

Abstract
The yeast mating response is one of the best understood heterotrimeric G protein signaling pathways. Yet, most descriptions of this system have been qualitative. We have quantitatively characterized the heterotrimeric G protein cycle in yeast based on direct in vivo measurements. We used fluorescence resonance energy transfer to monitor the association state of cyan fluorescent protein (CFP)-Gα and Gβγ-yellow fluorescent protein (YFP), and we found that receptor-mediated G protein activation produced a loss of fluorescence resonance energy transfer. Quantitative time course and dose–response data were obtained for both wild-type and mutant cells possessing an altered pheromone response. These results paint a quantitative portrait of how regulators such as Sst2p and the C-terminal tail of α-factor receptor modulate the kinetics and sensitivity of G protein signaling. We have explored critical features of the dynamics including the rapid rise and subsequent decline of active G proteins during the early response, and the relationship between the G protein activation dose–response curve and the downstream dose–response curves for cell-cycle arrest and transcriptional induction. Fitting the data to a mathematical model produced estimates of the in vivo rates of heterotrimeric G protein activation and deactivation in yeast.
Living organisms detect and respond to a variety of environmental cues through heterotrimeric G protein signal transduction pathways. Genetic or pharmacologic perturbation of these systems in humans can have significant medical consequences (1). Many pharmaceutical agents are directed against components of the G protein activation/deactivation cycle (1). Achieving a quantitative understanding of this cycle and of the relationship between G protein activation and downstream physiology can aid in drug development.
One of the best studied heterotrimeric G protein signaling systems is the yeast mating response in Saccharomyces cerevisiae (2, 3). Haploid yeast cells respond to a peptide pheromone from their partner by undergoing a series of events (e.g., cell-cycle arrest, synthesis of new proteins) in preparation for mating. In a cells, the pheromone α-factor, secreted by α cells, activates the G protein coupled α-factor receptor (Ste2p). Most of the components of this pathway have been identified, including the receptors Ste2p and Ste3p (a-factor receptor), the G protein subunits Gpa1p (Gα), Ste4p (Gβ), and Ste18p (Gγ), and the G protein regulator (RGS) Sst2p.
Yeast geneticists have isolated mutants possessing altered sensitivity to the pheromone α-factor (4). In particular, the following all confer an α-factor supersensitive phenotype: the deletion of BAR1 (bar1Δ), which encodes a secreted protease that degrades α-factor, the deletion of SST2 (sst2Δ), and the removal of the C-terminal tail of α-factor receptor STE2 (ste2300Δ). It is important to provide a quantitative description of how these mutations affect the G protein cycle to complement the existing qualitative understanding.
Until recently, the dynamics of heterotrimeric G protein activation have been primarily studied through in vitro systems (5), or through indirect in vivo readouts such as changes in cell membrane electrochemical properties (6). Devreotes and colleagues (7) used fluorescence resonance energy transfer (FRET) between the α-subunit and the βγ-subunits to measure directly in vivo G protein activation in Dictyostelium discoideum cells. Inactive heterotrimer produced a specific FRET signal, and the addition of the stimulating ligand adenosine 3′,5′-cyclic monophosphate (cAMP) led to a dose-dependent loss of FRET.
In this article, we performed a quantitative investigation of the in vivo dynamics and regulation of G protein activation in both wild-type yeast haploid a cells and mutant cells containing α-factor supersensitivity mutations. We constructed strains possessing genomic copies of the FRET pairs CFP-GPA1 (Gα) and STE18-YFP (Gγ), which replaced their cognate genes. We observed the dose-dependent loss of FRET on α-factor addition, which was quantitated by fluorometer. The kinetics and dose response of G protein activation were measured and compared with the pheromone responsiveness of two downstream events, cell-cycle arrest, and transcriptional activation of pheromone-inducible genes. Finally, we fit the data to a mathematical model that furnishes a detailed description of the yeast heterotrimeric G protein cycle (Fig. 1) and also enables quantitative explanations of the data.
Fig. 1.

Reaction diagram of heterotrimeric G protein cycle. The individual reactions comprising the key dynamics of heterotrimeric G proteins in yeast are represented along with the rate constants. The solid dot on the reaction arrow indicates that the reaction is catalyzed by the protein connected to the dot. The yellow circle attached to Gβγ represents YFP fused to Ste18p (Gγ) and the blue circle attached to Gα represents CFP fused to Gpa1p (Gα).
