

Abstract
Axin antagonizes the developmental effects of Wnt in vertebrates. We show here that Axin simultaneously binds two components of the Wnt pathway, β-catenin and its negative regulator glycogen synthase kinase-3β. In mammalian cells, Axin inhibits Wnt-1 stimulation of β-catenin/lymphoid enhancer factor 1-dependent transcription. Axin also blocks β-catenin-mediated transcription in colon cancer cells that have a mutation in the adenomatous polyposis coli gene. These findings suggest that Axin, by forming a complex with β-catenin and glycogen synthase kinase-3β, can block signaling stimulated by Wnt or by adenomatous polyposis coli mutations.
The Wnt/wingless family of secreted molecules regulates a variety of developmental processes in invertebrates and vertebrates (1, 2). During Drosophila development, wingless is required for patterning of the embryonic segments (3). In Xenopus embryos, Wnt induces a duplication of the embryonic axis if it is overexpressed at the dorsal side of the blastomere (4). One of the downstream effectors of the Wnt pathway is the armadillo gene product, the Drosophila homologue of β-catenin, which is posttranscriptionally regulated by wingless (3, 5). Mutations in armadillo gene mimic the developmental effects of mutations in wingless on segment polarity (3). In Xenopus, overexpression of β-catenin reproduces axis duplication induced by Wnt overexpression (6).
Genetic experiments in Drosophila have shown that β-catenin is regulated negatively by zeste-white-3, the Drosophila homologue of glycogen synthase kinase 3β (GSK-3β) (7). Although it has been proposed that β-catenin and GSK-3β interact directly, this physical interaction has never been demonstrated directly. Recent experiments in mammalian cells suggested that GSK-3β may phosphorylate the adenomatous polyposis coli (APC) tumor suppressor protein, which, in its hyperphosphorylated state, has a higher affinity for β-catenin. Once bound to APC, β-catenin is targeted for rapid degradation (8). In cell lines that have high levels of cytoplasmic β-catenin due to APC mutations, a complex has been found including GSK-3β, β-catenin, and mutated APC (8). GSK-3β or other kinases also may destabilize β-catenin by phosphorylation of sites on its amino terminus. Directed mutagenesis of the amino-terminal phosphorylation sites causes increased stability of β-catenin (9).
Wnt, β-catenin, and APC have been implicated in carcinogenesis. The Wnt-1 gene first was identified as a proto-oncogene that was activated in mouse mammary tumor tissues by retroviral insertion (10). In addition, overexpression of Wnt genes causes transformation of the mammary epithelial cell lines C57MG and RAC311 (11). Mutations in APC that increase the level of β-catenin protein have been found with high frequency in sporadic and hereditary colorectal cancer (12–14). Mutations in the β-catenin gene that directly stabilize the protein also have been associated with human colon cancer and melanoma cell lines (15–17). Although the mechanism by which Wnt and β-catenin enhance tumorigenesis is not known, it is likely that an effect of these proteins on transcription is important. Recently, β-catenin was shown to bind the HMG (high mobility group) box transcription factor Lef-1/Tcf (lymphocyte enhancer binding factor/T cell factor) and to enter the nucleus to regulate Lef-1-dependent transcription (18, 19). In cell lines containing either stabilizing mutations in β-catenin or mutations in APC that increase β-catenin stability, β-catenin/Tcf transcriptional activity is constitutively high (16, 17). Lef-1 overexpression in Xenopus also mimics Wnt overexpression by inducing axis duplication, and this activity is enhanced greatly by coexpression of β-catenin. In this paper, we propose that Axin, a molecule involved in embryonic axis formation of vertebrates, is a bridging molecule between GSK-3β and β-catenin and acts as a negative regulator of the Wnt pathway by inhibiting β-catenin-mediated Lef-1 activation.
MATERIALS AND METHODS
Materials.
Anti-human c-myc antibody was obtained from PharMingen. Anti-GSK-3β antibody was from Transduction Laboratories (Lexington, KY). Anti-β-catenin mAb was obtained from Zymed. pCG-Lef-1 and pGL3-fos-7LEF-luciferase were generous gifts from R. Grosschedl (University of California, San Francisco). Mouse Wnt-1 gene was a gift from I. Taylor (National Institutes of Health, Bethesda, MD). pTK–β-galactosidase (β-gal) vector was obtained from Promega. Luciferase activity was measured by using a luciferase assay system from Promega, and the β-gal assay was done by using the β-gal assay system II from CLONTECH.
