

Abstract
Loci detected by Southern blot hybridization of 3,977 expressed sequence tag unigenes were mapped into 159 chromosome bins delineated by breakpoints of a series of overlapping deletions. These data were used to assess synteny levels along homoeologous chromosomes of the wheat A, B, and D genomes, in relation to both bin position on the centromere-telomere axis and the gradient of recombination rates along chromosome arms. Synteny level decreased with the distance of a chromosome region from the centromere. It also decreased with an increase in recombination rates along the average chromosome arm. There were twice as many unique loci in the B genome than in the A and D genomes, and synteny levels between the B genome chromosomes and the A and D genome homoeologues were lower than those between the A and D genome homoeologues. These differences among the wheat genomes were attributed to differences in the mating systems of wheat diploid ancestors. Synteny perturbations were characterized in 31 paralogous sets of loci with perturbed synteny. Both insertions and deletions of loci were detected and both preferentially occurred in high recombination regions of chromosomes.
Hexaploid wheat Triticum aestivum L. (2n = 42, genome formula AABBDD) evolved by hybridization of three diploid (2n = 14) species: Triticum urartu Thum ex Gand. (the source of the A genome), Aegilops speltoides Tausch or a closely related species (the source of the B genome), and Aegilops tauschii Coss. (the source of the D genome) (1–4). Each of the 21 wheat chromosomes has been allocated to one of these ancestral genomes and one of seven homoeologous groups (5). Chromosomes within each homoeologous group are related by descent from a chromosome of the ancestor of the Triticum–Aegilops alliance.
Most wheat cytogenetic stocks, including those on which this article is based, were developed in the genetic background of cultivar Chinese Spring. Classical studies of chromosome pairing in hybrids suggested that the structure of Chinese Spring chromosomes closely corresponds to the structure of the chromosomes of the three wheat ancestors (6). Analyses of C-banded meiotic pairing configurations in hybrids and comparative linkage mapping refined the understanding of Chinese Spring chromosome structure (7–11). These studies revealed that the long arms of chromosomes 4A and 5A, and the short arm of chromosome 7B, were involved in an interchange. Chromosome 4A also suffered paracentic and a pericentric inversions. A translocation of short terminal segments involving few gene loci differentiated the short arms of chromosomes 2B and 6B. Except for these few changes, the relationships among Chinese Spring homoeologous chromosomes have not been perturbed by major translocations or inversions.
Comparative isozyme and restriction fragment length polymorphism (RFLP) mapping and studies of homoeologous relationships revealed that chromosome structure has been conserved not only among the wheat genomes but also among genomes of different genera across the tribe Triticeae, where the Triticum–Aegilops alliance belongs. Each of the diploid tall wheatgrass (Lophopyrum elongatum A. Löve, genomes EE) chromosomes or those of the more distantly related barley (Hordeum vulgare L., genomes HH) corresponds to a single-wheat homoeologous group (12–16).
A marked attribute of wheat genomes, and those of wheat diploid relatives in the tribe Triticeae, is the great abundance of duplicated gene loci. One quarter or more of all gene motifs present in these genomes are represented by paralogous sets of loci (16–20). The mechanism by which these duplicated loci have evolved is unknown. It appears, however, that they did not originate by polyploidy (20).
The availability of DNA sequence information for the rice (Oryza sativa L.) genome (www.ncbi.nlm.nih.gov) and deletion map position of a large number of wheat loci detected by EST unigenes (wheat.pw.usda.gov/NSF/progress_mapping.html) made it possible to obtain a detailed picture of homoeologous relationships between wheat and rice chromosomes (21). This information was used to determine, for 40 paralogous sets of wheat loci, which locus was ancestral and which was derived from it by duplication (20). Duplication-derived loci were most often located in the distal regions of wheat chromosomes, and their distribution was positively correlated with the gradient of recombination rate along the average wheat chromosome arm that increases with the square of the distance of a chromosomal region from the centromere (20, 22).
The set of gene loci located on a wheat chromosome constitutes a syntenic group of that chromosome. A structural change, such as a translocation, reduces the length of the syntenic group that the chromosome shares with a homoeologous chromosome. In contrast, deletions or insertions of individual loci, while not affecting the lengths of the syntenic groups shared by homoeologous chromosomes, affect the proportion of loci within shared syntenic groups having orthologues on both chromosomes. This proportion will be referred to as the synteny level, or briefly synteny, between homoeologous chromosomes. It will be assumed that the lengths of syntenic groups shared by wheat homoeologous chromosomes are equivalent to the lengths of the chromosomes. The structurally altered chromosome 4A, the short arm of chromosome 7B, and the long arm of chromosome 5A, all having a shortened syntenic group, were excluded from the analyses.
