

Abstract
The isolation of chemically induced mutations in forward genetic screens is one of the hallmarks of Drosophila genetics. However, mapping the corresponding loci and identifying the molecular lesions associated with these mutations are often difficult and labor-intensive. Two mapping methods are most often used in flies: meiotic recombination mapping with marked chromosomes and deficiency mapping. The availability of the fly genome sequence allows the establishment and usage of molecular markers. Single-nucleotide polymorphisms have therefore recently been used to map several genes. Here we show that thousands of molecularly mapped P element insertions in fly strains that are publicly available provide a powerful alternative method to single-nucleotide polymorphism mapping. We present a strategy that allows mapping of lethal mutations, as well as viable mutations with visible phenotypes, with minimal resources. The most important unknown in using recombination rates to map at high resolution is how accurately recombination data correlate with molecular maps in small intervals. We therefore surveyed distortions of recombination rates in intervals <500 kb. We document the extent of distortions between the recombination and molecular maps and describe the required steps to map with an accuracy of <50 kb. Finally, we describe a recently developed method to determine molecular lesions in 50-kb intervals by using a heteroduplex DNA mutation detection system. Our data show that this mapping approach is inexpensive, efficient, and precise, and that it significantly broadens the application of P elements in Drosophila.
Forward genetic screens in Drosophila using chemical mutagens or γ-ray irradiation have uncovered thousands of complementation groups (1, 2). However, identifying the molecular lesions that underlie the phenotypes has traditionally been labor-intensive and time-consuming. All mapping strategies depend on a detectable phenotype caused by the mutation (e.g., lethality) to establish its genetic linkage with known markers or failure to complement characterized deficiencies. One of the most common strategies is meiotic recombination mapping, and resolution and accuracy primarily depend on the availability of markers and the number of meiotic events that are scored.
The publication of the fly genome (3) made it possible to generate many more precisely mapped markers on the basis of single-nucleotide polymorphisms (SNPs). SNPs are single base-pair differences between homologous chromosomes. The use of SNPs as molecular markers is a two-step process. First, SNP maps have to be established or confirmed; second, SNPs have to be detected by either sequencing or restriction enzyme digestion. Several teams have recently shown that SNPs are present in sufficient numbers in the Drosophila genome to theoretically allow the mapping of every gene. They have also demonstrated the successful application of the method in a few cases (4–6). An alternative to SNPs are P elements with molecularly defined insertion sites. They are transposable elements that often have been engineered with a visible marker such as the white+ gene. The Berkeley Drosophila Genome Group Gene Disruption Project has generated >6,500 publicly available fly strains that carry molecularly mapped P elements. This implies that P elements are available, on average, every 20–30 kb. Here we test the applicability of these molecularly mapped P insertions as tools to map lethal complementation groups as an alternative to SNP mapping.
We have recently isolated numerous lethal complementation groups in a genetic screen by using ethyl methanesulfonate as a mutagen. To map these complementation groups in parallel, we compared the cost and effort of several mapping strategies: meiotic recombination with visible markers or molecularly mapped P insertions, male recombination, deficiency mapping, and SNP mapping. One of the major problems associated with deficiency mapping strategies and recombination mapping experiments by using visual markers is that they rarely provide a resolution of <300 kb in flies. This limitation is mostly due to a paucity of visible markers as well as to the incomplete coverage of the genome by deficiencies or the lack of information concerning the molecular breakpoints of the deficiencies. The male recombination technique (7) allows mapping to an interval between molecularly mapped P insertions and can map mutations to a small interval as closer P insertions are used. However, on the basis of our experience, male recombination has several major drawbacks that hamper a large-scale approach: (i) the necessity to recombine markers onto the chromosomes that carry the mutations to be mapped; (ii) the inability to define an approximate mapping position in each round (i.e., the mutation maps either to the left or the right of the P insertion, but no finer estimate is obtained); and (iii) the need to perform at least three mapping rounds. These drawbacks have led several teams to focus on SNP mapping approaches, especially for high-resolution mapping (4–6). However, SNP mapping requires costly molecular biology reagents and analyses. This restriction is especially an issue when one needs to establish high-resolution SNP maps. We therefore explored an alternative high-throughput and high-resolution mapping strategy by using the set of molecularly mapped P insertions that are publicly available (ref. 8; http://flypush.imgen.bcm.tmc.edu/pscreen).
