

Abstract
Nasopharyngeal carcinoma is a malignancy that is prevalent among populations from Southeast Asia. Epidemiological studies indicate that genetic predisposition, Epstein–Barr virus, and environmental conditions may play a role in determining incidence. Molecular studies have implicated a tumor suppressor gene(s) on the short arm of chromosome 3. In this study we provide functional evidence, via monochromosome transfer, for a tumor suppressor gene(s) activity in chromosome 3p21.3.
Nasopharyngeal carcinoma (NPC) is a cancer that is rare among most populations but is particularly prevalent among the southern Chinese (1). Several cofactors are believed to contribute to the development of this malignancy. These include Epstein–Barr virus, genetic factors, and certain dietary and environmental conditions (2–5). Evidence for a genetic contribution to the disease includes differential susceptibility to NPC among northern and southern Chinese, the continued increased risk of NPC among migrants to other countries, and the familial aggregation of the disease. Thus, the high incidence of NPC in a specific genetic population suggests a genetic predisposition to the disease.
Multistep progression, involving multiple genetic alterations, is a general feature of human malignancies (6). With the advent of molecular genetic technology the molecular events, involving both activation of oncogenes and inactivation of tumor suppressor genes, have been elucidated for initiation and progression of certain malignancies, e.g., colorectal cancer and head and neck cancers (7, 8). For tumor suppressor genes, the eventual isolation and identification of the relevant gene often involves the arduous task of positional cloning (6, 9). Thus, functional evidence for tumor suppression is an extremely useful correlative to the identification of loss of heterozygosity (LOH) at specific chromosome locations before candidate gene cloning. Monochromosome transfer has been shown to be useful in this regard (10–13).
Very little is known about the molecular genetic changes that are associated with NPC. Studies to date have shown that mutations in the p53 tumor suppressor gene (TSG) are relatively rare compared with other common malignancies (14). Also, no detectable retinoblastoma susceptibility gene alterations have been observed (15).
Cytogenetic analyses have been somewhat more revealing. Analyses of NPC biopsy specimens, NPC xenografts, and derivative cell lines have identified common abnormal markers, including those derived from chromosomes 1, 3, 9, 11, 12, and 17 (16, 17). An obvious candidate TSG on chromosome 9 is p16INK4, a cdk inhibitor that maps to 9p21 (18, 19). That possibility was found not to be the case (20); however, other candidate TSGs map to this region, e.g., p15 (21). It should be noted that decreased expression of the p16/MTS1 gene was seen in two NPC cell lines in the absence of point mutations (20), an epigenetic event that has been associated with gene silencing via methylation (22).
Chromosome 3 presents a promising target. NPC biopsy samples occasionally contain 3q+ markers (16, 17). However, probably the most frequent chromosome 3 abnormality observed involves 3p deletions. These have been observed both cytogenetically and by LOH analyses. In particular, LOH studies have indicated multiple regions of involvement, including 3p13–3p14.3, 3p23, and 3p25 (23–25). The most frequent deletions have been found in the chromosome 3p13–3p14.3 region. Multiple regions of LOH on chromosome 3p have been associated with a variety of malignancies, including lung, renal, and uterine cervix carcinomas (26–30). Candidate TSGs that have been cloned from these map locations include the Von Hippel Lindau gene that maps to 3p25 and the FHIT gene at 3p14.2 (31, 32).
We have used the technique of microcell fusion to transfer single normal chromosomes as a functional assay for the TSG activity (10–12). This has been particularly useful in confirming TSG functions associated with specific chromosomes where the map location is suspected but no candidate TSG has been cloned. Also, by transferring chromosomes possessing interstitial deletions, it has been possible to map the location of TSGs more precisely (33).
In this study we have used a series of deleted copies of human chromosome 3 derived from normal cells, with discrete interstitial deletions in the p arm, for transfer into the HONE1 nasopharyngeal carcinoma cell line.
MATERIALS AND METHODS
Cell Lines And Culture Conditions.
