

Abstract
Using serial analysis of gene expression (SAGE), we identified a SAGE tag that was present only in invasive breast carcinomas and their lymph node metastases. The transcript corresponding to this SAGE tag, dermcidin (DCD), encodes a secreted protein normally expressed only in the pons of the brain and sweat glands. Array comparative genomic hybridization, fluorescence in situ hybridization, and immunohistochemical analyses determined that DCD is overexpressed in ≈10% of invasive breast carcinomas; in some cases its overexpression is coupled with a focal copy number gain of its locus at 12q13.1, and its expression is associated with advanced clinical stage and poor prognosis. Expression of DCD in breast cancer cells promotes cell growth and survival and reduces serum dependency. Putative high- and low-affinity receptors for DCD are present on the cell surface of breast carcinomas and neurons of the brain. Based on these data we hypothesize that DCD may play a role in tumorigenesis by means of enhancing cell growth and survival in a subset of breast carcinomas.
Breast cancer is one of the most common neoplasms in women and is a leading cause of cancer-related deaths worldwide. Improved diagnostic tools have made it possible to detect breast cancers at early stages, leading to a significant decrease in breast cancer mortality rates over the past decades (1). However, mortality rates of advanced-stage cancer have not decreased significantly because of a lack of effective therapies, and ≈25% of breast cancer patients will die of the disease (1). Therefore, the development and application of new molecularly based diagnostic and prognostic tools and therapies are of utmost importance. The key to the development of such rational preventive and therapeutic approaches lies in the identification of genes and biochemical pathways involved in breast tumorigenesis. One approach to the discovery of novel diagnostic and prognostic markers and therapeutic targets is to compare the gene expression profiles of normal and cancer cells and identify genes or subsets of genes with expression levels that correlate with tumor stage or clinical outcome. Several comprehensive gene expression profiling studies have been performed in breast cancer, and several novel putative molecular markers have been identified (2–6). Most of these studies used array-based platforms and, therefore, were inherently limited to the analysis of known genes and ESTs. Serial analysis of gene expression (SAGE) is an alternative comprehensive gene expression profiling technique that does not require the a priori knowledge of the transcripts present in the cells. Thus, it allows for the identification of novel transcripts, making it particularly suitable for the discovery of new molecular targets (7, 8).
In this study we used the SAGE technology to determine the comprehensive gene expression profiles of normal breast tissue and breast carcinomas of all clinical stages with the aim of identifying genes involved in the initiation and progression of breast tumorigenesis. This approach led to the identification of a previously uncharacterized growth and survival factor that is overexpressed in a significant fraction of invasive breast carcinomas with poor prognostic features.
Methods
Cell Lines and Tissue Specimens. Breast cancer cell lines were obtained from American Type Culture Collection or were generously provided by Steve Ethier (University of Michigan, Ann Arbor), Gail Tomlinson (University of Texas, Austin), and Arthur Pardee (Dana–Farber Cancer Institute). Cells were grown in media recommended by the provider. Tumor specimens were obtained from Brigham and Women's and Massachusetts General Hospitals (Boston), Duke University, University Hospital Zagreb (Zagreb, Croatia), and the National Disease Research Interchange, snap frozen on dry ice, and stored at –80°C until use. All human tissue was collected using protocols approved by the institutional review boards. Tissue microarrays were (i) obtained from Imgenex (San Diego), Ambion (Austin, TX), Ardais Corporation, and Gentaur (Brussels); (ii) provided by the Cooperative Breast Cancer Tissue Resource; and (iii) generated at Johns Hopkins University and at Beth Israel Deaconess Medical Center following published protocols (9). Brain samples were collected from autopsies performed at Brigham and Women's Hospital and from subjects prospectively enrolled in the Rapid Autopsy Program of the Joseph and Kathleen Price Bryan Alzheimer Disease Research Center at Duke University Medical Center (10).
RNA Preparation, mRNA in Situ Hybridization, and Northern Blot Analysis. We performed RNA isolation, RT-PCR, and Northern blot analyses as described (11). We performed mRNA in situ hybridization using paraffin sections and digitonin-labeled riboprobes following a protocol developed by St. Croix et al. (12), and we hybridized frozen sections as described with minor modifications (13).