An insertion of a duplicated gene changes the level of synteny of the chromosome with its homoeologues. Because the distribution of duplication-derived loci is positively correlated with recombination rates along wheat chromosome arms, the level of synteny between homoeologous chromosomes should decline from proximal, low-recombination regions to distal high-recombination regions. This pattern was demonstrated between rice chromosome 1 and wheat chromosome 3B, which are homoeologous. Because of the great evolutionary distance between wheat and rice, comparisons of wheat and rice homoeologous chromosomes are intrinsically uncertain, because it cannot be excluded that the lengths of compared syntenic groups were not shortened by undetected translocations. Because these uncertainties are largely absent from comparisons among wheat homoeologous chromosomes, we elected to test this hypothesis with wheat homoeologous chromosomes. Synteny levels along chromosome arms of homoeologous chromosomes were quantified, and the correlation between synteny level and recombination rate was assessed. To obtain a detailed insight into the causes of synteny perturbations in wheat, the following characteristics were determined for 31 paralogous sets with perturbed synteny: (i) whether the cause of perturbed synteny was a duplication or deletion event; (ii) whether the duplication or deletion occurred in wheat or its diploid ancestors; (iii) whether a locus showing perturbed synteny was located in a high- or low-recombination region; (iv) which locus of a paralogous set was ancestral and which was derived; and (v) whether perturbed synteny involved an ancestral or derived locus.
Materials and Methods
ESTs and Their Mapping. From 1,000 to 11,000 clones were randomly selected from each of 41 cDNA libraries comprising most organs and developmental phases of the wheat plant. At the time of this analysis, ≈460 base pairs each of 80,000 cDNA clones had been sequenced and deposited in GenBank (www.ncbi.nlm.nih.gov). ESTs, their characteristics, and mapping status can be viewed at http://wheat.pw.usda.gov/NSF/progress_est.html.
A total of 101 deletion stocks, each containing one or several deletions, 24 ditelosomic stocks, and 21 nullisomic-tetrasomic stocks, all in the Chinese Spring genetic background, were used in mapping of loci detected by EST unigenes (23). The series of deletion breakpoints along chromosome arms defined physical regions called bins (23). DNA isolation from these stocks, the Southern hybridization of them, and subsequent analyses have been described (20, 23).
Recombination Rate. Recombination rates, expressed as coefficients of exchange (CEs, cM/Mb) (24), were derived from estimates of genetic lengths of bins in centimorgans (cM) divided by estimates of bin lengths in megabases (Mb). The estimation of genetic lengths and lengths in Mb of bins has been described (20). As in ref. 20, the relative distance of deletion breakpoints along the nucleolar organizing region (NOR) bearing chromosome arms 1BS and 6BS, as specified in http://wheat.pw.usda.gov/west/binmaps, were recalculated so that the satellites were included within the arms. Individual CE estimates were likely burdened by a sizable experimental error rate due to variation in determination of deletion breakpoints and linkage. We therefore grouped the 156 bins according to the relative physical distance of bin midpoint from the centromere into six equal intervals, and computed a mean CE for each interval. The coefficients of variation of these six means ranged from 0.5 to 1.3. Using these six mean CEs in correlation analyses reported here not only reduced the effect of variation due to measurement error but also averaged chromosome-arm specific recombination patterns, such as local recombination minima and maxima caused by recombination hotspots and crossover interference, differences in recombination rate gradients along chromosome arms because of arm lengths (22), heterochromatin (25), NOR loci (26), and other factors, across all 42 wheat chromosome arms.
Assessment of Synteny Level Between Homoeologous Chromosomes. The wEST-SQL database of 3,977 EST unigenes (http://wheat.pw.usda.gov/cgi-bin/westsql/westsql.cgi) was queried by using a script that extracted those EST unigenes for which all restriction fragments hybridizing with an EST unigene were mapped. This query produced a total of 1,059 EST unigenes. For each of these EST unigenes, the autoradiograms (http://wheat.pw.usda.gov/cgi-bin/westsql/map_locus.cgi) were examined to verify that no spurious artifacts and unmapped fragments were present. A total of 845 EST unigenes were retained for the assessment of synteny among the homoeologous chromosomes of the three wheat genomes. All analyses were done on a locus basis. If several restriction fragments within a bin hybridized with a single clone, it was assumed that a single locus was within the bin, because the number of loci detected by a clone within a bin could not be determined.