The NPC cell line HONE1, used in this study, was established from a poorly differentiated nasopharyngeal squamous cell carcinoma (34). It was used as the recipient for all of the microcell fusion experiments. The copies of the various intact and truncated copies of chromosome 3 were retained in mouse A9 microcell hybrids and were the only human chromosomal DNA in those cells (E.J.S., unpublished results and ref. 35). Retention of the neomycin-resistance-gene-tagged chromosomes was accomplished by culturing the cells in growth medium containing Geneticin (G418; 400 μg/ml). The HONE1–chromosome 3 microcell hybrids were also selected in growth medium containing G418 (400 μg/ml). Growth medium for all cell lines consisted of DMEM supplemented with 10% fetal calf serum. All cultures were regularly monitored for mycoplasma contamination and were uniformly negative. The copies of human chromosome 3 were all derived from normal diploid fibroblasts (E.J.S., unpublished results).
Microcell-Mediated Chromosome Transfer.
Transfer of the individual copies of human chromosome 3 was accomplished by microcell fusion as described (12). After fusion and a 24-h recovery period, the HONE1–chromosome 3 microcell hybrids were selected in growth medium containing G418 (400 μg/ml) and hypoxanthine/aminopterin/thymidine (HAT). The HAT selection was used to eliminate any contaminating neomycin-resistant hypoxanthine phosphoribosyltransferase-deficient A9–chromosome 3 donor cells.
DNA Slot Blot Assay.
This assay was used to detect any contaminating mouse DNA in the HONE1–chromosome 3 microcell hybrids. Five micrograms of genomic DNA from all cell lines was transferred directly to a nitrocellulose membrane with a Minifold II slot blot apparatus and hybridized to total mouse A9 genomic DNA (50 ng). Mouse DNA probes were radioactively labeled by random priming in the presence of [α-32P]dATP (36). Blots were hybridized at 65°C and washed for 15 min with 0.1× SSC/0.1% SDS, at 65°C for 10 min, before autoradiography.
Microsatellite Analysis.
Hybrid cell line DNAs were genotyped by using semi-automated fluorescent PCR-based analysis on an Applied Biosystems model 373 DNA sequencer (37). Primer sequences flanking the 28 chromosome 3p microsatellites used in this study were obtained from the Genome Data Base (http://gdbwww.gdb.org/gdb/map). Genetic and cytogenetic map positions were obtained from Genethon (38) and Genome Data Base, respectively. Markers were grouped into multiplex panels as described (37, 39) and coamplified with AmpliTaqGold on an Applied Biosystems model 877 Turbo catalyst workstation using the AmpliTaqGold linkage mapping set conditions recommended by the manufacturer. Coelectrophoresis of PCR products was carried out as described (37, 39). Presence or absence of a given marker product in the cell line DNAs was assessed by using genescan software and confirmed independently at least two times. Independent isolates of DNA from the hybrid line MCH924.4 were also assessed for the presence or absence of the 3p markers and eight additional markers mapping to 3q (D3S2459, D3S1769, D3S1273, D3S1744, D3S3053, D3S1754, D3S2436, and D3S3054) by using the same amplification conditions described above on an Applied Biosystems model 9600 GeneAmp PCR machine. PCR products were visualized on 2% agarose gels.
Fluorescent in Situ Hybridization (FISH).
Metaphase spreads were prepared by following published procedures (40). The chromosome 3 content of HONE1 cells, A9–chromosome 3 MCH donors, and HONE1–chromosome 3 MCH clones were determined by using a chromosome 3-specific library probe (Oncor). The probe was labeled with digoxigenin and hybridization was detected with fluorescein-conjugated anti-digoxigenin antibodies. The chromosome painting was done essentially by the technique of Gray et al. (41) with modifications suggested by Oncor. Slides were counterstained with 4,6-diamidino-2-phenylindole and the fluorescent signals were viewed with a Zeiss axiophot epifluorescence microscope equipped with a triple band filter. The images were captured and digitally enhanced by using the Oncor Imaging System and Adobe photoshop.
Tumorigenicity Assays.
Six- to 10-week-old female athymic nude mice were used to assay tumor formation. This assay has been repeatedly and successfully used for measuring the degree of tumor suppression in microcell hybrid populations (10–12). One × 107 cells suspended in 0.2 ml of DMEM were inoculated subcutaneously into each of six sites (two sites per animal). The animals were monitored regularly for tumor formation and palpable nodules were measured with calipers. If tumor formation was noted, then representative tumors were reconstituted into cell culture for FISH and microsatellite analyses.
RESULTS
Analysis of the A9–Chromosome 3 Microcell Hybrids.