Dermcidin (DCD) Expression in Mammalian Cells and Growth and Survival Assays. We generated an N-terminal alkaline phosphatase (AP) C-terminal DCD fusion protein using the AP-TAG-5 expression vector (GenHunter, Nashville, TN). We transfected mammalian cells with FuGENE6 (Roche), Lipofectamine, or Lipofectamine 2000 (Life Technologies, Rockville, MD) reagents. For mammalian expression, we subcloned the human DCD cDNA into the pBabe construct and confirmed DCD protein expression by immunoblot analysis. To determine the effect of DCD expression on cell growth, we plated 5,000 control (pBabe) or DCD-expressing (pBabe-DCD) cells per well in a 24-well plate, and 21NT cells were grown in either complete MCF10A medium (American Type Culture Collection) or MCF10A medium diluted 1:10 with basal MCF10A medium without growth factors added. Cells were counted (three wells per time point) on days 1, 3, 5, and 7 after plating. For menadione survival assays, 21NT pBabe and 21NT pBabe-DCD stable pools were plated (105 cells per well in a 24-well plate). At 6 h, cells were washed and medium was changed to serum-free DMEM-F12 medium with or without menadione (0, 100, and 200 μm; three wells per treatment), and cells were counted at 24 h. The experiment was repeated three times. For glucose deprivation assays, 21NT pBabe and 21NT pBabe-DCD stable pools were plated (5 × 104 cells per well in a 24-well plate). At 6 h, cells were washed and medium was changed to basal media with 5% or 0.5% horse serum and 0 or 4 mM glucose (three wells per treatment), and cells were counted at 48 h. The experiment was repeated three times.
Fluorescence in Situ Hybridization (FISH) and Array Comparative Genomic Hybridization (CGH). We obtained bacterial artificial chromosomes (BACs) containing the human DCD, CDK4, SAS, GLI, and MDM2 genes from Research Genetics (Huntsville, AL). The d1223 probe for identification of chromosome 12 was obtained from Vysis (Naperville, IL). We performed FISH to paraffin-embedded or frozen tissue and metaphase chromosomes from normal human lymphocytes as described (14). We performed BAC array CGH essentially as described (15). DNA copy number variations that deviated significantly (at least three times higher than the standard deviation of the overall fluorescence intensity of the tumor DNA) from background ratios measured in normal genomic DNA control hybridizations were considered real copy number variations. In the case of the BAC containing DCD, the average log fluorescence ratio was 0.3. The detailed results of the array CGH analysis of the 152 breast tumors will be reported elsewhere.
Antibodies and Immunoblot, Immunohistochemical, and Statistical Analyses. We generated an affinity-purified polyclonal anti-DCD antibody against a synthetic peptide (RQAPKPRKQRSS) corresponding to amino acids 53–64 of the human DCD protein (Zymed), and other antibodies used were obtained from sources previously described (6). We performed immunoblot analyses as described (11). We analyzed the expression of DCD in primary tumors by the use of immunohistochemistry to tissue microarrays that contained evaluatable paraffin-embedded specimens derived from ductal carcinoma in situ, primary invasive breast cancer and distant breast cancer metastases, pancreatic, gastric, prostate, kidney, and colon carcinomas, melanomas, lymphomas, and gliomas. Immunohistochemical and statistical analyses were performed as described (6).
Ligand Binding Assays. We performed in vivo and in vitro ligand binding assays on primary tissues and cell lines using AP-DCD essentially as described (16). Briefly, we fixed frozen sections of various human specimens, incubated with either AP-DCD fusion protein or AP control-conditioned medium, rinsed, and then incubated with AP substrate forming a blue/purple precipitate. For in vitro assays we incubated cells in suspension with conditioned medium containing either AP alone or AP-DCD fusion protein, rinsed, and then assayed for bound AP activity.