The deletion lines used for locus mapping were isolated in progenies of plants with a gametocidal chromosome, which introduces breaks into chromosomes (27). It is conceivable that the gametocidal chromosome could cause also small, cytologically undetectable deletions that would perturb synteny among deletion lines. Restriction profiles of deletion lines were therefore always compared with those of Chinese Spring ditelosomic and nullisomic-tetrasomic lines that were hybridized simultaneously with the deletion lines (23). Only synteny perturbations that were monomorphic in these lines were considered in analyses reported here.
There are two possible directions in which synteny level (S) between a pair of homoeologous chromosomes can be examined. Assume that loci mapped on an A genome chromosome are used as the basis for synteny assessment. In this case, synteny SB/A is defined as the probability that there is a B genome orthologue for a locus on the A genome chromosome. In the opposite direction, the loci mapped on the B genome chromosome are used as the basis for synteny assessment. Synteny, SA/B, is then defined as the probability that there is an A genome orthologue for a locus on the B genome chromosome.
The following strategy was used to estimate synteny levels along chromosome arms. Synteny SB/A will be used as an example. The centromere-telomere axis, on which the centromere is at 0.00 and the telomere is at 1.00, was divided into six equal intervals, each 0.17 of the relative arm length. Bins along each A genome chromosome (the rearranged chromosome 4A was excluded) were allocated into these six intervals on the basis of the position of bin midpoint on the centromere-telomere axis. The level of synteny in the ith interval (Si,B/A) is defined as
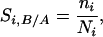 |
where Ni is the total number of loci in the ith interval in the A genome, and ni is the number of loci in the B genome that are orthologous to these loci. The arithmetic mean (S̄ B/A) of the six Si,B/As was used as the measure of total level of synteny between the A and B genomes. Statistical significance of differences between S̄ values was tested by paired t test using the six paired Sis as variables.
Relationship Between Synteny and Recombination Rate. For statistical analyses, estimates in which the same genome was used as the basis of synteny level assessment; e.g., Si,B/A and Si,D/A, were averaged (Si,B+D/A). The three possible mean synteny level estimates in the ith interval were designated Si,B+D/A, Si,A+D/B, and Si,A+B/D. The Pearson correlation coefficients (r) of mean synteny level and recombination rates along the average chromosome arm were computed. The probability that an r equals zero was determined from statistical tables using 4 degrees of freedom.
Characterization of 31 Paralogous Sets with Incomplete Synteny. The population of 845 EST unigenes was used to find those that detected loci with perturbed synteny in the wheat genomes, i.e., the absence of a locus in one or two of the three wheat homoeologous chromosomes. After verification of mapping results, 31 EST unigenes were chosen for further study (for a list of these 31 clones, see Table 3, which is published as supporting information on the PNAS web site, www.pnas.org). They were hybridized with Southern blots of EcoRI-digested DNAs of 19 L. elongatum (E genome) disomic substitution (DS) lines (12, 28, 29) and 6 disomic addition (DA) lines of Betzes barley chromosomes (H genome) in the Chinese Spring genetic background (30), and Betzes barley. Disomic substitution lines 4E(4B) and 5E(5A) and DA1H line were not available.
Loci located on homoeologous chromosomes in wheat, L. elongatum, or barley were assumed to be orthologous. In wheat paralogous sets containing two or more loci, the wheat locus that had an orthologue in L. elongatum or barley was assumed to be the ancestral locus and the locus (or loci), which did not have an orthologue either in L. elongatum or barley was assumed to have been derived by duplication during the radiation of the Triticum–Aegilops alliance. When there were three (or more) loci within wheat genomes, and two of them were detected in the L. elongatum and barley genomes, then it was concluded that the latter two loci were ancestral and the third locus (and additional loci) was derived by duplication.
If a locus was detected only on one or two of the three wheat homoeologues, but was on the L. elongatum or barley homoeologues, a deletion that occurred during the radiation of the Triticum–Aegilops alliance, or during polyploid wheat evolution, was assumed to be the most likely cause of incomplete synteny of the locus in wheat. If a locus was detected only on one or two of the three wheat homoeologues, but was not detected on the L. elongatum and barley homoeologues, a duplication that occurred either during the radiation of the Triticum–Aegilops alliance or during polyploid wheat evolution was the most likely cause of incomplete synteny of the locus in wheat.