The G banding analyses of the presumed intact and truncated copies of chromosome 3 retained within the clones of mouse A9 cells are shown in Fig. 1. Both MCH903.1 and MCH906.15 give a banding pattern consistent with an intact chromosome 3. Microcell hybrid MCH939.2 shows a discrete interstitial 3p deletion. MCH910.7 has a more significant deletion and MCH924.4 has a small metacentric chromosome. The identity of the arms of this latter truncated chromosome could not be ascertained.
Figure 1.
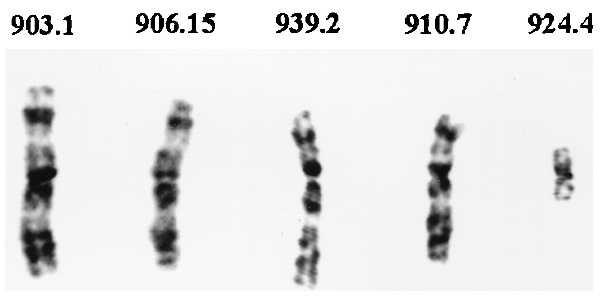
G banding of the intact and truncated copies of chromosome 3. In each case, the copy of chromosome 3 illustrated was present as the only human chromosome in a mouse A9 microcell hybrid clone (see ref. 35 for more detailed information).
The molecular analysis of these chromosomes, based upon multiplex PCR analyses of polymorphic microsatellites, was more informative (Fig. 2). MCH903.1 appeared to be intact by this analysis; however, MCH906.15 had suffered a discrete deletion that includes the D3S1211 and D3S1619 markers, a region that contains the gene for transforming growth factor β receptor type II (42). MCH939.2 also showed discrete deletions in two regions, flanked by the markers D3S1286 and D3S1266, and D3S1298 and D3S1295, respectively. Surprisingly, given the significant reduction in size of the chromatid arms revealed by G banding, the molecular deletions seen in MCH924.4, although multiple, leave a significant portion of the 3p chromosome arm apparently intact. To account for the reduced size of the transferred chromosome 3 in this cell line, we hypothesized that material from 3q, in addition to 3p, may also have been deleted. To test this possibility, eight markers from 3q spanning centimorgans 141.9 to 271.6 on the chromosome 3 genetic map (Chelsea Cooperative Human Linkage Centre Version 4.0 Sex-Averaged Recombination Minimization Maps) were assessed for the presence or absence of PCR products in the MCH924.4 cell line. Of the eight loci tested, four markers distal to centimorgan 196.7 were absent, suggesting deletion of at least half of the long arm of this chromosome.
Figure 2.
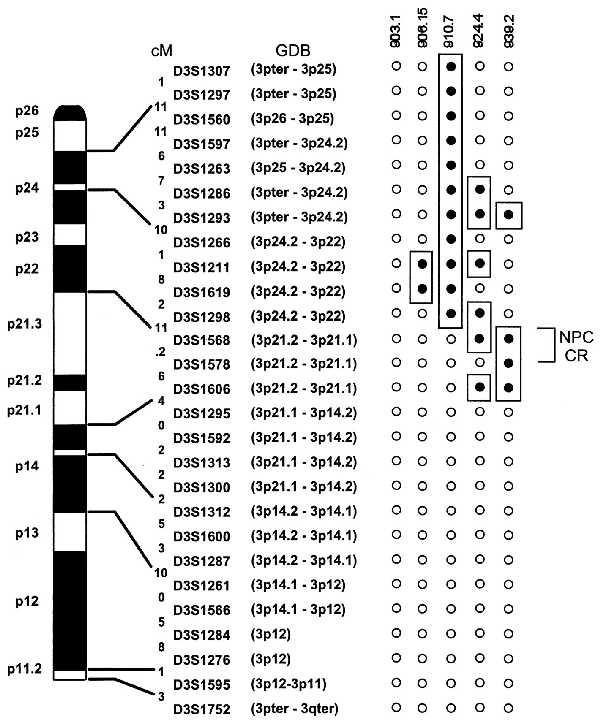
(Left) Ideogram of the short arm of chromosome 3. The physical order, genetic, and cytogenetic distances between the 28 microsatellite loci were derived from the 1996 Genethon Map (38) and Genome Data Base. Although the map position of locus D3S1568 is stated as 3p21.2–3p21.1 in Genome Data Base, a cosmid containing this marker has been assigned to 3p21.3 (S. Pack and M.I.L., unpublished results; see text). (Right) The genotype results of the five A9–chromosome 3 microcell hybrid DNAs are shown next to the ideogram. ○, Presence of a marker product; •, absence of a marker product. The extent of the chromosome 3p deletions in the cell lines is indicated by the open boxes. NPC CR, NPC critical region.
