

Abstract
Myelin oligodendrocyte glycoprotein (MOG) is a key CNS-specific autoantigen for primary demyelination in multiple sclerosis. Although the disease-inducing role of MOG has been established, its precise function in the CNS remains obscure. To gain new insights into the physiological and immunopathological role of MOG, we determined the 1.8-Å crystal structure of the MOG extracellular domain (MOGED). MOGED adopts a classical Ig (Ig variable domain) fold that was observed to form an antiparallel head-to-tail dimer. A dimeric form of native MOG was observed, and MOGED was also shown to dimerize in solution, consistent with the view of MOG acting as a homophilic adhesion receptor. The MOG35-55 peptide, a major encephalitogenic determinant recognized by both T cells and demyelinating autoantibodies, is partly occluded within the dimer interface. The structure of this key autoantigen suggests a relationship between the dimeric form of MOG within the myelin sheath and a breakdown of immunological tolerance to MOG that is observed in multiple sclerosis.
Multiple sclerosis (MS) is an inflammatory disease of the CNS characterized by localized myelin destruction and axonal degeneration (1). An autoimmune reaction against myelin antigens of the CNS contributes to the immunopathological mechanisms of this enigmatic disease. This concept is supported by observations in patients with MS such as elevated levels of lymphocytes reactive to myelin antigens, the presence of oligoclonal Ig in the cerebrospinal fluid, and perivascular inflammatory cell invasion of the CNS (2). Immune responses to abundant proteins of the CNS myelin sheath, proteolipid protein (PLP), and myelin basic protein (MBP) are thought to produce some of the pathological lesions in MS and its animal model, experimental autoimmune encephalomyelitis (EAE). However, there is another important autoantigen linked to the pathogenesis of both MS and EAE, the myelin oligodendrocyte glycoprotein (MOG), which is a minor component of the myelin sheath (≈0.05% of total myelin protein) (3). In susceptible animals, immunization with native or recombinant MOG, or MOG-derived peptides, or passive transfer of MOG-specific T cells and autoantibodies against MOG elicits a severe neurological disease that mimics many of the clinical, pathological, and immunological features of MS (2, 4, 5). A number of encephalitogenic epitopes of MOG (MOG1-22, MOG35-55, and MOG92-106) have been identified (4, 6, 7). MOG35-55, in particular, is a potent encephalitogen that, in contrast to MBP and PLP, elicits striking inflammatory and demyelinating immune responses in the CNS not only in various species but also across different strains of mice (5, 6, 8).
The ensuing MS-like disease is characterized by relapses and remissions, extensive neural inflammation and demyelination, axonal injury, and the production of antibodies to MOG (5, 7–10). Moreover, antibodies to MOG are directly associated with myelin damage in the marmoset model of EAE as well as in the CNS of patients with MS (10). The epitopes recognized by mAb 8-18C5 and other demyelinating mAbs to MOG are conformation-dependent (11), whereas antibodies against linear epitopes of MOG do not induce widespread demyelination (12).
MOG is a type I integral membrane protein possessing a single extracellular Ig variable domain (Ig-V) (3, 13, 14). The amino acid sequence of MOG is highly conserved among animal species (>90%), indicative of an important biological function. MOG is specifically expressed in the CNS on the outermost lamellae of the myelin sheath as well as the cell body and processes of oligodendrocytes (3). The developmentally late expression of MOG correlates with the later stages of myelinogenesis, suggesting that MOG has a role in the completion, compaction, and/or maintenance of myelin, further suggesting that MOG has an adhesive function within the CNS (3). Consistent with MOG's possible adhesive role in the CNS, a homodimeric form of MOG has not only been observed after isolation from the CNS but has additionally been observed in situ (15). In addition, MOG may also have an immune-related function because it binds C1q, and thus could be a regulator of the classical complement pathway (16).
We have determined the 1.8-Å crystal structure of MOG extracellular domain (MOGED) to further our understanding of the function of this protein and to elucidate its role in the pathogenesis of demyelinating diseases of the CNS.
Methods
Protein Expression and Refolding. MOGED (residues 1–117) of the mature mouse protein, containing an amino terminal sixhistidine tag, was produced in the Escherichia coli strain M15 (pREP4) by using the pQE9 expression vector (Qiagen, Melbourne, Australia). After expression and preparation of a cleared lysate per the manufacturer's instructions, MOGED was purified by using Ni-NTA Superflow (Qiagen) under denaturing conditions (6 M guanidine·HCl), per the manufacturer's instructions, on a BioLogic LP Chromatography System (Bio-Rad). The column was washed to remove nonspecifically bound material, and bound protein was refolded directly on the column by using linear gradients of denaturing versus nondenaturing solutions. After elution with 300 mM imidazole in sodium phosphate/Tris (pH 8.0), MOGED was dialyzed against 50 mM NaCl/5 mM Tris (pH 8.0) and concentrated to 1.3 mg/ml.