Results
Identification of Invasive Breast Cancer 1 (IBC-1)/DCD. To identify genes implicated in breast tumorigenesis we determined the gene expression profiles of normal mammary epithelial cells and in situ, invasive, and metastatic breast carcinomas using SAGE. Using this approach we identified a SAGE tag with no database match that was highly expressed only in a subset of invasive breast carcinomas (17, 18) designated IBC-1. Searching the human genome sequence with the IBC-1 SAGE tag and 5′ NlaIII site (5′-CATGACGTTAAAGAC-3′), we identified a genomic clone containing this tag and predicted (19) that it encodes a transcribed gene composed of five exons (Fig. 1A). Confirming the restricted expression pattern suggested by SAGE, based on Northern blot hybridization IBC-1 was expressed in only two regions of the brain: in the pons and, at a lower level, in the paracentral gyrus of the cerebral cortex, and not in 75 other normal human adult and fetal tissues (Fig. 1B). The predicted IBC-1 gene encodes a 110-aa protein with limited homology to lacritin and an EST containing an N-terminal signal peptide (Fig. 1 C and D). Further database searches using the predicted IBC-1 protein sequence revealed that IBC-1 nearly matches a 20-aa peptide derived from the mouse proteolysis-inducing factor or cachectic factor, and exactly matches a 30-aa neural survival-promoting peptide (20, 21) (Fig. 1B). While this work was in progress another group independently identified a cDNA from human sweat glands identical to IBC-1 and named it DCD (22). Thus, to avoid confusion due to multiple gene names, we renamed IBC-1 as DCD.
Fig. 1.

DCD and its homologues. (A) Genomic structure of the human IBC-1/DCD gene. Exon–intron boundaries, start-and-stop codons, and the SAGE tag that led to the identification of IBC-1/DCD are indicated. (B) Evaluation of DCD expression in 76 human adult and fetal tissues on a dot-blot expression array. High level of expression was detected only in the pons of the brain, whereas low-level expression was seen in the paracentral gyrus of cerebral cortex. (C) Human IBC-1/DCD cDNA and predicted amino acid sequence. Sequences of the peptides derived from cachectic factor and survival peptide are indicated by thick and thin underlining, respectively. An arrow marks the predicted secretory signal peptidase cleavage site. (D) Amino acid alignment of DCD, lacritin, and EST-AI12471 proteins. Amino acids identical to the consensus are shaded in gray. Comparison was made by using DNAStar and the clustal algorithm.
Expression of DCD in Breast Carcinomas and Correlation with Histopathologic Features. Next, we analyzed the expression of DCD in normal breast organoids, primary breast carcinomas, and breast cancer cell lines by Northern blot, RT-PCR, and mRNA in situ hybridization analyses and determined that it was expressed only in a subset of breast cancer cell lines and primary tumors (Fig. 2 A–C and data not shown). To determine the expression of DCD at the cellular level we performed mRNA in situ hybridization. Intense red or black (depending on hybridization protocol used) staining demonstrates that DCD is expressed in tumor cells and not in stromal cells (Fig. 2C). No signal was observed in adjacent normal mammary epithelial cells (Fig. 2C). In tumors 15 and 238 only a subset of cells showed high DCD expression indicating intratumoral heterogeneity (Fig. 2C).
Fig. 2.

Expression of DCD in normal and cancerous tissues. (A) Northern blot analyses of normal breast organoids, breast cancer cell lines, primary breast carcinomas, and corresponding normal breast tissue. High DCD expression is detected in only two tumors (15 and 236). The blot was rehybridized with β-actin to indicate equal loading. (B) RT-PCR analyses of breast cancer cell lines using DCD- and β-actin-specific primers. (C) mRNA in situ hybridization using digitonin-labeled DCD riboprobes on tissue sections (tumor and normal 236, red staining; tumors 15 and 238, black staining). Adjacent section stained with hematoxylin/eosin. (D) DCD immunostaining of normal breast tissue, ductal carcinoma in situ, a DCD-positive invasive breast carcinoma (IDC), and sweat gland of the skin. (E) Immunoblot analysis of human sweat and cells transfected with empty or DCD-expressing vector. An ≈11-kDa protein is detected in both transfected cells and sweat.
To evaluate the expression of the DCD protein we performed immunohistochemical analysis of several tissue microarrays composed of breast carcinomas (Fig. 2D). Correlating with our SAGE results we detected DCD expression in primary invasive breast carcinomas (48/558), and rarely in ductal carcinoma in situ (1/70) or distant metastases (1/49). Statistical analysis determined that DCD-positive breast tumors were more likely to be of advanced stage (tumor node metastasis stage 2 or 3, mostly due to higher T and N, P = 0.007) indicating that DCD expression correlates with larger tumor size and with the presence of metastatic lymph nodes. Because both of these tumor characteristics are known to predict a bad prognosis, we analyzed DCD expression in relation to overall and distant metastasis-free survival in a subset of breast tumors with clinical follow-up data. Patients with DCD-positive tumors appeared to have decreased overall and distant metastasis-free survival, but this decrease did not reach statistical significance (data not shown).