Theoretically, by using two molecularly mapped P insertions, a mutation can be mapped to a single nucleotide. The accuracy of this approach depends on the colinearity of the physical and recombinational maps and the number of progeny scored. Recombination rates (RRs) vary greatly over the length of the chromosome; they are several-fold lower close to the centromeres and telemores than in the middle of the chromosome arm (the centromere effect) (9, 10). However, not much is known about the variability of RRs over relatively short intervals (<500 kb). A recent study by Nairz et al. (6) using recombination mapping with SNPs showed a highly variable RR in a 350-kb region on the third chromosome. To determine the practically achievable accuracy of high-resolution recombination mapping using molecularly mapped P elements, we performed a survey of the recombination variability over short distances on Drosophila chromosomes for 15 sample regions of 200–1,600 kb. Our survey demonstrates considerable recombination distortions in genomic intervals <500 kb and provides an estimate for implicated mapping errors. On the basis of these results, we present a series of required steps to ensure a mapping accuracy of <50 kb [≈0.1 centimorgan (cM)]. Finally, we present a molecular mapping technique that allows the detection of point mutations in heteroduplex DNA for stretches that average 50 kb.
Materials and Methods
Drosophila Stocks. All P insertion lines used in this study are listed at http://flypush.imgen.bcm.tmc.edu/pscreen and are made available by the Bloomington Stock Center.
Calculation of Projected Molecular Position (PMP) and RRs. The PMP was calculated by using each pair of flanking P elements as in Fig. 1C. The RR of the region between a pair of P insertions that flank the mutation was calculated from the RDs:
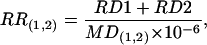 |
where RR(1,2) (cM/megabases) is the RR of the region between P elements 1 and 2, RD1 (cM) is the percentage of white-eyed flies from P insertion 1 crosses × 100, and RD2 (cM) is the percentage of white-eyed flies from P insertion 2 crosses × 100.
Fig. 1.

Schematic outline of the principle of P insertion mapping. (A) The three-step mapping procedure: rough and fine mapping are based on meiotic recombination mapping by using molecularly defined P element insertions. Mutation detection is based on temperature gradient capillary electrophoresis (TGCE), sequencing, and complementation tests with candidate genes. (B) Crossing scheme of rough and fine mapping. Mutant chromosomes are indicated by open bars, P insertion-containing chromosomes are in pink, and the balancer chromosomes are in black. The mutation sites (red stars) are marked with either 1 or 2 to indicate the different alleles. Note that all flies are in a w– background, meaning the P insertions are the only source of w+. Shown in the gray box are P and F1 crosses. Shown in the green box are the nonrecombinant offspring. Shown in the yellow box are the possible recombination events, which are color-coded and labeled A, B, and C, corresponding to the F1 female, where the three types of recombination events are marked likewise. (C) Calculation of mapping positions. PMD, projected molecular distance in base pairs; MD, molecular distance in base pairs; RD, recombination distance in cM. Note that the PMP can be calculated by using P1 (as shown) or P2 (PMDc).
For the region between P insertions that do not flank the mutation, the RR was calculated as the following. For example, if three P insertions were used and the mutation was between P insertions 1 and 2, RR(2,3) was calculated from RR(1,2) and RR(1,3).
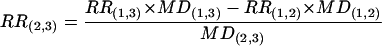 |
DNA Isolation, Primer Design, and PCR. Genomic DNA was prepared by homogenizing 10 heterozygous flies (mutant chromosome over isogenized chromosome) in 500 μl of squishing buffer (10 mM Tris, pH 8.0/1 mM EDTA/25 mM NaCl/0.2 mg/ml proteinase K), incubating at 55°C for 30–60 min, and inactivating proteinase K at 95°C for 4–5 min.
Primers were designed by using a modified version of the primer3 algorithm from the Whitehead Institute, Massachusetts Institute of Technology (11) (www-genome.wi.mit.edu/genome_software/other/primer3.html). Our implementation for automatic tiled primer pair design for larger regions is publicly available at http://flypush.imgen.bcm.tmc.edu/primer. This algorithm is run in a loop to generate primer pairs covering 600 bp with a 100-bp overlap. Hence, 100 primer pairs with highly homogenous characteristics such as melting temperature and GC content are generated to cover 50 kb.