Cell Culture, Transfection, and Immunological Procedures.
SW480 cell lines were obtained from American Tissue Culture Collection and grown in DMEM with 10% fetal calf serum; 293 cells were grown in DMEM with 10% fetal calf serum. All of the transfections were performed by Lipofectamine (GIBCO). To express in mammalian cells, Axin cDNAs were subcloned into pcDNA3.1 vector (Invitrogen). Cells were washed with PBS and lysed in lysis buffer (20 mM Tris⋅HCl, pH 7.5/1 mM EDTA/0.1% Triton X-100/0.15 mM NaCl/1 mM phenylmethylsulfonyl fluoride/10 μg/ml each of aprotinin and leupeptin). For immunoprecipitation, cell lysates were incubated with various antibodies for 4 h at 4°C, then added Dynabeads bound sheep Ig anti-mouse IgG1 (Dynal, Great Neck, NY) for 2 h. Enhance chemiluminescence reagents (Amersham) were used for detection of the immunoblots.
In Vitro Binding.
β-catenin was immunoprecipitated from Chinese hamster ovary cell lysate, immobilized to Dynabeads bound sheep Ig anti-mouse IgG1, and washed extensively with lysis buffer (20 mM Tris⋅HCl, pH 7.5/1 mM EDTA/0.1% Triton X-100/0.5 M NaCl/1 mM phenylmethylsulfonyl fluoride). 35S-labeled Axin was in vitro-transcribed and -translated in rabbit reticulocyte lysate (TNT system, Promega). Partially purified GSK-3β was obtained from Upstate Biotechnology (Lake Placid, NY). Combinations of the three proteins were mixed in binding buffer (20 mM Tris⋅HCl, pH 7.5/1 mM EDTA/0.1% Triton X-100/0.3 M NaCl/1 mM phenylmethylsulfonyl fluoride/10 μg/ml each of aprotinin and leupeptin) and incubated at 4°C for 4 h. Beads were collected and washed extensively with binding buffer, and protein complexes bound to the beads were analyzed by Western blotting (for β-catenin and GSK-3β) or autoradiograph (for 35S-labeled Axin).
Reporter Gene Assay.
For the reporter gene assay, 293 cells were seeded at 2 × 105 cells/well in 12-well culture plates. SW480 cells were seeded at 1 × 105 cells/well in 12-well plates. Cells were transfected with 0.2 μg of the luciferase reporter gene, 0.02 μg of Lef-1, 0.03 μg of pTK-β-gal as an internal control, the indicated amount of Axin cDNA or Wnt-1 cDNA, and pcDNA3.1 vector to a total amount of 0.4 μg of plasmids. Transfection was performed by Lipofectamine. Luciferase and β-gal activities were measured 48 h after transfection. Relative light units (RLU) were measured with a luminometer (Monolight 2010, Analytical Luminescence Laboratory, San Diego).
RESULTS AND DISCUSSION
To understand the mechanism of action of GSK-3β and β-catenin, we studied their ability to interact with other proteins. We used GSK-3β as bait in a yeast two-hybrid screen (20). After screening 2 × 106 independent cDNA clones in a murine T cell library, we obtained five positive clones. Four of those clones encoded proteins that lacked significant homology to known proteins. One clone appeared to be a partial cDNA (encoding amino acids 289–956) of Axin (21), a protein recently described as the gene product of the fused locus in mice. Mice that are homozygous for fused have axis duplications, a phenotype similar to that observed in frogs that overexpress the Wnt protein. Recently, Zeng et al. (21) showed that expression of Axin in Xenopus embryos could inhibit secondary axis formation induced by overexpression of Wnt or dominant-negative GSK-3β.
We studied the interaction of Axin and GSK-3β in mammalian cells. We used a Myc epitope-tagged, full length Axin (FL-Axin, amino acids 126–956 of form 1 transcript) and an amino terminus-truncated Axin (ΔN-Axin, amino acids 289–956) that corresponds to the clone we isolated from the yeast two-hybrid screen as shown in Fig. 1. Both forms of Axin bound to endogenous GSK-3β in SW480 cells (Fig. 2). Axin coprecipitated with either the wild-type or kinase-deficient form (KK → MI) (22) of GSK-3β (data not shown).
Figure 1.