To determine whether synteny of loci detected by an EST unigene was perturbed at the diploid level or polyploid level, each clone was hybridized with Southern blots of DNAs of nine lines of T. urartu, nine lines of Ae. speltoides, and seven lines of Ae. tauschii (for a description of the lines see Table 4, which is published as supporting information on the PNAS web site) digested with EcoRI, and restriction fragments were allocated to loci. Consider first a case in which it had been assumed that incomplete synteny was caused by a deletion. If a locus absent from a wheat genome was present in the diploid ancestor of that genome, it was assumed that the deletion occurred at the polyploid level. If the locus was absent both from the wheat genome and that of the diploid ancestor, then it was assumed that the deletion occurred at the diploid level. Consider now a case in which it had been assumed that incomplete synteny was caused by a duplication. If the duplicated locus was present in the diploid ancestor of the genome, the duplication was assumed to have occurred at the diploid level. If the duplicated locus was absent from the genome of the diploid ancestor, it was assumed that the duplication occurred at the polyploid level. This last inference assumed that the duplicated locus was not deleted from the diploid ancestor prior to the origin of polyploid wheat.
It has previously been shown that the recombination rate is low in the proximal two-thirds, and it rapidly increases in the distal one-third of the average chromosome arm (20). This division into the proximal two-thirds and distal one-third was used as an arbitrary division of the average chromosome arm into low-recombination and high-recombination regions. The ancestral loci and locus duplications and deletions were grouped according to their locations into these two intervals.
The randomness of the distribution of duplicated and deleted loci was statistically tested as follows: Akhunov et al. (20) used the distribution of restriction fragments detected by 730 EST unigenes as an estimate of gene distribution along wheat chromosome arms. From 2,743 restriction fragments, 51% were in the proximal two-thirds of the chromosome arms and 49% were in the distal one-third of the chromosome arms, which is a ratio of ≈1:1. The homogeneity of loci in the two intervals was tested by χ2 test or by Fisher's exact test, using a 1:1 ratio as the null hypothesis.
Results
Mean Synteny Levels. Mean synteny estimates based on loci mapped in the B genome (S̄A/B and S̄D/B) were 0.91 (n = 686, n = 624) and 0.92 (n = 686, n = 633), respectively (for data, see Table 5, which is published as supporting information on the PNAS web site). These two estimates were significantly lower (P values ranged from 0.008 to 0.07) than the estimates based on loci mapped in the A genome, S̄B/A = 0.95 (n = 593, n = 564) and S̄ D/A = 0.96 (n = 593, n = 569), or the estimates based on loci mapped in the D genome, S̄A/D = 0.94 (n = 746, n = 702) and S̄B/D = 0.96 (n = 746, n = 713). The latter four estimates did not significantly differ from each other.
Genome-Unique Loci. Of the total numbers of 593, 686, and 746 loci mapped in the A, B, and D genomes, 13 (2.2%), 41 (6.0%), and 19 (2.5%) loci were unique to the A, B, and D genomes, respectively. In the B genome, 21 unique loci were in the proximal two-thirds (low-recombination interval) and 20 were in the distal one-third (high-recombination interval). In the A and D genomes, there were 6 and 5 unique loci in the lowrecombination interval and 7 and 14 in the high-recombination interval, respectively.
Correlation Between Synteny Levels and Recombination Rates Along Chromosome Arms. Si,B/A and Si,D/A synteny decreased from the proximal to the distal interval along the six intervals of the average chromosome arm (Fig. 1A; for data see Table 5). Synteny Si,A/D and Si,B/D followed the same trend (Fig. 1C). In contrast, Si,A/B and Si,D/B synteny changed little on the centromere-telomere axis (Fig. 1B). All six synteny profiles along the average chromosome arm showed disproportionately high levels of synteny in the third intervals from the centromere (Fig. 1). Whether interstitial recombination minima present in some wheat chromosome arms or another cause was responsible for this variation could not be determined.
Fig. 1.

Synteny (S) along the average chromosome arm. The distance of an interval from the centromere is expressed as a fraction of the average chromosome arm length. The centromere of the average chromosome arm is at 0.00, and the telomere is at 1.00.
Both Si,B+D/A and Si,A+B/D synteny were negatively correlated (P = 0.04 and 0.01, respectively) with recombination rates along chromosome arms (Table 1). In contrast, correlation of Si,A+D/B synteny with recombination rate was not statistically significant, although also being negative (Table 1). Similar relationships were observed between the level of synteny and the location of an interval on the centromere-telomere axis (Table 1).
Table 1. Correlation (r) between synteny, recombination rate, and relative distance of an interval from the centromere.
|
r
|
||
|---|---|---|
| Synteny | Recombination rate | Relative distance from centromere |
| Si,B+D/A | -0.82 (0.04)* | -0.91 (0.01) |
| Si,A+D/B | -0.41 (0.42) | -0.38 (0.46) |
| Si,A+B/D | -0.93 (0.01) | -0.91 (0.01) |
P values are in parentheses.