Transfer of Chromosome 3 into NPC cells.
Microcell fusion products were selected in medium containing G418 (400 μg/ml). Colonies that grew in this selective medium were picked, expanded, and subjected to chromosomal analysis. Those clones in which a majority (>90%) of the cells contained the transferred chromosome, as ascertained by FISH and G banding analysis, were selected for further study. Representative FISH analyses are shown in Fig. 3. It should be noted that there is no intact normal copy of chromosome 3 in the parental HONE1 cells. Most of the identifiable chromosome 3 material was found to be translocated to other chromosomes. The most intact copy contained nonchromosomal 3 material at one of the ends. Because of the significant degree of rearrangement of chromosome 3 material noted by G banding analysis (data not shown), no effort was made to analyze these chromosomes further. In contrast, successful and stable transfer of the various donor copies of chromosome 3 was readily identified (arrowheads). Microsatellite analysis confirmed the transfer of the various donor chromosomes (data not shown). Two to three clones of each respective chromosome 3 transfer were selected for further study.
Figure 3.
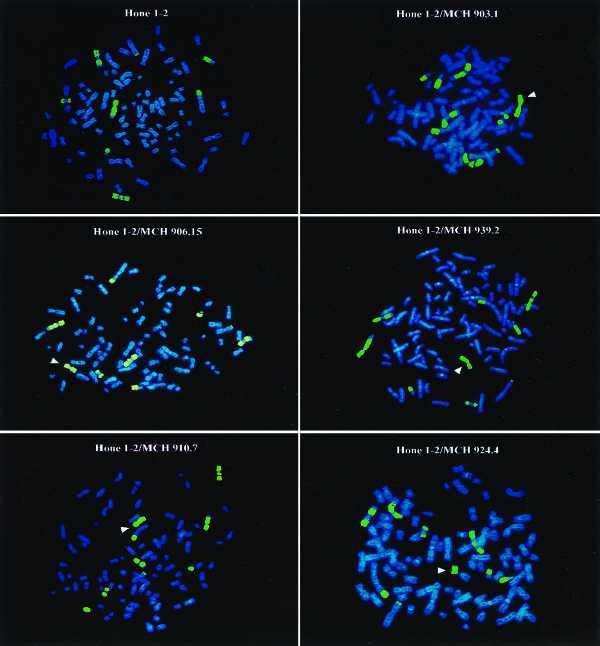
FISH analysis of parental HONE1 cells and HONE1–chromosome 3 microcell hybrids. The copies of chromosome 3 are visualized by in situ hybridization using a chromosome 3-specific library probe. The transferred chromosome 3 (via microcell fusion) is identified in each case by an arrowhead. The identity of the transferred chromosome is indicated by identifying the A9–chromosome 3 donor cell.
Tumorigenicity Assays.
No significant differences in in vitro growth kinetics were seen in any of the selected clones (data not shown). However, very significant differences in tumor-forming potential were seen (Table 1). The parental NPC line HONE1 is highly tumorigenic; palpable tumors formed within 15 days of inoculation of 1 × 107 cells into the subcutaneous site in 100% of animals. Tumorigenicity was strongly suppressed by an intact or truncated copy of chromosome 3 derived from MCH903.1, MCH906.15, and MCH910.7, respectively. There was either complete suppression of tumorigenicity or tumors arose after a significantly extended lag period. This is strongly indicative of an in vivo selection of tumorigenic segregants from a majority population of nontumorigenic cells. In contrast, transfer of the truncated copies of chromosome 3 derived from MCH924.4 and MCH939.2, respectively, had no effect on tumor-forming ability. The kinetics of tumor formation of the various HONE1–chromosome 3 clones are shown in Fig. 4.
Table 1.