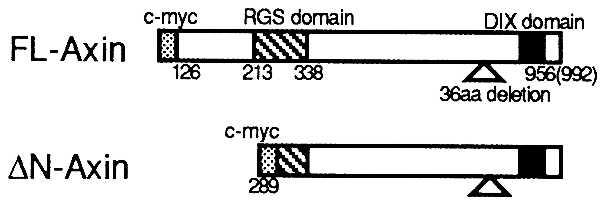
Axin cDNAs used in this study. Amino acid (aa) sequence position is based on previously published data (21). FL-Axin represent the longest ORF in form 1 starting from the amino acid position 125 (first methionine). Form 1 transcript lacks a 36-amino acid segment that was found in form 2. Amino terminus-deleted Axin (ΔN-Axin) was constructed by digesting with Asp1, corresponding to amino acid position 289.
Figure 2.
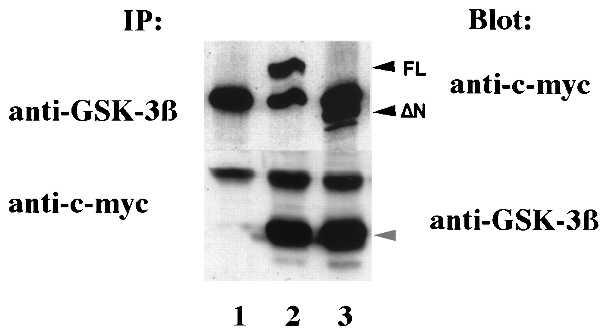
Interaction of Axin and GSK-3β in vivo. SW480 cells were transfected with pcDNA3.1 vector alone or the same vector containing FL-Axin or ΔN-Axin (7 μg each, lanes 1, 2, and 3 respectively). Cell lysates were immunoprecipitated and analyzed by Western blotting as indicated. Similar results were obtained from other cell lines, including 293 cells, Chinese hamster ovary cells, and COS7 cells.
Previously, it had been shown that GSK-3β binds to β-catenin in cell lines that contain high levels of β-catenin and mutated APC (8). However, GSK-3β immune complexes did not contain β-catenin in 293 cells that have wild-type APC (ref. 8; Fig. 3). It was surprising to find that, when either FL-Axin or ΔN-Axin was overexpressed, β-catenin formed a stable complex with GSK-3β in the presence of wild-type APC (Fig. 3). The expression level of GSK-3β protein was not changed by transfection of Axin (data not shown). These data suggested that Axin simultaneously binds to GSK-3β and β-catenin.
Figure 3.
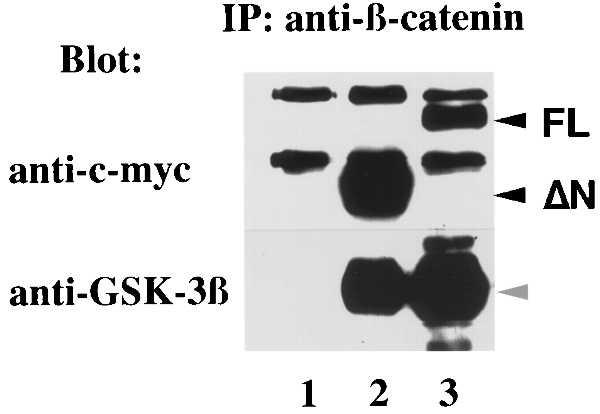
β-catenin immune complexes: 293 cells were transfected with pcDNA3.1 vector, ΔN-Axin, or FL-Axin (lanes 1, 2, or 3, respectively). Cell lysates were immunoprecipitated with β-catenin antibody and analyzed by immunoblot with antibody to c-myc or GSK-3β as shown.
To confirm the direct interaction of Axin, β-catenin, and GSK-3β, we translated and radiolabeled Axin in vitro. When immobilized β-catenin was mixed with the in vitro-translated Axin, it bound Axin directly (Fig. 4, lane 4). Immobilized β-catenin failed to bind purified GSK-3β (Fig. 4, lane 2); however the β-catenin/Axin complex did bind GSK-3β (Fig. 4, lane 3). This experiment showed the formation of a ternary complex including Axin, β-catenin, and GSK-3β. In the context of our other data, the findings suggested that β-catenin and GSK-3β can each interact with Axin.
Figure 4.