We also analyzed DCD expression in multiple human tumor types and found that 2/64 pancreatic carcinomas expressed DCD. Thus, DCD overexpression may occur in other human tumor types, but the determination of this will require the examination of large tumor sets from each tumor type. Although the staining of melanomas did not detect any DCD-positive tumor cells, adjacent sweat glands of the skin were strongly DCD-positive (Fig. 2D), confirming DCD expression in sweat glands (22).
Despite an extensive analysis of cell lines from various tumor types, we were not able to identify a cell line that endogenously expresses the DCD protein at levels detectable by Western blot analysis using our antibody (data not shown). Thus, to confirm that the DCD transcript we identified encodes a protein that exists in vivo, we performed immunoblot analysis of DCD-transfected cells and human sweat. Correlating with its predicted molecular weight, the exogenously expressed recombinant DCD protein migrates as a single ≈11-kDa protein, and a protein of approximately the same size is also detected in sweat (Fig. 2E). The slightly higher and lower molecular weight proteins recognized with our DCD antibody in the sweat may correspond to posttranslationally modified or partially proteolyzed DCD (Fig. 2E). These results confirm that a full-length DCD protein is expressed and secreted in vivo.
Focal Copy Number Gain of the DCD Locus in Breast Carcinomas. Based on the human genome sequence, DCD was localized to chromosome 12 in band q13.1, which we confirmed by FISH (Fig. 3B). Examination of the expression of all known and predicted genes in the vicinity (±5 megabases) of DCD determined that two genes localized next to DCD were expressed only in the same three breast carcinomas that expressed DCD and were not detected in any of the other >100 SAGE libraries (Fig. 3A and data not shown). This suggested that the overexpression of DCD in breast tumors may be due to gene amplification. To determine whether the DCD locus is amplified in the DCD-overexpressing tumors we performed FISH and detected moderate levels of DCD amplification in tumor I2 (Fig. 3B). We also analyzed several known oncogenes (CDK4, SAS, GLI1, and MDM2) localized to 12q and detected only CDK4 amplification in tumor I2 (Fig. 3B and data not shown). In three other tumors (I1, LN1, and 236) overexpressing DCD the FISH pattern was consistent with three to five copies of DCD and all of the other genomic regions tested, suggesting that a large part of chromosome 12 was gained (data not shown). However, based on SAGE, these oncogenes (MDM2, CDK4, SAS, etc.) were not overexpressed in DCD-positive breast tumors (Fig. 3A). To establish how frequently a gain of the DCD locus is detected in breast tumors, we analyzed an independent set of 152 breast tumors by using BAC array CGH and found a significant focal copy number increase of the DCD locus in 20 tumors (Fig. 3C).
Fig. 3.

Gain of DCD locus in breast carcinomas. (A) Idiogram of human chromosome 12 and the expression of genes adjacent to DCD in SAGE libraries generated from normal (N1and N2) breast tissue, and in situ (D1–8), invasive (I1–6), and metastatic (LN1–2 and M1) breast carcinomas. Genes closest to DCD (highlighted with yellow color), lacritin (LACRT), and, to a lesser extent, a phosphatase subunit (PPP1R1A) are expressed only in the three tumors with high levels of DCD, suggesting possible amplification of this chromosomal area. No other genes near DCD appear to be overexpressed in these breast tumors. (B) FISH analysis of DCD to normal metaphase chromosomes shows hybridization at 12q13 on both copies of chromosome 12 (Left). Hybridization of DCD (red) and an alpha-satellite probe to the centromere of chromosome 12 (green) reveals amplification of DCD and disomy of chromosome 12 in tumor I2 interphase cells (Center). Analysis of DCD (green) and CDK4 (red) reveals coamplification in tumor I2 interphase cells (Right). (C) A representative BAC array CGH profile demonstrating a gain of the DCD locus (arrow).