Tumorigenicity assays of parental HONE1 and HONE1–chromosome 3 microcell hybrid clones
| Cell line | Identification | Tumor formation, no. tumors/no. sites | Time to appearance of tumors, days |
|---|---|---|---|
| HONE1 | Parental NPC cells | 6/7 | 15–20 |
| MCH4.5 | HONE1 × MCH903.1 | 3/6 | 35 |
| MCH4.6 | HONE1 × MCH903.1 | 2/6 | 40–45 |
| MCH4.8 | HONE1 × MCH903.1 | 1/6 | 40–45 |
| MCH8.4 | HONE1 × MCH906.15 | 0/6 | — |
| MCH8.8 | HONE1 × MCH906.15 | 3/6 | 40–55 |
| MCH8.12 | HONE1 × MCH906.15 | 1/6 | 60 |
| MCH12.1 | HONE1 × MCH910.7 | 0/6 | — |
| MCH12.7 | HONE1 × MCH910.7 | 0/6 | — |
| MCH12.10 | HONE1 × MCH910.7 | 3/6 | 50–100 |
| MCH11.3 | HONE1 × MCH939.2 | 6/6 | 25–35 |
| MCH11.8 | HONE1 × MCH939.2 | 6/6 | 30–35 |
| MCH5.1 | HONE1 × MCH924.4 | 6/6 | 15–20 |
| MCH5.2 | HONE1 × MCH924.4 | 6/6 | 20–25 |
| MCH5.5 | HONE1 × MCH924.4 | 6/6 | 25 |
Figure 4.
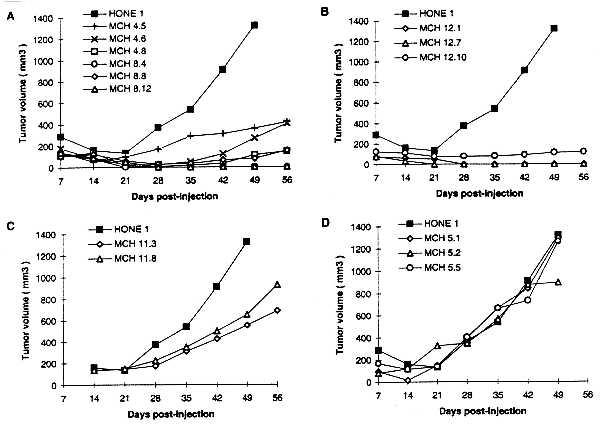
Kinetics of tumor formation of parental HONE1 cells and HONE1–chromosome 3 microcell hybrid clones. The curves represent an average of the tumor volumes of all sites inoculated for each cell population. The identity of the cell lines tested is in Table 1, as well as more detailed information on the degree of tumor suppression.
By comparing the regions of deletion in the copies of the transferred chromosome 3 homologs that either suppressed (MCH-903.1, -906.15, and -910.7) or did not suppress (MCH-924.4 and -939.2) tumor formation, the likely map position of the NPC tumor suppressor gene(s) could be assigned (see Fig. 2). Comparison of the hybrid lines MCH906.15, MCH910.7, and MCH939.2, which contained one or at most two discrete deletions, placed the tumor suppressor activity in a 21.2-centimorgan region proximal to D3S1298 and distal to D3S1295. The addition of MCH924.4, which had suffered multiple discrete deletions along 3p and which like MCH939.2 did not suppress tumorigenicity, suggested that the position of the NPC gene(s) was contained within an 11.2-centimorgan region bounded by D3S1298 and D3S1578 and centered around D3S1568. FISH analysis using a cosmid containing sequences of the D3S1568 marker showed that this locus maps to 3p21.3 (S. Pack and M.I.L., unpublished results), which is consistent with the reported map positions of the flanking markers D3S1298 and D3S1295 within band 3p21 (43).
Cytogenetic and Molecular Analysis of Tumor Segregants.
Representative tumors that grew in the athymic nude mice were reconstituted into culture and expanded. As soon as there were sufficient cells (two or three passages), cytogenetic and genotype analyses were carried out. In the vast majority of cases, the tumor reconstitutes were shown to have retained the transferred chromosome by FISH analysis (data not shown). For those tumors that had not been suppressed (MCH5 and MCH 11 clones), this was to be expected. When tumor suppression was seen, the tumor segregants are assumed to have grown due to the loss of tumor suppressor activity. This could be due to chromosomal loss, discrete deletion of the putative tumor suppressor gene, or epigenetic silencing due to methylation or some other gene silencing mechanism. PCR analysis of select tumorigenic segregant populations with markers in the D3S1578–D3S1606 region were uninformative; either the polymorphism was the same as one of the parental HONE1 alleles or one or more of the alleles were indistinguishable among the different microcell hybrid cell lines (data not shown). Thus, although total chromosomal loss was excluded, the other two mechanisms of loss of tumor suppressor activity remain possibilities to be explored.