PCR was performed in a 20-μl volume including 1 μl of DNA and 10 pmol of each primer in a 96-well format. For the regions screened so far, 95–100% of the PCRs successfully amplified the DNA in the first round.
TGCE. Mutation detection analysis was carried out on a SpectruMedix (State College, PA) Reveal TGCE apparatus SCE9610 in a 96-well format. For sample preparation, PCR products were diluted 1:5 in PCR buffer followed by heteroduplex formation performed in a thermal cycler by using the following temperature profile: 3 min at 95°C; decrease from 95°C to 80°C at 3°C/min; from 80°C to 55°C at 1°C/min; 20 min at 55°C; decrease from 55°C to 45°C at 1°C/min; and decrease from 45°C to 25°C at 2°C/min (12). Samples were then subjected to TGCE, and data were analyzed by using revelation mutation detection software from SpectruMedix. Positive signals were tested by sequencing heterozygous DNA.
Results
Meiotic Recombination by Using Molecularly Mapped P Insertions. The speed and efficiency of a recombination-based mapping approach largely depend on the ease with which the marker(s) can be scored. Most P insertions of the Berkeley Drosophila Genome Group Gene Disruption Project are marked with white+, arguably the easiest marker to score. Hence, molecularly defined P insertions with a visible marker allow molecular mapping by scoring eye color alone. Fig. 1 summarizes the overall strategy. It is based on meiotic recombination mapping for the first two mapping steps: rough and fine (Fig. 1 A). The crossing scheme is illustrated in Fig. 1B. We use two independently isolated alleles (1 and 2) that fail to complement each other to avoid mapping errors due to second-site hits. The issue of second-site hits has to be kept in mind when mapping complementation groups consisting of single alleles. Indeed, when 15 to 25 mM ethyl methanesulfonate is used, ≈50% of all chromosome arms bearing lethals carry a single lethal hit if a Poisson distribution is assumed (9).
In brief, mutant allele 1 of an essential complementation group is crossed to a homozygous viable w+-marked P insertion strain. Recombination occurs in F1 females heterozygous for allele 1 and the P insertion-bearing chromosome. These females are crossed to males heterozygous for allele 2 (or a deficiency strain that fails to complement allele 1) balanced over an hs-hid balancer (13). A single heat shock at 38°Cfor1h4dafter setting up the cross kills all of the progeny that carry the balancer. Hence, in the F2 offspring, the only progeny that survive if no crossing-over occurs have the genotype mutant allele 2/P{w+m}. These progeny are red-eyed (Fig. 1B, green box). White-eyed flies are the result of recombination events between the P insertion and mutant allele 1 (Fig. 1B, yellow box). The percentage of white-eyed flies in the F2 progeny therefore represents the RD in cM between the mutation and P insertion (Fig. 1C). Because the P insertions are molecularly mapped, the molecular distance (MD in base pairs) between any two P insertions can easily be calculated from their insertion sites. Hence, for every pair of P element insertions, a PMP of the mutation can be defined (Fig. 1C). Viable mutations can be mapped similarly if the phenotype of the homozygous mutants can easily be distinguished from the recombinant white-eyed flies.
For rough mapping, we have selected sets of 7–10 P insertions spanning each autosome arm shown in Table 2, which is published as supporting information on the PNAS web site, www.pnas.org. Mutant allele 1 is crossed to all P insertions in parallel, and at least 500 and, if possible, >1,000 F2 progeny from each P insertion cross are scored to achieve an accuracy of <1 cM, which corresponds to an average interval of 500 kb (≈25 megabases/≈55 cM per chromosome arm). Given the variable RRs in different regions of the chromosomes and the presence of double crossover events, we consider the PMP calculated from the two closest-flanking P insertions to be the most accurate. The rough mapping interval is determined as PMP ± 1 cM. Lower RRs at centromeric and telomeric regions and higher RRs in the middle of the chromosome arms are known from low-resolution maps (9, 10). Correspondingly, we found that 1 cM in the telomeric cytological division 21 (2L) corresponds to 1,550 kb, whereas 1 cM in division 95 (3R) corresponds to 102 kb. The variability in RR over the entire chromosome does not, however, significantly affect the accuracy of mapping in cM but does affect the size of the mapping interval.