Characterization of 31 Paralogous Sets with Perturbed Synteny. The strategy used to infer which locus was ancestral, which was derived, and whether synteny was perturbed at the diploid level or polyploid level is illustrated in Fig. 2. Hybridization of clone WHE0825–0828_L16_L16 with Southern blots of DNAs of L. elongatum DS lines indicated that this EST unigene detected a single locus in the wheat B and D genomes, but detected four loci in the A genome. In the A genome, loci were detected on chromosomes 1A, 2A, 4A, and 6A. The clone detected a locus only on chromosome 1E in the L. elongatum genome. Therefore, the locus in homoeologous group 1 is the ancestral locus of this paralogous set, whereas the loci on 2A, 4A, and 6A are duplication derived. The absence of any additional restriction fragment in the profiles of the six barley DA lines was consistent with this conclusion, but direct evidence was not available because DA1H was missing from the set. Betzes barley showed a single restriction fragment, indicating that there was a single locus hybridizing with this clone in barley (data not shown). All six wheat loci were found in the genomes of wheat ancestors. Whereas only one locus each was present in the Ae. speltoides and Ae. tauschii genomes, four loci were found in each line of T. urartu, suggesting that all duplicated loci originated in the T. urartu phylogenetic lineage.
Fig. 2.

Southern blot hybridization of cDNA clone WHE0825–0828_L16_L16 with EcoRI-digested genomic DNA of Chinese Spring (CS), L. elongatum (E genome) disomic substitution lines in Chinese Spring, disomic addition lines of barley (H genome) chromosomes in Chinese Spring and lines of T. urartu (lanes 1–9), Ae. tauschii (lanes 10–16), and Ae. speltoides (lanes 17–25). In the designations of disomic substitution lines, the wheat chromosome replaced by its L. elongatum homoeologue is indicated in parentheses. The chromosomal locations of the restriction fragments are shown on the left. The four A-genome loci found in wheat were found in the T. urartu genome, whereas only one locus each was found in the genomes of Ae. tauschii and Ae. speltoides. Restriction fragments shared by Chinese Spring D and B genomes and Ae. tauschii and Ae. speltoides, respectively, are marked by arrows.
By using this strategy, it was possible to determine which locus was ancestral and which was derived, for 29 of 31 investigated paralogous sets. In two sets, duplication occurred before the divergence of Triticeae, and an ancestral locus could not be identified; we therefore concluded that each of these sets had two ancestral loci. Of these 33 ancestral loci, 16 were located in the proximal two-thirds of chromosome arms (the lowrecombination interval) and 17 were located in the distal one-third of chromosome arms (the high-recombination interval). The frequencies of ancestral loci in the two intervals did not significantly differ (P = 0.73) from the 1:1 ratio of all loci in these two intervals.
A total of 33 loci at which synteny was perturbed were characterized (Table 2). Twenty-three were caused by duplications and 10 were caused by deletions of loci. Four of the 10 deleted loci were duplication derived, 4 were ancestral loci, and for 2 the status was unknown. All four deleted ancestral loci were located in the distal interval. Twenty-five of these events originated at the diploid level and 8 (24%) originated at the polyploid level.
Table 2. Ploidy level and interval in which events perturbing synteny in 31 paralogous sets occurred.
| No. assigned to a chromosome
|
Ploidy level
|
Interval (diploid level)*
|
Interval (polyploid level)*
|
||||
|---|---|---|---|---|---|---|---|
| Event | Diploid | Polyploid | Low recombination | High recombination | Low recombination | High recombination | |
| Duplication | 23 | 19 | 4 | 6 | 13 | 1 | 3 |
| Deletion | 10 | 6 | 4 | 0 | 6 | 0 | 4 |
| Total | 33 | 25 | 8 | 6 | 19 | 1 | 7 |
Low-recombination interval is defined as the proximal two-thirds of the average chromosome arm, and high-recombination interval is defined as the distal one-third of the average chromosome arm.
Of the 33 loci at which synteny was perturbed, 7 were located in the proximal two-thirds of the average chromosome arm, and 26 were located in the distal one-third of the average chromosome arm. The frequencies of loci in these two intervals significantly differed from the 1:1 distribution of loci in these intervals (P = 0.001).