DISCUSSION
NPC is a cancer that is prevalent among southern Chinese (1). This is a particularly interesting cancer from a molecular epidemiological standpoint because it appears to involve genetic predisposition, Epstein–Barr virus infection, and environmental exposures (2–5). There is a strong correlation between Epstein–Barr virus and NPC, but the role that this virus plays in initiation or progression is unclear. Unlike many other human cancers, very little is known about the potential roles of known oncogenes and tumor suppressor genes in NPC. For example, there is no consistent association between any oncogene or a number of TSGs, including p53, pRB, and p16/MTS1 (14, 15, 18). However, cytogenetic analyses and allelotyping studies have identified a number of chromosome regions that may harbor a TSG. This is particularly true of the short arm of chromosome 3, where several regions have been implicated (16, 17).
We have tested the possibility that tumor suppressing activity can be restored to the NPC cell line HONE1 via transfer of a normal chromosome 3. Furthermore, when this was successfully accomplished, we regionalized the TSG activity to the 3p21.3 region by using truncated chromosomes, a method that has been used to identify a second Wilms’ tumor suppressor locus (33). Interestingly, the most common regions of LOH identified in NPC tumors, namely, 3p14 and 3p25 (16, 17), did not contain a TSG activity for the HONE1 cell line. The 3p14 region contains the candidate TSG FHIT (32). However, we found no evidence of loss or rearrangement of the FHIT gene in HONE1 cells (G.H., unpublished observations). Also, we found no evidence for involvement of the Von Hippel Lindau TS gene, which maps to 3p25 (31). Involvement of the 3p21.3 region has been noted in LOH studies (23) and is confirmed by the functional analysis described herein.
The chromosome 3p21.3 region has been implicated as a region of loss in various human malignancies, including lung, breast, cervix, and renal carcinoma (26–30). Functional studies, using transfection of P1 clones containing an 80-kb fragment of DNA from the 3p21.3 region and transfer of the same or similar chromosome 3 fragments via microcell fusion, have resulted in tumor suppression of the A9 mouse fibrosarcoma cell lines (44–46). Candidates for such a tumor suppressing activity include a number of genes including a member of the semaphorin family and BAP-1 (ref. 26; F. Rauscher, personal communication). Equally as probable, of course, is the existence of another TSG. Given the frequent involvement of the chromosome 3p21.3 in multiple human cancers, it suggests that this region may harbor multiple TSGs that are involved in many different cancers.
Although we have identified a candidate TSG in chromosome 3p21.3 that is defective in HONE1 cells, this does not preclude other TSGs at other regions of the short arm of chromosome 3 that may play a role in NPC. The functional studies described herein should prove useful in this regard when independently derived NPC cell lines are used. The identification of the candidate TSG on chromosome 3p21.3 should be assisted by molecular analysis of HONE1–chromosome 3 microcell hybrids that have regained their tumor-forming properties. These studies are currently in progress.
Acknowledgments
We thank Ronald Glaser for the HONE1 cell line. We are grateful for the assistance of Haiyan Ge, Ulla Bengtsson, and Amy Larson. These studies were supported by National Cancer Institute Grant CA19401 (to E.J.S.) and the Research Grants Council of Hong Kong.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: TSG, tumor suppressor gene; NPC, nasopharyngeal carcinoma; LOH, loss of heterozygosity; FISH, fluorescent in situ hybridization.
References
- 1.Waterhouse, J., Muir, C., Shanmugaratham, K., Powell, J., Peacham, D. & Whelan, S. (1982) IARC Sci. Publ. 4, no. 42.