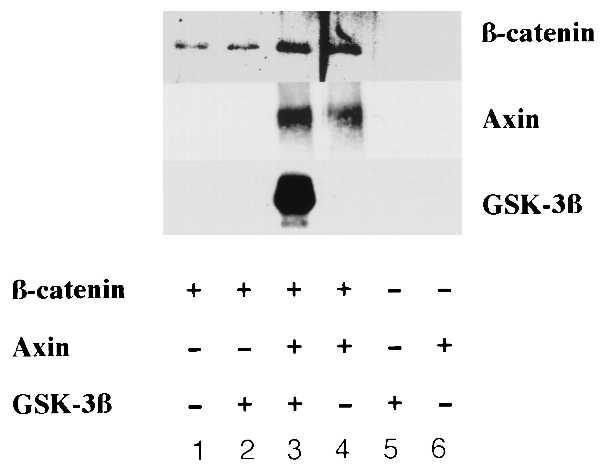
In vitro binding of β-catenin, Axin, and GSK-3β. Three proteins were mixed as indicated. The rabbit reticulocyte lysates were added in lanes 1, 2, and 5. There were Dynabeads and anti-β-catenin antibody alone in lanes 5 and 6.
Next, we examined the effect of Axin on the Wnt pathway in mammalian cells by using a luciferase reporter construct containing multiple Lef-1 binding DNA sequences and a minimal c-fos promoter. The HMG (high mobility group) box transcription factor Lef-1/Tcf has been shown to bind β-catenin and strongly activate this reporter gene. Studies of the Wnt pathway in mammalian culture cells have been limited by the lack of a convenient measurement of Wnt-induced signals and have been confined to the cell lines that exhibited a morphological change in response to Wnt (2, 11). However, we were able to measure the activation of Lef-1 reporter constructs by expression of Wnt-1 in several mammalian cell lines including 293 cells. In these cells, Wnt-1 activates Lef-1 reporter gene transcription four- to sixfold when compared with mock-transfected cells (Fig. 5A). Both FL-Axin and ΔN-Axin inhibited the effects of Wnt-1 on the Lef-1 reporter. Neither FL-Axin nor ΔN-Axin affected Lef-1 reporter activity in the absence of Wnt. To verify the effect of Axin on β-catenin-mediated transcription, we examined SW480 cells that contain a mutation of APC that is known to stabilize β-catenin in absence of Wnt signal (23). We found that expression of FL-Axin and ΔN-Axin inhibited the Lef-1 reporter gene transcript in SW480 cells (Fig. 5B). This suggests that Axin inhibits the Wnt pathway by inhibiting β-catenin function either at the level of β-catenin or downstream of β-catenin. Previously, Zeng et al. (21) showed that the effects of overexpressed β-catenin in Xenopus embryos could not be blocked by Axin. Although our findings appear to be different in mammalian cells, it is possible that, in the Xenopus injection experiment, a higher level of β-catenin overexpression was achieved and this might have overcome the effects of Axin. We could not assess accurately whether Axin expression changes β-catenin levels because only a small fraction of the cells was transfected.
Figure 5.
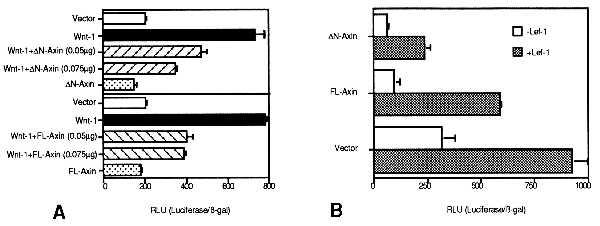
Inhibition of Lef-1 reporter activity by Axin. The assays were performed as duplicate transfections. Representative data from three independent experiments are shown. Luciferase activity was normalized to transfection efficiency by using β-gal activity. There were no significant differences in β-gal activity in each transfection. Luciferase and β-gal activity were measured 48 h after transfection. (Bars = SEM.) (A) 293 cells were seeded and transfected in 12-well culture plates. The Wnt-1 gene (0.075 μg) was transfected. (B) SW480 cells were seeded and transfected in 12-well culture plates. The inhibition of the Lef-1 reporter gene was constantly higher in ΔN-Axin-transfected cells than in FL-Axin-transfected cells. This may depend on the difference of protein expression level (Fig. 2).
