scholar]
- 46.Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. J Biol Chem. 1996;271:27205–27308. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- 47.Bokemeyer D, Sorokin A, Yan M, Ahn N G, Templeton D J, Dunn M J. J Biol Chem. 1996;271:639–642. doi: 10.1074/jbc.271.2.639. [DOI] [PubMed] [Google Scholar]
- 48.Misra-Press A, Rim C S, Yao H, Roberson M S, Stork P J S. J Biol Chem. 1995;270:14587–14596. doi: 10.1074/jbc.270.24.14587. [DOI] [PubMed] [Google Scholar]
- 49.Keyse S M, Emslie E A. Nature (London) 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 50.Kwak S P, Hakes D J, Martell K J, Dixon J E. J Biol Chem. 1994;269:3596–3604. [PubMed] [Google Scholar]
- 51.Charles C H, Abler A S, Lau L F. Oncogene. 1992;7:187–190. [PubMed] [Google Scholar]
- 52.Beltman J, McCormick F, Cook S J. J Biol Chem. 1996;271:27018–27024. doi: 10.1074/jbc.271.43.27018. [DOI] [PubMed] [Google Scholar]
- 53.Magi-Galluzzi C, Mishra R, Fiorentino M, Montironi R, Yao H, Capodieci P, Wishnow K, Kaplan I, Stork P J S, Loda M. Lab Invest. 1997;76:37–51. [PubMed] [Google Scholar]