- 2.Henle W, Henle G. In: Advances in Viral Oncology. Klein G, editor. New York: Raven; 1985. pp. 201–238. [Google Scholar]
- 3.Chan S H, Day N E, Kunaratnam N, Chua K B, Simons M J. Int J Cancer. 1983;32:171–176. doi: 10.1002/ijc.2910320206. [DOI] [PubMed] [Google Scholar]
- 4.Yu M D, Garabrant D H, Huang T B, Henderson B E. Int J Cancer. 1990;45:1033–1039. doi: 10.1002/ijc.2910450609. [DOI] [PubMed] [Google Scholar]
- 5.Lu S J, Day N E, Degos L, Lepage V, Wang P C, Chan S H, Simons M, McKnight B, Easton D, Zeng Y, De Thé G. Nature (London) 1990;346:470–471. doi: 10.1038/346470a0. [DOI] [PubMed] [Google Scholar]
- 6.Stanbridge E J. Annu Rev Genet. 1990;24:615–657. doi: 10.1146/annurev.ge.24.120190.003151. [DOI] [PubMed] [Google Scholar]
- 7.Fearon E, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 8.Sidransky D. Curr Opin Oncol. 1995;7:229–233. doi: 10.1097/00001622-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Kinzler K, Nilbet M D, Su L-K, Vogelstein B, Bryan T M, Levy D B, Groffen J, Boguski M S, Altschul S F, Horii A, et al. Science. 1991;253:661–664. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 10.Saxon P J, Srivatsan E S, Stanbridge E J. EMBO J. 1986;5:3461–3466. doi: 10.1002/j.1460-2075.1986.tb04670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weissman B E, Saxon P J, Pasquale S R, Jones G R, Geiser A G, Stanbridge E J. Science. 1987;236:175–180. doi: 10.1126/science.3031816. [DOI] [PubMed] [Google Scholar]
- 12.Goyette M D, Cho K, Fasching C L, Levy D B, Kinzeler K W, Paraskeva C, Vogelstein B, Stanbridge E J. Mol Cell Biol. 1992;12:1387–1395. doi: 10.1128/mcb.12.3.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satoh H, Lamb P W, Dong J T, Everitt J, Boreiko C, Oshimura M, Barrett J C. Mol Carcinogen. 1993;7:157–164. doi: 10.1002/mc.2940070306. [DOI] [PubMed] [Google Scholar]
- 14.Effert P, McCoy R, Abdel-Hamid M, Flynn K, Zhang Q, Busson P, Tursz T, Liu E, Raab-Traub N. J Virol. 1992;66:3768–3775. doi: 10.1128/jvi.66.6.3768-3775.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y, Hegamyer G, Colburn N H. Oncogene. 1993;8:791–795. [PubMed] [Google Scholar]
- 16.Huang D P, Ho J H C, Chan W K, Lau W H, Lui M. Int J Cancer. 1989;43:936–939. doi: 10.1002/ijc.2910430535. [DOI] [PubMed] [Google Scholar]
- 17.Mitelman F, Mark-Vendel E, Mineur A, Giovanella B, Klein G. Int J Cancer. 1983;32:651–655. doi: 10.1002/ijc.2910320602. [DOI] [PubMed] [Google Scholar]
- 18.Huang D P, Lo K-W, van Hassett C A, Woo J K S, Choi P H K, Leung S-F, Sheung S-T, Cairns P, Sidransky D, Lee J C K. Cancer Res. 1996;54:4003–4006. [PubMed] [Google Scholar]
- 19.Kamb A, Gruis N A, Weaver-Feldhaus J, Liu Q, Harshman K, Tartigian S V, Stockert E, Day R S, III, Johnson B E, Skolnick M H. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Hildesheim A, Lanier A E P, Cao Y, Yao K-T, Raab-Traub N, Yang C-S. Oncogene. 1995;10:785–788. [PubMed] [Google Scholar]
- 21.Hannon G J, Beach D. Nature (London) 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 22.Merlo A, Herman J G, Mao L, Lee D J, Garielson E, Burger P C, Baylin S B, Sidransky D. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 23.Huang D P, Lo R-W, Shoi P H K, Ng A Y T, Tsao S-Y, Yiu G K C, Lee J C K. Cancer Genet Cytogenet. 1991;54:91–99. doi: 10.1016/0165-4608(91)90035-s. [DOI] [PubMed] [Google Scholar]
- 24.Lo R-W, Tsao S-W, Leung S-F, Choi P H K, Lee J C K, Huang D P. Int J Oncol. 1994;4:1359–1364. doi: 10.3892/ijo.4.6.1359. [DOI] [PubMed] [Google Scholar]