We initially cloned Axin as a molecule that binds to GSK-3β. We demonstrated that Axin can bind to GSK-3β and β-catenin simultaneously and that it forms a stable ternary complex. Accordingly, Axin dramatically enhanced the coprecipitation of β-catenin and GSK-3β. These findings suggest that Axin may function as a bridge between β-catenin and its negative regulator, GSK-3β. It is likely that APC is also in this complex because we could detect APC protein in Axin immune complexes from SW480 cell lysates (data not shown). Overexpression of Axin inhibited the activation of β-catenin/Lef-1-dependent transcription by Wnt. We also demonstrated that Axin was able to inhibit β-catenin/Lef-1-dependent transcription in human cancer cell lines that overexpress β-catenin because of an APC mutation. Therefore Axin, blocks β-catenin-induced transcription that results either from APC mutations or by Wnt signaling. Our findings clarify the step at which Axin inhibits the Wnt pathway, suggesting that the main effect of Axin is to inhibit the function of β-catenin. This regulation may involve the recruitment of GSK-3β into a complex with β-catenin. Further studies will be required to show conclusively that the effect of Axin on β-catenin function requires GSK-3β. Our findings also predict that mutations in Axin might stabilize β-catenin and should therefore mimic mutations in APC. It will be important to explore further the role of Axin in cell transformation, in the Wnt pathway, and in the regulation of both β-catenin and GSK-3β-mediated signals.
Acknowledgments
We thank Drs. R. Grosschedl and S. Hsu for the Lef-1 reporter assay, Drs. T. Sun and S. Harrison for their helpful discussions, Drs. L. Molz, K. Ramer, and T. Quinn for their review comments on the manuscript, and Ms. B. Cheung for her assistance with the preparation of this manuscript. This work was supported partially by funds from the National Institutes of Health/National Heart, Lung, and Blood Institute/Program of Excellence PO HL43821 and by an unrestricted award from the Howard Hughes Medical Institute.
ABBREVIATIONS
- GSK-3β
glycogen synthase kinase 3β
- APC
adenomatous polyposis coli
- Lef-1
lymphoid enhancer factor 1
- β-gal
β-galactosidase
References
- 1.McMahon A P. Trends Genet. 1992;8:236–242. [Google Scholar]
- 2.Nusse R, Varmus H E. Cell. 1992;69:1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 3.Wieschaus E, Riggleman R. Cell. 1987;49:177–184. doi: 10.1016/0092-8674(87)90558-7. [DOI] [PubMed] [Google Scholar]
- 4.McMahon A P, Moon R T. Cell. 1989;58:1075–1084. doi: 10.1016/0092-8674(89)90506-0. [DOI] [PubMed] [Google Scholar]
- 5.Van Den Heuvel M, Nusse R, Johnsyon P, Lawrence P A. Cell. 1989;59:739–749. doi: 10.1016/0092-8674(89)90020-2. [DOI] [PubMed] [Google Scholar]
- 6.Funayama N, Fagotto F, McCrea P D, Gumbiner B M. J Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peifer M, Sweeton D, Casey M, Wieschaus E. Development. 1994;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- 8.Rubinfeld R, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 9.Yost C, Torres M, Miller R R, Huang E, Kimmelman D, Moon R T. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 10.Nusse R, van Ooyen A, Cox D, Fung Y K, Varmus H E. Nature (London) 1984;307:131–136. doi: 10.1038/307131a0. [DOI] [PubMed] [Google Scholar]
- 11.Brown A M C, Wildin R A, Prendergast T J, Varmus H E. Cell. 1986;46:1001–1009. doi: 10.1016/0092-8674(86)90699-9. [DOI] [PubMed] [Google Scholar]
- 12.Bodmer W, Bailey C, Bodmer J, Bussey H, Ellis A, Gorman P, Lucibell F, Murday V, Rider S, Scambler P. Nature (London) 1987;328:614–616. doi: 10.1038/328614a0. [DOI] [PubMed] [Google Scholar]
- 13.Kinzler K W, Nilbert M C, Su L-K, Vogelstein B, Bryan T M, Levy D B, Smith K J, Preisinger A C, Hedge P, McKechnie D, et al. Science. 1991;253:661–664. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 14.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P, et al. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 15.Korinek V, Barker N, Morin P J, van Wichen D, deWeger R, Kinzler K W, Vogelstein B. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 16.Morin P J, Sparks A B, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler K W. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 17.Rubinfeld B, Robbins P, ElGamil M, Albert I, Porfiri E, Polakis P. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 18.Behrens J, Von Kries J P, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Nature (London) 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 19.Molenaar M, Van de Wetering M, Oosterwegel M, Petersonmaduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 20.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 21.Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek T J, Perry W L, Lee J J, Tilghman S M, Gumbiner B M, Constantini F. Cell. 1997;90:181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 22.He X, Saintjeannet J P, Woodgett J, Varmus H E, Dawid I B. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- 23.Smith K J, Johnson K A, Bryan T M, Hill D E, Markowitz S, Willson J K, Paraskeva C, Petersen G M, Hamilton S R, Vogelstein B, et al. Proc Natl Acad Sci USA. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]