Of the 25 synteny perturbations that occurred during the evolution of wheat diploid ancestors, six were in the proximal two-thirds of the average chromosome arm and 19 were in the distal one-third of the average chromosome arm. Eighteen of these synteny perturbations were monomorphic among the nine lines of T. urartu or seven lines of Ae. tauschii, depending on the genome location of locus at which synteny was perturbed in Chinese Spring wheat. As illustrated in Fig. 2, sufficient numbers of T. urartu and Ae. tauschii restriction fragments were shared with profiles of Chinese Spring so that they could be allocated to loci in these species on the basis of deletion mapping in Chinese Spring. In contrast, very few Ae. speltoides restriction fragments were shared with Chinese Spring. This fact and frequent variation for number of restriction fragments present among the nine lines often precluded unequivocal allocation of restriction fragments to loci. Therefore, homogeneity of the distribution of loci at which synteny was perturbed at the diploid level was statistically tested only in T. urartu and Ae. tauschii. Of 18 loci, 2 were in the proximal two-thirds of the average chromosome arm, and 16 were in the distal one-third of the average chromosome arm, which significantly differed from the 1:1 frequency of loci in these two intervals (Fisher's exact test, P = 0.015).
Discussion
Analyses of the presence and absence of orthologous loci detected by 845 EST unigenes in wheat homoeologous chromosomes revealed that synteny between homoeologous chromosomes has been perturbed by frequent insertions and deletions of loci. Although the perturbations were discovered in polyploid wheat, the characterization of 31 paralogous sets with perturbed synteny indicated that 75% of these perturbations actually occurred during the evolution of wheat diploid ancestors.
Synteny perturbations were not equal among the three genomes. Whereas 8–9% of the B genome loci defied syntenic relations, only 4–6% of the A and D genome loci defied syntenic relations. A greater differentiation of the B genome chromosomes from their A and D genome homoeologues, than that of the A and D genome chromosomes from each other, is inversely related to the levels of chiasmate metaphase I (MI) pairing between wheat homoeologues in the absence of the Ph1 locus. MI pairing between the A and D genome homoeologues was higher than that between the A and B or D and B genome homoeologues (31). This relationship suggests an intriguing possibility that there may be a link between chromosome differentiation described here and MI chiasmate pairing between homoeologous chromosomes.
It was shown earlier that duplication-derived loci accumulated preferentially in distal, high-recombination regions of wheat chromosomes (20). Because an insertion of a duplicated gene or gene fragment creates a unique locus, it was hypothesized that the level of synteny between wheat homoeologous chromosomes should be lower in distal, high-recombination regions than in proximal, low-recombination regions. The level of synteny was found here to correlate negatively with interval distance from the centromere and recombination rate along homoeologous chromosome arms, which was consistent with this hypothesis. However, the correlations were strong and statistically significant, only when loci mapped in the A and D genomes were used as the basis of synteny assessments (Si,B/A, Si,D/A, Si,A/D, and Si,B/D synteny). They were weak and not significant when loci mapped in the B genome were used as the basis of synteny assessment (Si,A/B and Si,D/B synteny). However, it does not mean that synteny is decaying evenly along the B genome chromosomes. Employing a longer evolutionary time scale, steep synteny gradients were observed in both chromosome arms between wheat chromosome 3B and rice chromosome 1 (20), which is homoeologous with 3B (21).
We offer the following explanation of the B genome anomaly based on a parallel between synteny levels along chromosome arms and Nei's gene diversity measured by RFLP along chromosome arms. The wheat B genome was most likely contributed by Ae. speltoides or its extinct ancestor (refs. 5, 32, and 33; for review, see ref. 34). The ratio of Nei's gene diversity in distal, high-recombination regions to that in proximal, low-recombination regions (Hd/Hp) was 1.3 in Ae. speltoides, but was 2.6 in T. urartu and 2.2 in Ae. tauchii (35, 36). This difference was largely caused by a disproportionately greater value for Nei's gene diversity in the proximal, low-recombination region in Ae. speltoides than that in T. urartu and Ae. tauschii. In parallel, the Si,A/B and Si,D/B synteny gradients were less steep than the Si,B/A, Si,D/A, Si,A/D, and Si,B/D synteny gradients. This result was caused by disproportionately lower Si,A/B and Si,D/B synteny in proximal, low-recombination regions than Si,B/A, Si,D/A, Si,A/D, and Si,B/D synteny in these regions (Fig. 1). There were three to four times as many unique loci in the proximal, low-recombination interval in the B genome than in the A and D genomes.
In Aegilops and Triticum, RFLP gradients along chromosome arms are related to recombination rate gradients along chromosome arms, possibly reflecting the strength of indirect selection along chromosome arms (35). Indirect selection (hitchhiking and background selection) reduces the effective population size in the vicinity of a selected locus (37, 38). Variation in recombination rates along a chromosome arm causes variation in the strength of indirect selection on neutral loci linked to selected loci. Hence, the levels of neutral polymorphism, such as RFLP, are more effectively reduced in low-recombination regions than in high-recombination regions. Because self-pollination reduces the magnitude of recombination per generation, in addition to reducing the effective population size, low recombination was likely also the principal cause of lower Nei's gene diversity (H) measured by RFLP in the self-pollinating T. urartu and Ae. tauschii, as compared with the crosspollinating Ae. speltoides (35). It is therefore likely that differences in S̄, and in Si gradients along the average chromosome arm in the B genome, as compared with the A and D genomes, reflect differences in the mating system of the ancestors of these genomes, and that synteny and RFLP are related because both are affected by the same factor; i.e., the recombination rate.