Once the rough mapping interval (≈1 cM) is established, several strategies can be pursued. Deficiencies can be used to confirm and further define the mapping position. However, we favor a fine mapping strategy that uses the same principle as for the rough mapping strategy, because it provides highly consistent results. We use four to five P insertions spanning the interval defined by the rough mapping interval and score 10,000 F2 progeny for each P insertion to achieve an accuracy of ≈0.1 cM. The PMP of the mutation can then be determined as illustrated above (Fig. 1C). We find this strategy more accurate than deficiency mapping due to higher fidelity in scoring and not depending on deficiency coverage.
RRs Vary Greatly in Small Genomic Intervals. The accuracy of the mapping of a point mutation site depends on the RR. In other words, the PMP will be accurate only if the RRs in the interval between the mutation to be mapped and the two P insertions are identical. Unfortunately, very little is known about the variation of RRs in regions of 500 kb or fewer. We have mapped the lethality of a total of 15 complementation groups on chromosome arms 2L, 2R, and 3R to a 250- to 1,600-kb interval. We then carried out fine mapping (Fig. 2). Each diagram represents a mapping experiment, where PMPs obtained from each pair of flanking P insertions are indicated. We then used this data set of 15 genomic regions to survey the variability of RRs. Our analysis shows highly variable RRs (>2-fold difference within the region) in ≈50% of the cases (Fig. 2 B, F–J, N, and O). For example, Fig. 2 H and I show ≈6.6- and 12-fold differences in RRs in proximity of the mapped gene. Highly variable RRs in adjacent intervals have a strong effect on the accuracy of fine mapping. In contrast, when the RR is similar across the region spanned by the P insertions, the PMP becomes restricted to a smaller interval (PMP interval), as observed for groups A and I. In A, the RRs over a 493-kb region are very similar; therefore, all PMPs using different pairs of P insertions are within a 4-kb interval. In I, all PMPs are within 26 kb of an 876-kb region. In this example, the variation of RRs was never observed to be >2.08-fold. As the RR variability increases, so does the PMP interval. In our survey, we analyzed the RR variation of segments between pairs of neighboring P insertions in 15 fine mapping regions (eight examples are shown in Table 1). We found that, on average, the RR of a segment between a pair of P insertions varies by 34.7% when compared with the mean RR of the whole interval (n = 47). This variability did not show significant differences for varying lengths of segments or cytological locations. The variation in recombination is clearly due to the presence of recombinational “hot spots” and “cold spots” throughout the genome and is inherent to all methods based on meiotic recombination. If only the flanking P insertions of a mutation are used, however, the differences in RRs compared with surrounding areas become less relevant, and the distortion becomes much smaller the closer the flanking P elements are. For the eight genes mapped in this study, the PMP was, on average, 17 kb from the gene, and three of eight PMPs precisely pinpointed the gene.
Fig. 2.

Fine mapping results for 15 representative complementation groups. Panels represent mapping experiments, and cytological positions of each region are listed next to each panel label. The x axis is the distance between the P insertions in kilobases. The first P insertion site is normalized to 0, and the relative insertion sites of the other P elements are listed on the x axis. The number of flies counted is indicated above each P insertion (white-eyed flies/total). The red crosses mark the PMP by using pairs of P insertions, and their intervals (in kilobases) are indicated at the bottom. The y axis represents the RR (in cM/megabases) of each segment between two neighboring P insertions. Identified genes bearing the mutations are indicated by red bars on the x axis, and their relative genomic regions are listed in the red box above the gene in examples (A–H).
Table 1. Summary of mapping results for eight genes.
| Gene label (as in Fig. 2) | Gene size, kb | Length of gene-bearing segment, kb | Distance between gene and PMP,* kb | Recombination variation,† % |
|---|---|---|---|---|
| A | 1 | 97 | 10 | 6.92 |
| B | 85 | 172 | 0 | 10.12 |
| C | 17 | 182 | 51 | 20.91 |
| D | 4 | 254 | 9 | 2.81 |
| E | 6 | 362 | 42 | 30.25 |
| F | 7 | 148 | 0 | 41.11 |
| G | 18 | 83 | 0 | 8.14 |
| H | 4 | 338 | 23 | 9.16 |
PMP is obtained with the two closest-flanking P insertions.