To further investigate the association between gain of the DCD locus and the overexpression of the DCD protein, we performed immunohistochemical analysis on eight breast tumors that showed a 12q13.1 focal copy number increase of the DCD locus based on BAC array CGH. Five of these eight tumors expressed the DCD protein, which is a much higher fraction than expected (P = 0.0003) based on the frequency of DCD positivity in unselected tumors (48/558). Thus, this result further suggests that at least in some cases DCD overexpression in breast tumors is due to a gain of the 12q13.1 chromosomal area.
DCD Is a Growth and Survival Factor for Breast Cancer Cells. To analyze the effect of DCD overexpression on breast cancer cell growth and survival we established derivatives of the 21NT breast cancer cell line, chosen based on its features resembling DCD-expressing primary breast tumors, that stably overexpressed DCD. Next we compared the growth of pools of control, empty vector transfected cells with that of cells expressing DCD and found that DCD-expressing 21NT cells grew significantly faster than controls, especially in reduced serum-containing medium (Fig. 4A). Similar results were obtained in DCD-expressing VA13-transformed fibroblasts and C2C12 myoblasts, whereas preliminary data suggest that DCD has no effect on immortalized mammary epithelial cells (data not shown).
Fig. 4.

DCD function and receptors. (A) Growth curves of control (pBabe) and DCD-expressing (pBabe-DCD) 21NT breast cancer cells in high (5%) or low (0.5%) serum-containing media. DCD-expressing cells grew significantly faster in both conditions. A representative experiment is shown. (B) Survival data showing reduced susceptibility of 21NT cells expressing DCD to cell death induced by menadione or glucose deprivation. Data are the mean of three experiments with three determinations each. (C) In situ staining for DCD receptor in breast and brain tissue. Sections of breast tumors, normal breast tissue, and brain were incubated with AP control or AP-DCD fusion protein. Purple staining detects the presence of a putative DCD receptor. Faint brownish coloring of neurons of the locus ceruleus and substantia nigra in the control AP sections is due to natural pigment (melanin) present in these cells. (D) Scatchard transformation of binding analysis of AP-DCD to 21NT breast cancer cells. (Inset) The actual binding curve. (E) Growth curves of cells treated with purified AP-DCD.
To determine the effect of DCD expression on cell survival after oxidative stress, we treated control and DCD-expressing 21NT cells with varying concentrations of menadione, a potent inducer of mitochondrial reactive oxygen species (ROS) production. As depicted in Fig. 4B Left, DCD-expressing cells were significantly more resistant to menadione-induced cell death than control 21NT cells. To establish whether the DCD-mediated protection from ROS-induced cell death is important in a more physiologic oxidative stress-inducing condition, we analyzed the effect of glucose deprivation on control 21NT and DCD-expressing cells. Cancer cells are known to be particularly sensitive to the withdrawal of glucose that leads to increased mitochondrial ROS production and subsequent cell death (23). Similar to the results obtained with menadione, DCD-expressing 21NT cells survived growth in glucose-free medium significantly better than control cells, with the most pronounced difference seen in low-serum-containing medium (Fig. 4B Right).
Cell Surface DCD Binding. The DCD protein is predicted to be secreted, suggesting that DCD is likely to execute its function through binding to a cell surface receptor. To determine whether there is a DCD-binding cell surface protein(s), we generated an AP-DCD fusion protein to be used as a ligand in receptor binding assays (16). Conditioned medium containing AP-DCD or control AP was used to stain normal and cancerous mammary tissue sections. Intense purple staining indicated the presence of a DCD-binding protein in tumor 236, but not in normal mammary epithelial and stromal cells, whereas low-intensity staining was observed in tumor 19 (Fig. 4C). These results suggested the presence of a cell surface DCD-binding protein(s) in cancerous, but not normal, mammary epithelial cells, and are consistent with an autocrine and/or paracrine mechanism of DCD action.
Because of its expression pattern in normal human brain, we also tested whether neurons bind DCD (Fig. 4C). Weak DCD binding to almost all neurons was seen in human adult brain (data not shown), whereas the strongest DCD binding was detected in neurons of the locus ceruleus, nucleus raphe pontis, substantia nigra, and the lateral hypothalamic nuclei (Fig. 4C).