The inverse relationship between the level of synteny and recombination rate could therefore be caused by weaker indirect selection in the high-recombination regions as compared with low-recombination regions of chromosomes. Indirect selection may eliminate some of the gene insertions from low-recombination regions of chromosomes when they are in the polymorphic state. The persistence of polymorphisms for locus duplication in high-recombination regions may enhance an opportunity for the evolution of genes with new functions that could then be fixed due to positive selection. The ultimate result of these processes would be a faster loss of synteny in the high-recombination regions of genomes than in low-recombination regions.
Alternatively, the inverse relationship between synteny level and recombination rate could be attributed to preferential transposition of genes and gene fragments into chromosome regions with high homoeologous recombination rates. This possibility has been discussed in ref. 20, where it was pointed out that experimental observations are inconsistent with this assumption.
In a paralogous set composed of one ancestral and one duplication-derived locus, either locus can theoretically be deleted. Deletions detected here involved equal numbers of ancestral and duplication-derived loci; however, all deleted ancestral loci were located in high-recombination regions of chromosomes. The colocation of duplication-derived loci and locus deletions predisposes duplication-derived loci to deletion in chromosomes with distinct recombination gradients, unless a duplicated locus is favored by natural selection. In species with steep recombination gradients along chromosome arms, particularly in the lineages of self-pollinating species, this colocation of duplications and deletions in the same regions results in higher net gene turnover in distal, high-recombination regions than in proximal, low-recombination regions.
This article suggests the following genome evolution trends in the Triticum–Aegilops alliance. High-recombination regions of homoeologous chromosomes lose synteny faster than do lowrecombination regions, and self-pollination magnifies this disparity. If everything else is equal, homoeologous chromosomes of crosspollinating species lose synteny faster than do homoeologous chromosomes of their self-pollinating relatives. This trend effectively causes differences in the rates of genome differentiation between closely related self- and crosspollinating lineages. We suggest that variation in recombination rates is the causal agent in these trends.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Plant Genome Research Program Cooperative Agreement DBI-9975989.
Abbreviation: RFLP, restriction fragment length polymorphism.
References
- 1.Kihara, H. (1944) Agric. Hortic. 19, 13–14. [Google Scholar]
- 2.McFadden, E. S. & Sears, E. R. (1946) J. Hered. 37, 81–89, 107–116. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar, P. & Stebbins, G. L. (1956) Am. J. Bot. 43, 297–304. [Google Scholar]
- 4.Dvorak, J., di Terlizzi, P., Zhang, H. B. & Resta, P. (1993) Genome 36, 21–31. [DOI] [PubMed] [Google Scholar]
- 5.Sears, E. R. (1966) in Chromosome Manipulations and Plant Genetics, eds. Riley, R. & Lewis, K. R. (Oliver & Boyd, Edinburgh), pp. 29–44.
- 6.Kimber, G. & Riley, R. (1963) Can. J. Genet. Cytol. 5, 83–88. [Google Scholar]
- 7.Naranjo, T., Roca, A., Goicoechea, P. G. & Giraldez, R. (1987) Genome 29, 873–882. [Google Scholar]
- 8.Gale, M. D., Atkinson, M. D., Chinoy, C. N., Harcourt, R. L., Jia, J., Li, Q. Y. & Devos, K. M. (1993) in Proceedings of the 8th International Wheat Genetics Symposium, eds. Li, Z. S. & Xin, Z. Y. (China Agricultural Scientech Press, Beijing), Vol. 1, pp. 29–40. [Google Scholar]
- 9.Devos, K. M., Dubcovsky, J., Dvorak, J., Chinoy, C. N. & Gale, M. D. (1995) Theor. Appl. Genet. 91, 282–288. [DOI] [PubMed] [Google Scholar]
- 10.Mickelson-Young, L., Endo, T. R. & Gill, B. S. (1995) Theor. Appl. Genet. 90, 1007–1011. [DOI] [PubMed] [Google Scholar]
- 11.Devos, K. M., Millan, T. & Gale, M. D. (1993) Theor. Appl. Genet. 85, 784–792. [DOI] [PubMed] [Google Scholar]
- 12.Dvorak, J. (1980) Can. J. Genet. Cytol. 22, 237–259. [Google Scholar]
- 13.Hart, G. E. & Tuleen, N. A. (1983) Genet. Res. 41, 181–202. [Google Scholar]
- 14.Hart, G. (1995) in Methods of Genome Analysis in Plants, ed. Jauhar, P. P. (CRC, Boca Raton, FL), pp. 195–209.