Recombination variation is calculated by dividing the difference between the RR of the gene-bearing segment and the RR of the entire fine mapping region by the RR of the entire region.
When RRs vary greatly and the PMP intervals are large, several strategies can be pursued. First, small deficiencies and mutant alleles of candidate genes often allowed us to narrow the region significantly or even identify the mutant gene (Fig. 2 B, C, and E). Second, an additional round of fine mapping can be carried out with two or more P insertions around the PMP interval. This strategy has proven to be rather effective, and the density of P insertion is sufficient in most genomic regions (8). Finally, a high-resolution SNP map could be established to use recombinants from the P recombination mapping for SNP mapping.
To date, we have identified the molecular lesions in eight mutant genes of the 15 fine mapping examples shown in Fig. 2 (Table 1, examples A–H), either by failure to complement a mutation in a known gene (Table 1, examples B, C, and E) or by molecular mutation detection (Table 1, examples A, D, and F–H). Table 1 summarizes the mapping information of these genes, including the size of each gene, the distance between the gene and the PMP by using the closest P insertions, the length of the segment between the closest P insertions, and the recombination variation of the segment of the whole mapping region. The RRs over the region surrounding the mutation are relatively constant for A, F, and G, and, therefore, the PMP intervals are within 50 kb. The molecular lesions were identified by mutation detection (see below) and are point mutations located 10 kb (group A), <1 kb (group F), and 1.5 kb (group G) away from the PMP by using the closest-flanking P insertions, respectively. For groups C and E, the mutations were found to be outside the PMP interval. For group E, complementation tests with small deficiencies narrowed the region to the left of the PMP interval (data not shown). The candidate region of group C is close to the telomere (division 21), where RRs are very low. In this particular case, 1 cM corresponds to ≈1.5 megabases. We therefore performed complementation tests with all of the available mutant alleles in the 150-kb region surrounding the PMP interval. This revealed that the mutation mapped 51 kb to the right of the PMP. We conclude that mapping positions with an accuracy of <50 kb can generally be achieved with one round of fine mapping in regions of low RR variation and two rounds of fine mapping in regions with high RR variation.
Mutation Detection by Using TGCE. The result of fine mapping is a PMP interval calculated from different pairs of P element insertions. This interval serves as a guidepost for the following mutation detection step. When a candidate region is obtained from fine mapping results, various techniques can be used alone or in combination to further delimit the region or uncover the molecular lesion. Complementation tests with small deficiencies or alleles of candidate genes are usually the first tests to be performed.
When a 50-kb or smaller region is defined, we use TGCE to detect point mutations. This system is based on the principle that the heteroduplex DNA has a slightly lower melting temperature (Tm) than its corresponding homoduplex DNA due to a base-pair mismatch (14). Heteroduplex DNA therefore reaches its Tm earlier in an increasing temperature gradient. This slight difference in time can be displayed by capillary electrophoresis, as shown in Fig. 3 A, C, E, and G. The optimal size of DNA fragments to be analyzed with this method is between 400 and 600 bp (12). To cover a 50-kb region with overlapping DNA fragments of this size, we developed a program for automatic generation of equidistant primer pairs that allow amplification of DNA by using the same conditions for all DNA fragments (see Materials and Methods). The PCR products are in 96-well plates and are loaded simultaneously on a SpectruMedix Reveal TGCE apparatus. The TGCE results are compared among alleles and against the homozygous unmutagenized chromosome as a control. A difference in the trace of only one allele, but not the control, indicates a unique mutation, which is subsequently sequenced. SpectruMedix Reveal TGCE traces of two example alleles and their corresponding point mutations in sequences are shown in Fig. 3.
Fig. 3.

Molecular identification of lethal mutations by TGCE (SpectruMedix Reveal) and sequencing. TGCE was performed in a 96-well format with controls and all alleles in adjacent wells. Results of TGCE and sequencing are shown for two alleles of one complementation group that was fine-mapped with P insertion mapping. The DNA for sequencing and TGCE are from the same preparation of heterozygous flies (mutant chromosome over isogenized chromosome). Controls are homozygous for the isogenized chromosome. The same primers were used for TGCE and sequencing. The heteroduplexes in C and G could clearly be differentiated as double peaks when compared with the control traces in A and E. Sequencing of the heterozygous DNA revealed G-to-A transitions for both cases shown in D and H (forward and complement of reverse sequence shown). These transitions can appear as N, A, or G in the heterozygous sequence, depending on the intensity of the partial signals.