Materials and Methods
Plasmids and Strains. All yeast strains used were isogenic derivatives of W303. Genetic techniques were performed according to standard methods (8). Details on strain construction are presented in Supporting Text and Table 2, which are published as supporting information on the PNAS web site, www.pnas.org.
FRET Measurements. Exponentially growing cells were treated with α-factor for a specified period, then ≈4 OD600 units of cells were removed from the YPAD medium [yeast extract/peptone/dextrose (YPD) medium supplemented with adenine] and either (i) chilled, spun down, washed once with PBS, and resuspended in 100 μl of PBS, or (ii) fixed with ice-cold formaldehyde-PBS solution (3.7% formaldehyde in PBS) for 8 min, washed once with PBS, then resuspended in 100 μl of PBS. Formaldehyde fixation did not have a significant effect on the FRET signal; the spectra for FRET strain TMY101 were similar under the two preparation conditions. The cells were then placed into a 96-well plate and were read by using a Gemini XS SpectraMAX fluorometer. Quantification was performed on single wave-length readings with excitation at 434 nm and emissions monitored at 475 and 530 nm. For the representative spectra data presented in Fig. 2, the excitation was at 430 nm and the emissions were scanned from 466 to 600 nm.
Fig. 2.

G protein activation results in a loss of FRET. Shown is a representative fluorescence spectra of TMY101 showing FRET. Cells were excited at 430 nm, and the emission was detected by scanning from 466 to 600 nm. The control (dashed-dot line) was the combined signal from TMY102 (CFP-Gpa1p) and TMY103 (Ste18p-YFP). FRET was detected in TMY101 (solid line) as a decrease in the CFP emission signal (475 nm) and an increase in the YFP emission spectra (530 nm) relative to the control. Treating the TMY101 cells with α-factor for 1 min resulted in a loss of FRET (dashed line).
Time Course and Dose–Response FRET Experiments. In the time course experiments, 1 μM α-factor was added to cells in YPAD medium (30°C) at t = 0. At different time points, the cells were quenched by placing in the ice-cold formaldehyde-PBS solution, and single-wavelength fluorescence measurements were made at 475 and 530 nm.
In the dose–response experiments, a single time point was picked near the peak of the time course response for the relevant range of α-factor concentrations. This time point was 1 min for TMY101, 3 min for TMY111, 2 min for TMY112, and 1 min for TMY113. Different doses of α-factor were added to separate cultures, and the cells were then quenched in the formaldehyde-PBS solution after the specified time.
Pheromone Response Assays. A pheromone-responsive reporter gene (PFUS1-GFP) was constructed by using the promoter from the FUS1 gene fused to GFP on the single-copy pRS316 plasmid. This plasmid was transformed into the various strains. The cells in YPAD medium were treated with the different doses of α-factor for 2 h. Approximately 1 OD600 unit of cells was harvested, resuspended in 100 μl of PBS, placed into a 96-well plate, and read by using the Gemini fluorometer with the excitation at 470 nm and emission at 510 nm.
Cell-cycle arrest was quantitated by treating exponentially growing cells with different doses of α-factor for 4 h and then measuring the cell density (OD600) in a spectrophotometer.
Calculations to Determine Free Gβγ from FRET Data. The FRET efficiency, Ei, was calculated from the raw 475/530 ratio, ri, by using the following formula:
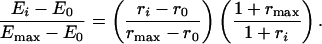 |
For TMY101, E0 = 0.3 was the FRET efficiency and r0 = 0.52 was the 475/530 ratio in the absence of α-factor. Emax = 0.18 was the FRET efficiency and rmax = 0.59 was the 475/530 ratio 30 s after the addition of 1 μM α-factor (see Supporting Text). Then, we used the following simple linear relationship to determine the fraction of free Gβγ:
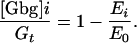 |
Model Structure and Parameter Estimation. The model represents the reactions depicted in Fig. 1 and was used to estimate the rates of G protein activation and deactivation. The units of concentration for the proteins are numbers of molecules per cell; the concentration of the ligand α-factor is in moles. The model is comprised of four ordinary differential equations (ODEs) describing the dynamics of the four state variables: free receptor (R), receptor bound to ligand (RL), inactive heterotrimeric G protein (G), and active Gα-GTP (Ga). The output is the level of free Gβγ ([Gbg] = Gt – [G]) and the input is the concentration of α-factor (L). The level of Gα-GDP (Gd) is determined by the conservation relationship [Gd] = Gt – [G] – [Ga]. The ODEs of the model are as follows: (i) d[R]/dt = –kRL [L][R] + kRLm [RL] – kRd0 [R] + kRs; (ii) d[RL]/dt = kRL [L][R] – kRLm [RL] – kRd1[RL]; (iii) d[G]/dt = –kGa [RL][G] + kG1 [Gd][Gbg]; and (iv) d[Ga]/dt = kGa [RL][G] – kGd [Ga]. Note that the rate constant for G protein deactivation kGd = kGd1 for SST2+ strains and kGd = kGd0 for the sst2Δ strain.