- 25.Hu L-F, Eriksdottir G, Lebedeva T, Kholodnyuk I, Alimov A, Chen F, Luo Y, Zaborovsky E R, Ingvarsson S, Klein G, Ernber I. Gene Chromosomes Cancer. 1996;17:118–126. doi: 10.1002/(SICI)1098-2264(199610)17:2<118::AID-GCC7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Roche J, Boldog F, Robinson M, Robinson L, Varella-Garcia M, Swanton M, Waggoner B, Fishel R, Franklin W, Gemmill R, Drabkin H. Oncogene. 1996;12:1289–1297. [PubMed] [Google Scholar]
- 27.van den Berg A, Buys C H C M. Genes Chromosomes Cancer. 1997;19:59–76. doi: 10.1002/(sici)1098-2264(199706)19:2<59::aid-gcc1>3.3.co;2-o. [DOI] [PubMed] [Google Scholar]
- 28.Wistuba I I, Montellano F d, Michgrub S, Virmani A K, Behrens C, Chen H, Ahmadian M, Nowak J A, Muller C, Minna J D, Gazdar A F. Cancer Res. 1997;57:3154–3158. [PubMed] [Google Scholar]
- 29.Wei M-H, Latif F, Bader S, Kashuba V, Chen J Y, Dub F M, Sekido Y, Lee C C, Geil L, Kuzmin I, et al. Cancer Res. 1996;56:1487–1492. [PubMed] [Google Scholar]
- 30.Larson A A, Liao S-Y, Stanbridge E J, Cavenee W K, Hampton G M. Cancer Res. 1997;57:4171–4176. [PubMed] [Google Scholar]
- 31.Latif F, Tory K, Gnarra J, Yao M, Duh F M, Orcutt M L, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 32.Sozzi G, Veronese M L, Negrini M, Baffa R, Cotticelli M G, Inoue H, Tornielli S, Pilotti S, De Gregorio L, Pastroino U, et al. Cell. 1996;85:17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- 33.Dowdy S F, Fasching C L, Scanlon D J, Araujo D, Livanos E, Lai K-M, Weissman B E, Stanbridge E J. Science. 1991;254:293–295. doi: 10.1126/science.254.5029.293. [DOI] [PubMed] [Google Scholar]
- 34.Yao K, Zhang H-Y, Zhu H-C, Wang F-X, Li G-Y, Wen D-S, Li Y-P, Tsai C-H A, Glaser R. Cancer. 1990;45:83–89. doi: 10.1002/ijc.2910450116. [DOI] [PubMed] [Google Scholar]
- 35.Imreh S, Kholodnyuk I, Allikmetts R, Stanbridge E J, Zaborovsky E R, Klein G. Genes Chromosomes Cancer. 1994;11:235–237. doi: 10.1002/gcc.2870110406. [DOI] [PubMed] [Google Scholar]
- 36.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 37.Hampton G M, Larson A L, Baergen R N, Felts R L, Kern S, Cavenee W K. Proc Natl Acad Sci USA. 1996;93:6704–6709. doi: 10.1073/pnas.93.13.6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Die C, Faure S, Fizames C, Samson D, Druot N, Vignal A, Millaseau P, Marc S, Hazan J, Seboun E, et al. Nature (London) 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 39.Larson A L, Kern S, Curtiss S, Gordon R, Cavenee W K, Hampton G M. Cancer Res. 1997;57:4082–4090. [PubMed] [Google Scholar]
- 40.Rooney D E, Czepulkowski B H. Human Cytogenetics: A Practical Approach. Washington, DC: IRL; 1986. [Google Scholar]
- 41.Gray J W, Pinkel D, Brown J M. Radiat Res. 1994;137:275–289. [PubMed] [Google Scholar]
- 42.Lin H Y, Wang X-F, Ng-Eaton E, Weinberg R A, Lodish H F. Cell. 1992;68:775–785. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- 43.van den Berg A, Kooy R F, Hulsbeek M M F, de Jong D, Kok K, van der Veen A Y, Buys C H C M. Cytogenet Cell Genet. 1996;72:225. doi: 10.1159/000134196. [DOI] [PubMed] [Google Scholar]
- 44.Killary A M, Wolf M E, Giambernardi T A, Naylor S L. Proc Natl Acad Sci USA. 1992;89:10877–10881. doi: 10.1073/pnas.89.22.10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Todd M D, Xiang R-C, Garcia D K, Kerbachev K E, Moore S L, Hensel C H, Liu P, Siciliano M J, Klok K, van den Berg A, et al. Oncogene. 1996;13:2387–2396. [PubMed] [Google Scholar]
- 46.Kholodnyuk I, Kost-Alimova M, Kashuba V, Gizatulin R, Szeles A, Stanbridge E J, Zaborovsky E R, Klein G, Imreh S. Genes Chromosomes Cancer. 1997;18:200–211. [PubMed] [Google Scholar]