To further test the binding characteristics of AP-DCD, we performed in vitro ligand binding assays using various cell lines. Low-level AP-DCD binding was detected in all cell lines tested, with stronger binding observed in human 21NT breast cancer cells (data not shown). To further characterize the AP-DCD-putative DCD-receptor interaction, we performed more detailed binding assays in 21NT breast cancer cells. Scatchard plot analysis showed two binding slopes in 21NT cells (Fig. 4D): one with a moderately high affinity (Kd = 1.5 × 10–8 M) and another with much lower affinity (Kd = 2.1 × 10–7 M). Further proving that DCD's effect is mediated through a cell surface receptor and that the AP-DCD fusion protein is a functional ligand for the putative DCD receptor, 21NT cells incubated in conditioned medium containing AP-DCD, or treated with purified AP-DCD, grew faster than controls (Fig. 4E and data not shown).
Discussion
Based on SAGE analysis of breast tumors of different clinical stages we identified DCD, a novel growth and survival factor for breast cancer cells. DCD encodes a secreted protein with limited homology to lacritin and an EST. Lacritin is a secretion-enhancing and growth-promoting factor recently identified from lacrimal gland (24). The EST is expressed in the cerebral cortex, and it encodes an uncharacterized protein containing a repetitive sequence (ETPA) found in several secreted proteins. In addition, two small proteolytic peptides identified as a cancer cachexia factor and a neural survival-promoting peptide, respectively, were likely to be derived from DCD (20, 21). The cachexia- and proteolysis-inducing factor was identified as a 24-kDa glycoprotein produced by the cachexia-inducing MAC 16 murine colon adenocarcinoma in mice and was shown to be present in the urine of cachectic cancer patients (25–27). The neural survival-promoting peptide was identified from the media of mouse HN33.1 hippocampal neurons and human Y79 retinoblasts treated with hydrogen peroxide and was shown to enhance neural survival after an oxidative insult (21, 28).
Based on FISH and array CGH analysis we determined that the overexpression of DCD in breast tumors is due to the focal copy number increase of the DCD locus. The low level of gain observed in array CGH could be due to the fact that the breast tumors used for this analysis were not microdissected; thus, contaminating stromal cells with two copies of the DCD locus may decrease the hybridization signal. In addition, as depicted in Figs. 2C and 3B, the expression and gain of DCD are heterogeneous in most breast tumors, with only a fraction of tumor cells being positive; thus, even a significant copy number gain may be detected only as a low level gain when the tissue is analyzed in bulk by using array CGH. Correlating with this, cDNA array CGH analysis of the tumors with significant DCD gain based on FISH revealed no significant copy number increase (data not shown). The minimum region of chromosomal gain based on BAC aCGH is 4.2 megabases, because the two flanking BACs that do not show gain are that far apart. This region encompasses DCD, CDK4, and SAS. However, based on SAGE, we did not see overexpression of any of these oncogenes (MDM2, CDK4, SAS, etc.) in DCD-positive breast tumors (Fig. 3A).
Consistent with being a putative oncogene, the overexpression of DCD in breast cancer cells enhanced cell growth and survival and reduced serum dependency. Because in the cell survival experiments performed with menadione cell viability was determined 24 h after plating the cells, the observed difference seen in live cell numbers is unlikely to be the effect of DCD on cell growth. Conversely, the effect of DCD on cell growth cannot fully be explained by its ability to protect against ROS generated because of culturing the cells under supraphysiologic (21% atmospheric) oxygen concentrations, as a similar effect was seen in cells grown at physiologic (3%) oxygen concentration (data not shown). Thus, the growth and survival-promoting effects of DCD appear to be distinct, although to conclusively prove this will require the detailed characterization of the DCD-signaling pathway. Correlating with the observed in vitro effect of DCD on breast cancer cell growth and survival, DCD-expressing primary breast tumors were larger and more likely to have metastatic lymph nodes. Based on these results, it is likely that the overexpression and copy number gain of DCD confer a selective advantage for breast tumor cells.
Based on in vivo ligand binding studies performed with an AP-DCD fusion protein, we detected strong cell surface DCD binding to breast cancer cells and neurons of the brain. Interestingly, catecholaminergic (noradrenergic and dopaminergic) neurons of the brain that strongly bound AP-DCD are particularly susceptible to oxidative stress because the biosynthesis of these neurotransmitters from tyrosine requires molecular oxygen. Moreover, the autooxidization of catecholamines, the end product of which is melanin that accumulates in neurons of the substantia nigra and locus ceruleus, leads to the generation of ROS (H2O2, 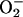 , and OH·). The strong binding of DCD to these neurons is consistent with the roles of a 30-aa neural survival factor (21) and a cachexia factor possibly derived from DCD (20). Definition of the relationships among these peptides requires further studies.