The model contains 10 parameters. We determined the values of seven of the parameters from direct experimental measurements and information in the literature. These include the rates of ligand-receptor association (kRL = 2·106 M–1·s–1) and dissociation (kRLm = 1·10–2 s–1), receptor synthesis (kRs = 4 molecules per cell s–1) and degradation (kRd0 = 4·10–4 s–1) with a faster rate for ligand-bound receptor (kRd1 = 4·10–3 s–1) that represents in aggregate the ligand-stimulated receptor down-regulation reactions, and G protein heterotrimerization (kG1 = 1 (molecule per cell)–1 s–1). We estimated the total number of G proteins per cell (Gt) to be ≈10,000 per cell from quantitative Western blots and fluorescence quantification.
The rates of G protein activation [kGa = 1·10–5 (molecules per cell)–1 s–1], Sst2p catalyzed G protein deactivation (kGd1 = 0.11 s–1), and uncatalyzed G protein deactivation (kGd0 = 0.004 s–1) were identified from the FRET time course and dose–response data. We selected the least-squares criterion as the error norm. An optimization procedure, the simplex algorithm, was used to find the parameter values that minimized the square error between the model simulations and the data. We did not attempt to characterize the distribution of our parameter estimates.
Results
Monitoring G Protein Activation by FRET. Janetopoulos et al. (7) demonstrated that inserting cyan fluorescent protein (CFP) into Gα1 from D. discoideum, and attaching yellow fluorescent protein (YFP) to Gβγ, produced FRET when the Gα1 was bound to Gβγ in the inactive heterotrimeric state. Examining the crystal structures of heterotrimeric G proteins (9), we observed that the N terminus of Gα is in close proximity to the C terminus of Gγ. We positioned CFP directly after the N-terminal myristoylation site (10) of Gα (CFP-Gpa1p) and we inserted YFP just in front of the prenylation consensus sequence (11) near the C terminus of Gγ (Ste18p-YFP). Both proteins were functional as demonstrated by their ability to complement their respective deletion strains.
To ensure that each cell contained a single copy of both FRET constructs under the appropriate transcriptional regulation, we replaced the genomic copies of GPA1 and STE18 with CFP-GPA1 and STE18-YFP, respectively, in a haploid a cell background containing the bar1Δ mutation (TMY101). Western blots demonstrated that the steady-state concentrations of the fluorescent fusion proteins were comparable to the levels of the wild-type proteins (data not shown). The pheromone response was also not altered significantly as measured by cell-cycle arrest (halo assay), and by the induction of PFUS1-GFP.
The reporter strain TMY101 exhibited a strong FRET signal in the absence of α-factor when measured using a fluorometer. Excitation of CFP in TMY101 resulted in a stronger YFP emission signal and weaker CFP signal compared with the combined spectra from the CFP-GPA1 strain (TMY102) and the STE18-YFP strain (TMY103), indicating the presence of FRET (Fig. 2). We estimated the FRET efficiency to be ≈30%.
The addition of α-factor resulted in a loss of the FRET signal in a dose-dependent fashion that required the presence of functional α-factor receptor. Removal of α-factor led to the recovery of the FRET signal (data not shown). These data argue against the nonspecific dimerization between CFP and YFP being responsible for the FRET signal.
We attributed the α-factor-induced loss of FRET to G protein activation that resulted in the dissociation of CFP-Gpa1p from Ste18p-YFP. We quantified the level of active G proteins by using the ratio of the emission signal at 475 nm (CFP) to the emission signal at 530 nm (YFP) after excitation at 434 nm. Increased G protein activation increased this ratio. Confocal imaging showed that the loss of FRET occurred primarily on the cell surface (data not shown).
In Vivo Kinetics of G Protein Activation. Time course experiments were performed to follow the in vivo kinetics of the formation of active G proteins. On addition of saturating concentrations of α-factor (1 μM), there was near maximal G protein activation at the first time point of 10 s (Fig. 3A). Levels of active G proteins peaked at ≈30 s, and the levels then underwent a decline that bottomed out at the 7.5-min time point. The 475/530 ratios in Fig. 3 were normalized by first subtracting the baseline ratio at t = 0, r0, to obtain the baseline adjusted ratio (ri – r0) and then dividing by the adjusted ratio at its maximum (rmax – r0), when t = 30 s, to obtain the normalized ratio (ri – r0)/(rmax – r0).