Discussion
Here, we illustrate a high-resolution mapping approach based on classical meiotic recombination by using a unique set of 6,500 P insertions as molecularly defined markers. The mapping strategy presented here uses the same principle as classic recombination mapping methods with marked chromosomes but offers much higher resolution and greater accuracy due to the large collection of molecularly defined P element insertions and the availability of the sequence of the Drosophila genome. Our high-resolution mapping experiments demonstrate that recombination and physical maps of molecularly characterized regions can be difficult to align in regions spanning <500 kb. Although these distortions often preclude a theoretical resolution of <1 kb, a resolution of <50 kb can often be achieved, especially when an additional round of fine mapping is pursued. When compared with other mapping methods, we find that this method is very powerful, cheap, and quick.
Comparison of Available High-Resolution Mapping Strategies: SNPs and P Insertions as Molecular Markers. Compared with SNP mapping, the P insertion mapping strategy offers several advantages. For SNP mapping, a low-density SNP map has to be generated or confirmed for each isogenized chromosome that is mutagenized. Subsequently, for high-resolution SNP mapping, individual maps have to be generated for the region where the mutation is located. The P insertion mapping strategy, however, uses publicly available P element insertion lines that can be used in any white– genetic background. Second, the scoring of recombination events in SNP mapping requires molecular analysis, e.g., by sequencing or restriction enzyme digestion, to identify restriction fragment-length polymorphisms associated with SNPs. In contrast, recombination events in P insertion mapping are easily recognized by eye color. A fundamental difference in established SNP mapping techniques and P insertion mapping as presented in this article is the use of recombination information. If sufficient numbers of SNPs are identified (for example, 1 SNP per ≈10 kb), mapping to either side of individual SNPs (similar to male recombination) can provide the resolution necessary to map to a single gene. If the SNPs are far apart, the binary result (either left or right) will not provide a high enough resolution to map to a single gene. In contrast, our strategy uses the recombination ratios to calculate an actual distance between the mutation and flanking P insertions. Furthermore, P insertion mapping is an extremely low-cost technique, because no molecular reagents are required; it can therefore be performed with the most basic laboratory setup. In addition, one round of rough mapping and one round of fine mapping can be accomplished in 8–10 wk. Finally, P insertion mapping is a highly flexible approach and can be combined with any of the other mapping methods, depending on expertise and reagents. In summary, the versatility, accuracy, low cost, and high speed of the P insertion mapping strategy make it highly efficient and effective.
The Impact of Recombination Variation on the Accuracy of P Insertion Mapping. In theory, meiotic recombination mapping should allow mapping to a single nucleotide. Indeed, in a few early studies, different alleles of the same locus were ordered relative to each other on the basis of recombinational mapping without the knowledge of the molecular nature of the affected genes (15–17). Likewise, high accuracy can be achieved through recombinational mapping by using molecularly mapped P insertions. The primary obstacle is the translation of this recombinational map in cM into the physical map in base pairs, which primarily depends on the colinearity of the two maps over short distances.
In our survey of 15 sample regions, we have found considerable variations in RRs in some small genomic intervals. Although these variations are likely to be primarily due to recombination hot spots and cold spots in the genome, it is also possible that P element insertions affect the recombination frequency in their vicinity. The RRs we observed may thus differ from those between wild-type and mutant chromosomes. When highly variable RRs are observed, a second round of fine mapping with P insertions around the PMP interval is, in our hands, more efficient and less expensive than other methods.
From Fine Mapping to Mutation Detection. In an effort to establish a standard method applicable in a high-throughput manner for 50-kb regions, we used a new mutation detection system based on TGCE. To screen a 50-kb genomic interval, the region has to be subdivided by using tiled primer pairs. We implemented a web-based system that automatically designs primer pairs covering large intervals of DNA. This system proved very reliable and is publicly available at http://flypush.imgen.bcm.tmc.edu/primer. The actual mutation detection on a SpectruMedix Reveal TGCE system proved suitable for a large-scale approach in both handling and data evaluation. Although we did find point mutations with this system so far in four cases, we cannot yet quantify the accuracy of the system. One drawback is the rather large number of false positives (the heterozygote mutant fragment showed a difference in Tm from the control homozygote fragment, yet it failed to reveal a point mutation by sequencing). False positives are an inherent problem of TGCE that can, for example, be caused by repetitive DNA sequences or the high sensitivity of the system. Complementary techniques like single-stranded nuclease digestion should improve the ratio of correct to false calls significantly. However, the number of candidate fragments that have to be sequenced is reduced by more than an order of magnitude when using TGCE as an intermediate step, resulting in significant savings. Obviously, the larger the number of available alleles in a complementation group, the higher the likelihood of detecting the point mutations. When TGCE is not available, sequencing of candidate genes is an option.