, and OH·). The strong binding of DCD to these neurons is consistent with the roles of a 30-aa neural survival factor (21) and a cachexia factor possibly derived from DCD (20). Definition of the relationships among these peptides requires further studies.
In summary, DCD is a novel growth and survival factor that is overexpressed in ≈10% of primary invasive breast carcinomas, and its overexpression, at least in some cases, is associated with a gain of its locus at 12q13.1. Based on its function and restricted expression pattern in normal adult tissues, DCD is a candidate cancer therapeutic target. The secreted nature and extracellular mechanism of DCD action make it even more attractive for such a purpose.
Neurons are particularly sensitive to ROS, whereas tumor cells themselves produce large amounts of ROS (29). Therefore, the high expression of DCD in these cell types may be essential for their survival. Thus, therapeutic activation of the DCD-signaling pathway may be beneficial in certain neurodegenerative diseases involving catecholaminergic neurons such as Parkinson's disease, and its therapeutic inhibition may be an effective treatment of tumors with DCD expression.
Acknowledgments
We thank Drs. Massimo Loda, Marcus Bosenberg, and Gabriela Lodeiro for their help with this study. We are indebted to Wen-Lin Kuo, Fred Waldman, Sandy DeVries, and Joe W. Gray (University of California, San Francisco) for providing their BAC array CGH data before publication. This work was supported in part by the National Cancer Institute Cancer Genome Anatomy Project and Specialized Programs of Research Excellence in Breast Cancer at Dana–Farber/Harvard Cancer Center (to K.P.), Johns Hopkins University (to Y.K.B. and E.G.), the University of California, San Francisco (to Joe W. Gray), National Institute on Aging Grant AG05128 (to C.M.H.), and U.S. Army postdoctoral fellowships (to P.S., A.M.-S., and D.P.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AP, alkaline phosphatase; FISH, fluorescence in situ hybridization; BAC, bacterial artificial chromosome; CGH, comparative genomic hybridization; DCD, dermcidin; IBC-1, invasive breast cancer 1; ROS, reactive oxygen species; SAGE, serial analysis of gene expression.
References
- 1.Greenlee, R. T., Murray, T., Bolden, S. & Wingo, P. A. (2000) CA Cancer J. Clin. 50, 7–33. [DOI] [PubMed] [Google Scholar]
- 2.Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., Pollack, J. R., Ross, D. T., Johnsen, H., Akslen, L. A., et al. (2000) Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- 3.Sorlie, T., Perou, C. M., Tibshirani, R., Aas, T., Geisler, S., Johnsen, H., Hastie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Vijver, M. J., He, Y. D., van't Veer, L. J., Dai, H., Hart, A. A., Voskuil, D. W., Schreiber, G. J., Peterse, J. L., Roberts, C., Marton, M. J., et al. (2002) N. Engl. J. Med. 347, 1999–2009. [DOI] [PubMed] [Google Scholar]
- 5.van't Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., Peterse, H. L., van der Kooy, K., Marton, M. J., Witteveen, A. T., et al. (2002) Nature 415, 530–536. [DOI] [PubMed] [Google Scholar]
- 6.Porter, D., Lahti-Domenici, J., Keshaviah, A., Bae, Y. K., Argani, P., Marks, J., Richardson, A., Cooper, A., Strausberg, R., Riggins, G. J., et al. (2003) Mol. Cancer Res. 1, 362–375. [PubMed] [Google Scholar]
- 7.Velculescu, V. E., Zhang, L., Vogelstein, B. & Kinzler, K. W. (1995) Science 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 8.Polyak, K. & Riggins, G. J. (2001) J. Clin. Oncol. 19, 2948–2958. [DOI] [PubMed] [Google Scholar]