Fig. 3.

Kinetics of G protein activation. (A) Time course of G protein activation. TMY101 cells were treated with 1 μM α-factor, and the FRET emission ratio 475/530 was measured at different time points from 0 to 10 min (black). These values were normalized by subtracting the baseline ratio at t = 0, and then dividing by the peak baseline adjusted ratio at t = 30 s. In a second experiment, the protein synthesis inhibitor cycloheximide was added with α-factor at the start (red). The time course of G protein activation at later times up to 60 min is also shown (Inset). The SE from three independent measurements are represented. (B) Time course of G protein activation in sst2Δ and ste2300Δ cells. TMY111 (sst2Δ, blue) and TMY112 (ste2300Δ, red) cells were treated with 1 μM α-factor, the FRET 475/530 ratio was measured from 0 to 10 min, and the baseline was adjusted and then normalized to the ratio at the 30-s time point. The data from TMY101 are redrawn in black for comparison.
Interestingly, when monitored over a period of one hour, the 475/530 ratio continued to rise after 7.5 min (Fig. 3A Inset). We suspected that this increase reflected the polarized, pheromone-induced synthesis of receptor, Gα, and Gβγ. To test this hypothesis, the protein synthesis inhibitor cycloheximide was added along with α-factor at t = 0. The cycloheximide-treated cells exhibited the rapid activation and deactivation phases, but no recovery of the 475/530 ratio after the decline; instead the normalized ratio stayed close to zero (Fig. 3A). As further evidence, the kinetics of the α-factor stimulated synthesis of Gpa1p were measured by using a GFP-GPA1 strain. The time course of the increase in levels of GFP-Gpa1p paralleled the rise in the 475/530 ratio (data not shown).
Our hypothesis was that the decline in active G proteins was caused at least in part by ligand-stimulated receptor endocytosis. Receptor internalization is important for adaptation in many G protein systems (12). The decay in the 475/530 ratio for the cycloheximide-treated cells was fitted to an exponential with a half-time of ≈3 min (Fig. 3A, red dashed line). This value is in rough agreement with, but slightly faster than, the rate of receptor endocytosis (t0.5 = ≈4.5 min) measured by labeled α-factor internalization (13). To further investigate this issue, the wild-type α-factor receptor was replaced with a mutant version lacking the C-terminal tail beyond residue 300 so that ligand-stimulated endocytosis was severely impaired (14). G protein activation was not detrimentally affected, but G protein deactivation was dramatically curtailed in the mutant (Fig. 3B). The truncated receptor protein possesses other abnormalities, such as not being hyperphosphorylated (15), which are likely to contribute to this effect along with the endocytosis defect.
The time course of G protein activation was also monitored in an sst2Δ strain (TMY111), which was expected to exhibit slower G protein deactivation. Indeed, the decrease in the 475/530 signal was considerably more modest than for the SST2+ strain. Because receptor endocytosis was not affected in the sst2Δ strain (data not shown), we concluded that in the absence of Sst2p, the rate of G protein deactivation was sufficiently slow to be rate limiting for the reassociation of the heterotrimer. Thus, the kinetics of G protein activation were rather complex: the initial fast rise was followed by a fall that required both Sst2p and the C-terminal tail of Ste2p, and the subsequent long-term increase in the 475/530 signal depended on new protein synthesis.
Dose–Response Curves to α-Factor in Wild-Type and Mutant Cells. Understanding and manipulating dose–response curves are key challenges in pharmacology. To what extent do the kinetics of ligand-binding and the dynamics of G protein activation/deactivation determine the dose response of events further down the signaling pathway? The typical measured outputs for the yeast pheromone response are cell-cycle arrest and transcriptional activation; there are many intervening reactions including the mitogen-activated protein (MAP) kinase cascade between these processes and the G protein cycle, which could modulate the sensitivity of the response.
The dose response to α-factor was measured at four different stages in this signaling pathway: (i) receptor binding (ligand-binding assay); (ii) G protein activation (loss of FRET); (iii) transcriptional induction (PFUS1-GFP reporter gene); and (iv) cell-cycle arrest (cell density). The dose–response curves were normalized by dividing by the response to saturating levels of α-factor (1 μM). Ligand-binding and G protein activation represent the early responses to pheromone (within a few minutes). We assayed transcriptional activation 2 h after treatment and cell-cycle arrest 4 h after pheromone addition.