In summary, we believe that the mapping approach presented in this study provides very valuable tools to map chemically induced mutations with minimum resources and is applicable in a high-throughput manner.
Supplementary Material
Acknowledgments
We thank Kathy Matthews, Kevin Cook, Koen Venken, and Sunil Mehta for critical reading of this manuscript and helpful discussions. We thank Ulrich Tepass for helpful discussions of experimental procedures. We further thank Mengfei Huang, Adam Xu, and Sven Vilain for assistance in the mapping experiments. Stocks with hs-hid balancers were a gift from Ruth Lehmann (New York University, New York). R.G.Z., P.R.H., K.L.S., H.J.-N., and K.K.N. are supported by the Howard Hughes Medical Institute. P.R.H. was further supported by a European Molecular Biology Organization postdoctoral fellowship; K.K.N., H.P., and H.J.B. are supported by the Pediatric Tumor Foundation; and M.P.G. is supported by a National Research Service Award. This work was further supported by the National Institutes of Health. H.J.B. is an investigator of the Howard Hughes Medical Institute.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SNP, single-nucleotide polymorphism; PMP, projected molecular position; RD, recombination distance; TGCE, temperature gradient capillary electrophoresis; cM, centimorgan; RR, recombination rate.
References
- 1.Lindsley, D. L. & Zimm, G. G. (1992) The Genome of Drosophila melanogaster (Academic, San Diego).
- 2.The FlyBase Consortium (2003) Nucleic Acids Res. 31, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., Scherer, S. E., Li, P. W., Hoskins, R. A., Galle, R. F., et al. (2000) Science 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 4.Berger, J., Suzuki, T., Senti, K. A., Stubbs, J., Schaffner, G. & Dickson, B. J. (2001) Nat. Genet. 29, 475–481. [DOI] [PubMed] [Google Scholar]
- 5.Martin, S. G., Dobi, K. C. & St. Johnston, D. (August 30, 2001) Genome Biol. 2, research0036.1–0036.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nairz, K., Stocker, H., Schindelholz, B. & Hafen, E. (2002) Proc. Natl. Acad. Sci. USA 99, 10575–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, B., Chu, T., Harms, E., Gergen, J. P. & Strickland, S. (1998) Genetics 149, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spradling, A. C., Stern, D., Beaton, A., Rhem, E. J., Laverty, T., Mozden, N., Misra, S. & Rubin, G. M. (1999) Genetics 153, 135–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashburner, M. (1989) Drosophila: A Laboratory Handbook and Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
- 10.True, J. R., Mercer, J. M. & Laurie, C. C. (1996) Genetics 142, 507–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozen, S. & Skaletsky, H. (2000) Methods Mol. Biol. 132, 365–386. [DOI] [PubMed] [Google Scholar]
- 12.Li, Q., Liu, Z., Monroe, H. & Culiat, C. T. (2002) Electrophoresis 23, 1499–1511. [DOI] [PubMed] [Google Scholar]
- 13.Moore, L. A., Broihier, H. T., Van Doren, M., Lunsford, L. B. & Lehmann, R. (1998) Development (Cambridge, U.K.) 125, 667–678. [DOI] [PubMed] [Google Scholar]
- 14.Howley, P. M., Israel, M. A., Law, M. F. & Martin, M. A. (1979) J. Biol. Chem. 254, 4876–4883. [PubMed] [Google Scholar]
- 15.Green, M. M. & Green, K. C. (1949) Rec. Genet. Soc. Am. 18, 91–92. [Google Scholar]
- 16.Lefevre, G. (1973) Genetics 74, S154. [Google Scholar]
- 17.Gelbart, W., McCarron, M. & Chovnick, A. (1976) Genetics 84, 211–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.