- 9.Kononen, J., Bubendorf, L., Kallioniemi, A., Barlund, M., Schraml, P., Leighton, S., Torhorst, J., Mihatsch, M. J., Sauter, G. & Kallioniemi, O. P. (1998) Nat. Med. 4, 844–847. [DOI] [PubMed] [Google Scholar]
- 10.Hulette, C. M., Welsh-Bohmer, K. A., Crain, B., Szymanski, M. H., Sinclaire, N. O. & Roses, A. D. (1997) Arch. Pathol. Lab. Med. 121, 615–618. [PubMed] [Google Scholar]
- 11.Krop, I. E., Sgroi, D., Porter, D. A., Lunetta, K. L., LeVangie, R., Seth, P., Kaelin, C. M., Rhei, E., Bosenberg, M., Schnitt, S., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9796–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.St. Croix, B., Rago, C., Velculescu, V., Traverso, G., Romans, K. E., Montgomery, E., Lal, A., Riggins, G. J., Lengauer, C., Vogelstein, B. & Kinzler, K. W. (2000) Science 289, 1197–1202. [DOI] [PubMed] [Google Scholar]
- 13.Qian, Y., Fritzsch, B., Shirasawa, S., Chen, C. L., Choi, Y. & Ma, Q. (2001) Genes Dev. 15, 2533–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuchinka, B. D., Kalousek, D. K., Lomax, B. L., Harrison, K. J. & Barrett, I. J. (1995) Mod. Pathol. 8, 183–186. [PubMed] [Google Scholar]
- 15.Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., Collins, C., Kuo, W. L., Chen, C., Zhai, Y., et al. (1998) Nat. Genet. 20, 207–211. [DOI] [PubMed] [Google Scholar]
- 16.Flanagan, J. G. & Leder, P. (1990) Cell 63, 185–194. [DOI] [PubMed] [Google Scholar]
- 17.Lal, A., Lash, A. E., Altschul, S. F., Velculescu, V., Zhang, L., McLendon, R. E., Marra, M. A., Prange, C., Morin, P. J., Polyak, K., et al. (1999) Cancer Res. 59, 5403–5407. [PubMed] [Google Scholar]
- 18.Porter, D. A., Krop, I. E., Nasser, S., Sgroi, D., Kaelin, C. M., Marks, J. R., Riggins, G. & Polyak, K. (2001) Cancer Res. 61, 5697–5702. [PubMed] [Google Scholar]
- 19.Burge, C. & Karlin, S. (1997) J. Mol. Biol. 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 20.Cariuk, P., Lorite, M. J., Todorov, P. T., Field, W. N., Wigmore, S. J. & Tisdale, M. J. (1997) Br. J. Cancer 76, 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham, T. J., Hodge, L., Speicher, D., Reim, D., Tyler-Polsz, C., Levitt, P., Eagleson, K., Kennedy, S. & Wang, Y. (1998) J. Neurosci. 18, 7047–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schittek, B., Hipfel, R., Sauer, B., Bauer, J., Kalbacher, H., Stevanovic, S., Schirle, M., Schroeder, K., Blin, N., Meier, F., et al. (2001) Nat. Immunol. 2, 1133–1137. [DOI] [PubMed] [Google Scholar]
- 23.Spitz, D. R., Sim, J. E., Ridnour, L. A., Galoforo, S. S. & Lee, Y. J. (2000) Ann. N.Y. Acad. Sci. 899, 349–362. [DOI] [PubMed] [Google Scholar]
- 24.Sanghi, S., Kumar, R., Lumsden, A., Dickinson, D., Klepeis, V., Trinkaus-Randall, V., Frierson, H. F., Jr., & Laurie, G. W. (2001) J. Mol. Biol. 310, 127–139. [DOI] [PubMed] [Google Scholar]
- 25.McDevitt, T. M. & Tisdale, M. J. (1992) Br. J. Cancer 66, 815–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todorov, P. T., McDevitt, T. M., Cariuk, P., Coles, B., Deacon, M. & Tisdale, M. J. (1996) Cancer Res. 56, 1256–1261. [PubMed] [Google Scholar]
- 27.Todorov, P., Cariuk, P., McDevitt, T., Coles, B., Fearon, K. & Tisdale, M. (1996) Nature 379, 739–742. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham, T. J., Jing, H., Akerblom, I., Morgan, R., Fisher, T. S. & Neveu, M. (2002) Exp. Neurol. 177, 32–39. [DOI] [PubMed] [Google Scholar]
- 29.Szatrowski, T. P. & Nathan, C. F. (1991) Cancer Res. 51, 794–798. [PubMed] [Google Scholar]