We also investigated the effect on these dose–response curves of three mutations possessing an α-factor supersensitive phenotype: (i) sst2Δ;(ii) ste2300Δ (deletion of the C-terminal tail beyond residue 300); and (iii) bar1Δ. It is thought that these mutations mainly exert their effect by perturbing the G protein activation/deactivation cycle by means of one of the three principal mechanisms for down-regulating G protein signaling: (i) G protein deactivation; (ii) receptor desensitization; and (iii) removal of the ligand α-factor.
TMY101 cells (STE2+ SST2+ bar1Δ) were treated with a range of α-factor concentrations from 0.1 nM to 1 μM. We measured the loss of FRET after 1 min, which was close to the peak response for the different doses. Interestingly, the four dose–response curves for receptor affinity, G protein activation, transcriptional induction, and cell-cycle arrest overlapped quite closely at ≈5 nM (Fig. 4A). One explanation is that receptor affinity is the key determinant of the dose–response behavior for this system.
Fig. 4.

Overlap of different dose–response curves in G protein signaling. (A) Pheromone dose–response behavior for TMY101 cells. α-Factor was added at different doses from 0.1 nM to 1 μM. Four different pheromone dose–response readouts were measured: (i) receptor affinity (black triangles), (ii) G protein activation (red circles), (iii) transcriptional induction of PFUS1-GFP (green squares), and (iv) cell-cycle arrest (blue diamonds). The data points from each readout were normalized to the response at 1 μM and were then fit with a Hill curve. (B) Pheromone dose–response behavior of TMY111 (sst2Δ) cells. α-Factor was added at different doses from 0.03 nM to 1 μM. The data from the four different dose–response readouts are presented as described above.
Results from the sst2Δ cells (TMY111) ruled out this hypothesis; in these cells, the receptor affinity was unchanged at 5 nM, whereas the other three curves were shifted to the left ≈20-fold to ≈0.3 nM (Fig. 4B). These data reflect the increased sensitivity to α-factor resulting from a slower rate of G protein deactivation caused by the absence of Sst2p. Thus, perturbing the dynamics of G protein activation/deactivation was sufficient to modulate the downstream dose response, independent of any changes in receptor affinity. Furthermore, these data provide support for the view that the G protein cycle is the primary site of action for Sst2p, a large protein containing multiple domains, during the early stages of the pheromone response.
The ste2300Δ cells (TMY112) also displayed increased sensitivity to α-factor as demonstrated by the shift to the left of the G protein activation, transcriptional induction, and cell-cycle arrest dose–response curves to a K0.5 of ≈0.6 nM (Table 1). The measured Kd for α-factor binding to truncated receptor was elevated to 15 nM, representing a decrease in receptor affinity. Because tail-less Ste2p is endocytosed at a slower rate (14), the steady-state numbers of receptors on the cell surface were increased from ≈10,000 molecules per cell to ≈60,000 molecules per cell. Raising the number of surface receptors may partially explain the more sensitive response. Thus, removal of the C-terminal tail of α-factor receptor had a direct effect on the G protein cycle, and once again the downstream processes were closely attuned to these changes in active G protein levels.
Table 1. Summary of pheromone dose–response behavior.
|
K0.5, nM
|
||||
|---|---|---|---|---|
| Strain | FRET | Transcriptional activation | Cell-cycle arrest | Receptor binding |
| TMY101 | 4 ± 1 | 5 ± 0.3 | 6 ± 1 | 6 ± 3 |
| sst2Δ | 0.3 ± 0.03 | 0.3 ± 0.03 | 0.2 ± 0.02 | 5 ± 1 |
| ste2300Δ | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.3 ± 0.03 | 15 ± 3 |
| BAR1+ | 7 ± 1 | 1000 ± 300 | 1500 ± 200 | ND |
The K0.5 values are shown from the dose—response curves of four strains for four outputs: G protein activation (FRET), transcriptional activation, cell-cycle arrest, and ligand binding to receptor. Also displayed are the SE for at least three independent measurements. ND, not determined.
All of the above strains were bar1Δ so that α-factor would not be degraded after administering the dose. Testing a BAR1+ strain (TMY113), we found that early G protein activation was relatively unchanged from the bar1Δ TMY101 cells, whereas the later responses were ≈100-fold less sensitive (Table 1). We reasoned that this difference arose because it takes time for the protease Bar1p to remove the α-factor. Indeed, the 475/530 ratio for BAR1+ cells treated with 100 nM α-factor steadily decreased over a period of 30 min to near its baseline pretreatment value (data not shown).