- 15.Dvorak, J., McGuire, P. E. & Mendlinger, S. (1986) Plant Syst. Evol. 144, 209–220. [Google Scholar]
- 16.Dubcovsky, J., Luo, M. C., Zhong, G. Y., Bransteitter, R., Desai, A., Kilian, A., Kleinhofs, A. & Dvorak, J. (1996) Genetics 143, 983–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graner, A., Jahoor, A., Schondelmeier, J., Siedler, H., Pillen, K., Fischbeck, G., Wenzel, G. & Herrman, R. G. (1991) Theor. Appl. Genet. 83, 250–256. [DOI] [PubMed] [Google Scholar]
- 18.Anderson, J. A., Ogihara, Y., Sorrells, M. E. & Tanksley, S. D. (1992) Theor. Appl. Genet. 83, 1035–1043. [DOI] [PubMed] [Google Scholar]
- 19.Kleinhofs, A., Kilian, A., Maroof, M. A. S., Biyashev, R. M., Hayes, P., Chen, F. Q., Lapitan, N., Fenwick, A., Blake, T. K., Kanazin, V., et al. (1993) Theor. Appl. Genet. 86, 705–712. [DOI] [PubMed] [Google Scholar]
- 20.Akhunov, E. D., Goodyear, J. A., Geng, S., Qi, L.-L., Echalier, B., Gill, B. S., Miftahudin, Gustafson, J. P., Lazo, G., Chao, S., et al. (2003) Genome Res. 13, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorrells, M. E., La Rota, C. M., Bermudez-Kandianis, C. E., Greene, R. A., Kantety, R., Munkvold, J. D., Miftahudin, Mahmoud, A., Gustafson, J. P., Qi, L., et al. (2003) Genome Res. 13, 1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukaszewski, A. J. & Curtis, C. A. (1993) Theor. Appl. Genet. 84, 121–127. [DOI] [PubMed] [Google Scholar]
- 23.Qi, L., Echalier, B., Friebe, B. & Gill, B. S. (2003) Funct. Integr. Genomics 3, 39–55. [DOI] [PubMed] [Google Scholar]
- 24.Lindsley, D. L. & Sandler, L. (1977) Philos. Trans. R. Soc. London B 277, 295–312. [DOI] [PubMed] [Google Scholar]
- 25.John, B. & Miklos, G. L. G. (1979) Int. Rev. Cytol. 58, 1–114. [DOI] [PubMed] [Google Scholar]
- 26.Luo, M. C., Yang, Z. L. & Dvorak, J. (1998) Genetics 145, 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Endo, T. R. (1988) J. Hered. 79, 366–370. [DOI] [PubMed] [Google Scholar]
- 28.Dvorak, J. & Chen, K. C. (1984) Can. J. Genet. Cytol. 26, 128–132. [Google Scholar]
- 29.Tuleen, N. A. & Hart, G. E. (1988) Genome 30, 519–524. [Google Scholar]
- 30.Islam, A. K. M. R., Shepherd, K. W. & Sparrow, D. H. B. (1981) Heredity 46, 161–174. [Google Scholar]
- 31.Naranjo, T. (1992) Hereditas 116, 219–223. [Google Scholar]
- 32.Dvorak, J. & Zhang, H. B. (1990) Proc. Natl. Acad. Sci. USA 87, 9640–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, G.-Z., Miyashita, N. & Tsunewaki, K. (1997) Proc. Natl. Acad. Sci. USA 94, 14570–14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dvorak, J. (1998) in Proceedings of the 9th International Wheat Genetics Symposium, ed. Slinkard, A. E. (University Extension Press, Univ. of Saskatchewan, Saskatoon, SK, Canada), Vol. 1, pp. 8–11. [Google Scholar]
- 35.Dvorak, J., Luo, M.-C. & Yang, Z.-L. (1998) Genetics 148, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dvorak, J., Yang, Z.-L. & Luo, M.-C. (1999) in 7th International Plant and Animal Genome Conference, ed. Heller, S. R. (Scherago International, San Diego), pp. P420.
- 37.Maynard Smith, J. & Haigh, J. (1974) Genet. Res. 23, 23–35. [PubMed] [Google Scholar]
- 38.Charlesworth, B. (1994) Genet. Res. 63, 213–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}