Rate Constants for G Protein Activation and Deactivation. The rates of G protein activation and deactivation were estimated by fitting the time course and dose–response data to a model of the heterotrimeric G protein cycle describing the reactions in Fig. 1. The complexity of the model was minimized so that it did not dwarf the available data, while capturing the essential dynamics of this process. The model consisted of four ordinary differential equations and 10 parameters, 7 of which were specified based on our own direct measurements as well as data reported in the literature (see Materials and Methods). The remaining three parameters, the rate of G protein activation (kGa), and the Sst2p-catalyzed (kGd1) and uncatalyzed (kGd0) rates of G protein deactivation, were estimated from the FRET data.
Changes in FRET efficiency provided a direct measure of the fraction of G proteins that were associated as inactive heterotrimers. We converted the raw 475/530 emission ratios into FRET efficiencies (see Materials and Methods), thus processing the data into a form suitable for fitting the model. As it turned out, the 475/530 ratio was roughly proportional to the level of free Gβγ.
Optimization techniques were applied to identify the parameter values for kGa and kGd that minimized the least-square error between the model simulations and the data. By using the FRET time course and dose–response data for TMY101, we obtained estimates of kGa = 1·10–5 (molecules per cell)–1 s–1 and kGd1 = ≈0.1 s–1 for the catalyzed rate of G protein deactivation when Sst2p is present at wild-type levels. The fitted model reproduced the fast rise, subsequent decline, and peak amplitude of G protein activation (Fig. 5A), as well as the K0.5 of the dose–response data (Fig. 5B). The model captured the general trends of the data, but not the finer features, which presumably would require a more detailed model.
Fig. 5.

Fitting the mathematical model to the TMY101 data. (A) Experimental data and model simulations describing the time course of G protein activation in TMY101. Two of the model parameters (kGa and kGd1) were fit to the data. The normalized 475/530 data were processed into the fraction of total G proteins that are active (•). A simulation of the fitted model in response to an input of 1 μM α-factor is presented (black line). (B) Experiments and modeling describing the G protein activation dose–response behavior of TMY101. The data (•) and simulations (black line) were normalized to the output value at 1 μM α-factor. A time point of t = 60 s was used. The error bars represent the SE from three independent measurements.
To estimate the uncatalyzed rate of G protein deactivation, we assumed that all of the parameter values were the same in the sst2Δ strain TMY111 as in TMY101, with the exception of kGd = kGd0, which was fitted to the time course and dose–response data of TMY111. The best estimate of kGd0 was 0.004 s–1. As an independent check, we measured the decay constant of the decline in G protein activation in TMY111 treated with cycloheximide to be ≈0.002 s–1 (data not shown). From the estimates of kGd0 and kGd1, we calculated that the stimulation of G protein deactivation by Sst2p was ≈25-fold, which is consistent with in vitro data in which Sst2p produced an “at least 20-fold” increase in GTP hydrolysis by Gpa1p (16). RGS proteins typically accelerate the GTPase activity of Gα by 10- to 1000-fold (17, 18).
Discussion
FRET was used to monitor the association state of the G protein heterotrimer. There were multiple pieces of evidence supporting the quantitative correlation between the decrease in the FRET signal, measured as the 475/530 ratio, and the levels of active G proteins: (i) the increase in the 475/530 ratio was dose-dependent on α-factor; (ii) the time course of the 475/530 ratio was consistent with existing indirect data on the kinetics of G protein activation and deactivation; (iii) mutations expected to perturb the G protein cycle had pronounced effects on the kinetics and dose response of the 475/530 ratio; and (iv) the dose response of the 475/530 ratio closely matched the dose response of two downstream readouts in both wild-type and mutant strains.
The dynamics of the activated G proteins were complex. After treatment with saturating concentrations of α-factor, there was an initial fast rise in the 475/530 ratio, followed by a decline, which we attributed to receptor endocytosis and other regulatory processes that depend on the C-terminal tail of Ste2p, and then a more gradual recovery that required new protein synthesis. In contrast, in Dictyostelium there was a fast rise in G protein activation, which reached a plateau without any decline over a period of 18 min (7). Consistent with this species difference is the observation that cAMP receptor continues to activate G proteins, even after phosphorylation (7).
Dose–Response Behavior and Pharmaceutical Implications. The yeast pheromone response system stretches from the G protein cycle at the start to downstream processes such as cell polarization and cell-cycle arrest. There are several pieces of evidence that argue that much of the control of this system occurs at the front end in the G protein cycle. First, mutations with the most dramatic pheromone supersensitive/resistant phenotypes affect genes involved in the G protein dynamics (4, 15, 19–21). Here, we demonstrated quantitatively that three of these mutations (sst2Δ, ste2300Δ, and bar1Δ) did indeed affect G protein activation/deactivation in a direct and substantial manner. Second, we have shown that perturbations in the sensitivity of the G protein cycle were transmitted to the downstream processes without any attenuation. Finally, many of the mating partner discrimination mutations that reduced the ability of a cells to distinguish the α cell producing the highest level of pheromone, were found in genes encoding for components of the heterotrimeric G protein cycle (22).
An interesting finding was the substantial overlap of the various dose–response curves. The K0.5 values for G protein activation, transcriptional induction, and cell-cycle arrest were approximately the same for each strain. One consequence is that there is greater coordination between upstream and downstream processes in terms of responding to the same critical range of ligand concentration. In mammalian G protein systems, the good correlation between the Kd for receptor-antagonist binding and the IC50 values for inhibiting the corresponding physiologically significant downstream process suggests that upstream and downstream dose–response curves are aligned as in yeast (23).
Many therapeutic agents are directed against G protein-coupled receptors or against proteins that modulate the levels of the ligand, such as transporters. This work presents quantitative evidence that the overall G protein cycle dynamics determines the dose response of G protein systems, not just the receptor-ligand dynamics. Thus, both RGS proteins and proteins that down-regulate receptor activity (e.g., G protein receptor kinases) should prove to be equally effective targets for future generations of drugs.
Evaluating the Rate Constants for G Protein Activation and Deactivation. We estimated the in vivo rates of G protein activation and deactivation by fitting a model of the G protein dynamics to the FRET time course and dose–response data. There have been no in vitro measurements of receptor-catalyzed G protein activation in yeast. Although Dohlman and colleagues (16) have measured the in vitro rates of both Sst2p-catalyzed and uncatalyzed GTP hydrolysis by yeast Gα, Gpa1p, it is difficult to make a direct comparison to the in vivo data (30°C), because the in vitro experiments were performed at 4°C.
A 25-fold enhancement of G protein deactivation in the presence of SST2 was observed. This finding is consistent with the approximate 20-fold stimulation of Gpa1p GTPase activity by Sst2p measured in vitro (16). It is important to note that this degree of enhancement occurred at wild-type levels of Sst2p. Data in the literature indicate that overexpressing Sst2p further accelerates G protein deactivation (19), suggesting that the rate of G protein deactivation depends on the concentration of Sst2p.
The visual phototransduction cascade is the most quantitatively characterized G protein signaling system (24). The initial rate of G protein activation was estimated to be ≈100 G proteins per second per activated rhodopsin (25), which is ≈1,000-fold faster than the yeast rate, which is 0.1 G proteins per second per activated receptor. Likewise, the rates of G protein deactivation were ≈10-fold faster than the corresponding yeast values: the GTPase activity of transducin was 1 s–1 in the presence of RGS9 and 0.02 s–1 in the absence of RGS9 (26). Future experiments are needed to determine whether the slow G protein activation kinetics in yeast are the consequence of intrinsically inefficient receptor or regulatory mechanisms designed to restrain the activation rate.
Linderman and colleagues (27) have proposed a mathematical model of G protein dynamics in human neutrophils in response to N-formyl peptide. The simple structure of our model is similar to their model. Interestingly, many of the estimates of the kinetic constants for the neutrophil system were not too different from the yeast values, including a G protein deactivation rate of 0.2 s–1 (kGd1 = ≈0.1 s–1) and a ligand-stimulated receptor internalization rate of 3.3·10–3 s–1 (kRd1 = 4·10–3 s–1).
Finally, a priority for the future is to investigate the feedback regulation of the heterotrimeric G protein cycle, especially mechanisms acting through Sst2p and modification of the receptor tail. Feedback control is necessary to ensure the robustness of the response in the presence of disturbances.
Supplementary Material
Acknowledgments
We thank Drs. Ray Deshaies and Chris Rao for critical reading of the manuscript; the Deshaies laboratory for yeast strains and vectors; and the Simon laboratory for valuable discussions. This work was supported by the Exploratory Research for Advanced Technology Kitano Symbiotic Systems Project of the Japanese Science and Technology Corporation.
Abbreviations: CFP, cyan fluorescent protein; YFP, yellow fluorescent protein; RGS, G protein regulator; FRET, fluorescence resonance energy transfer.
References
- 1.Hardman, J. G. & Limbird, L. E. (1996) Goodman & Gilman's The Pharma-cological Basis of Therapeutics, ed. Gilman, A. G. (McGraw–Hill, New York).
- 2.Sprague, G. F., Jr., & Thorner, J. W. (1992) in The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression (Cold Spring Harbor Lab. Press, Plainview, NY), Vol. 2, pp. 657–744. [Google Scholar]
- 3.Dohlman, H. G. & Thorner, J. W. (2001) Annu. Rev. Biochem. 70, 703–754. [DOI] [PubMed] [Google Scholar]
- 4.Chan, R. K. & Otte, C. A. (1982) Mol. Cell. Biol. 2, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wieland, T. & Jakobs, K. H. (1994) Methods Enzymol. 237, 1–12. [DOI] [PubMed] [Google Scholar]
- 6.Chen, C. K., Burns, M. E., He, W., Wensel, T. G., Baylor, D. A. & Simon, M. I. (2000) Nature 403, 557–560. [DOI] [PubMed] [Google Scholar]
- 7.Janetopoulos, C., Jin, T. & Devreotes, P. (2001) Science 291, 2408–2411. [DOI] [PubMed] [Google Scholar]
- 8.Guthrie, C. & Fink, G. R. (1991) Guide to Yeast Genetics and Molecular Biology (Academic, San Diego).
- 9.Lambright, D. G., Sondak, J., Bohm, A., Skiba, N. P., Hamm, H. E. & Sigler, P. B. (1996) Nature 379, 311–319. [DOI] [PubMed] [Google Scholar]
- 10.Song, J., Hirschman, J., Gunn, K. & Dohlman, H. G. (1996) J. Biol. Chem. 271, 20273–20283. [DOI] [PubMed] [Google Scholar]
- 11.Wedegaertner, P. B., Wilson, P. T. & Bourne, H. R. (1995) J. Biol. Chem. 270, 503–506. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson, S. S. G. (2001) Pharmacol. Rev. 53, 1–24. [PubMed] [Google Scholar]
- 13.Hicke, L. & Riezman, H. (1996) Cell 84, 277–287. [DOI] [PubMed] [Google Scholar]
- 14.Rohrer, J., Benedetti, H., Zanolari, B. & Riezman, H. (1993) Mol. Biol. Cell 4, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reneke, J. E., Blumer, K. J., Courchesne, W. E. & Thorner, J. (1988) Cell 55, 221–234. [DOI] [PubMed] [Google Scholar]
- 16.Apanovitch, D. M., Slep, K. C., Sigler, P. B. & Dohlman, H. G. (1998) Biochemistry 37, 4815–4822. [DOI] [PubMed] [Google Scholar]
- 17.Watson, N., Linder, M. E., Druey, K. M., Kehrl, J. H. & Blumer, K. J. (1996) Nature 383, 172–174. [DOI] [PubMed] [Google Scholar]
- 18.Hepler, J. R. (1999) Trends Pharmacol. Sci. 20, 376–382. [DOI] [PubMed] [Google Scholar]
- 19.Dohlman, H. G., Apaniesk, D., Chen, Y., Song, J. & Nusskern, D. (1995) Mol. Cell. Biol. 15, 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dohlman, H. G., Song, J., Ma, D., Courchesne, W. E. & Thorner, J. (1996) Mol. Cell. Biol. 16, 5194–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konopka, J. B., Jenness, D. D. & Hartwell, L. H. (1988) Cell 54, 609–620. [DOI] [PubMed] [Google Scholar]
- 22.Jackson, C. L. & Hartwell, L. H. (1990) Cell 63, 1039–1051. [DOI] [PubMed] [Google Scholar]
- 23.Snyder, S. H. (1996) Drugs and the Brain (Scientific American Library, New York).
- 24.Pugh, E. N., Jr., & Lamb, T. D. (1993) Biochim. Biophys. Acta 1141, 111–149. [DOI] [PubMed] [Google Scholar]
- 25.Leskov, I. B., Klenchin, V. A., Handy, J. W., Whitlock, G. G., Govardovskii, V. I., Bownds, M. D., Lamb, T. D., Pugh, E. N., Jr., & Arshavsky, V. Y. (2000) Neuron 27, 525–537. [DOI] [PubMed] [Google Scholar]
- 26.He, W., Cowan, C. W. & Wensel, T. G. (1998) Neuron 20, 95–102. [DOI] [PubMed] [Google Scholar]
- 27.Adams, J. A., Omann, G. M. & Linderman, J. J. (1998) J. Theor. Biol. 193, 543–560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.