Crystallization. Crystallization trials were conducted by using the hanging-drop vapor diffusion technique at room temperature. Tetragonal-shaped crystals were grown by mixing equal volumes of MOGED with the reservoir buffer (14–18% polyethylene glycol 4000/0.1 M sodium acetate, pH 4.6). The crystals were flash frozen before data collection by using 10% glycerol. All data were collected in-house on an R-AXIS IV++ detector with CuKα x-rays generated by a Rigaku (Tokyo) RU-H3RHB rotating anode generator and focused by using Osmic (Auburn Hills, MI) mirrors. The diffraction data were processed and analyzed by using d*trek (17) and programs from the ccp4 suite (18). The crystals diffracted to 1.8 Å and belong to space group I41 with unit cell dimensions a = b = 65.7 Å and c = 67.6 Å, consistent with one monomer per asymmetric unit. A summary of statistics is provided in Table 1.
Table 1. Data collection and refinement statistics.
| Data collection | |
| Temperature, K | 100 |
| Space group | I41 |
| Cell dimensions, Å (a, b, c) | 65.7, 65.7, 67.6 |
| Resolution, Å | 1.8 |
| Total no. of observations | 33,022 |
| No. of unique observations | 13,229 |
| Multiplicity | 2.5 |
| Data completeness, % | 99.0 (100.0) |
| Data >2 σI, % | 79.6 |
| I/σI | 6.9 (1.8) |
| Rmerge,* % | 6.5 (24.6) |
| Refinement | |
| Nonhydrogen atoms | |
| Protein | 934 |
| Water | 215 |
| Sulfate ions | 2 |
| Resolutions, Å | 50-1.8 |
| Rfactor,† % | 20.1 |
| Rfree,† % | 23.7 |
| rms deviations from ideality | |
| Bond lengths, Å | 0.005 |
| Bond angles, ° | 1.35 |
| Dihedrals, ° | 26.3 |
| Impropers, ° | 0.7 |
| Ramachandran plot | |
| Most favored and allowed region, % | 99 |
| B factors, Å2 | |
| Average main chain | 20.3 |
| Average side chain | 24.1 |
| Average water molecule | 40.3 |
| rms deviation in bonded B factors | 1.8 |
The values in parentheses are for the highest-resolution bin (approximate interval 0.1 Å).
Rmerge = Σ|IhkI - 〈IhkI〉|/ΣIhkI.
Rfactor = ΣhkI∥Fo| - |Fc∥/ΣhkI|F o| for all data except for 3% that was used for the Rfree calculation.
Structure Determination and Refinement. The structure was solved by using a combination of molecular replacement (amore), manual rebuilding in o (19), and the arp/warp procedure (20). The progress of refinement was monitored by the Rfree value (3% of the data). The starting model for molecular replacement was a domain from B7.2, with all differences mutated to alanine and the majority of loops removed. The highest peak in the rotation function also gave the highest peak in the translation function that packed well within the unit cell. Inspection of the electron density map suggested that the solution was correct although the phase information was poor. Given the high resolution of the data, arp/warp was tried; however, this procedure failed initially. Regardless, manual model rebuilding was initiated, interspersed with refinement within cns (21). When a sufficient core of the model was built, arp/warp was reused and proceeded to successfully build and refine the model. A final round of refinement was undertaken in cns.
The final model, comprising residues 2–117, 215 water molecules, and two sulfate ions, has an Rfactor of 20.1% and an Rfree of 23.7% (Table 1). One residue, His-103, although in the disallowed area of the Ramachandran plot, fits the electron density very well.
Western Blot Analysis. Myelin was prepared from the brains of adult wild-type and MOG-deficient mice (unpublished data) and human brain by using sucrose gradient centrifugation. Native MOG was purified from human brain by affinity chromatography with the MOG-specific monoclonal antibody 8-18C5 (22). Myelin proteins (2.5 μg per lane) and purified native MOG were resolved on 12% SDS/PAGE gels. MOG was detected by Western blot analysis as described (15), using 8-18C5 and enhanced chemiluminescence detection per the manufacturer's instructions (ECL, Amersham Biosciences).
Cross-Linking. Protein for cross-linking was buffer-exchanged into PBS (50 mM phosphate/150 mM NaCl, pH 7.2) by running the protein on a Superdex 75 10/30 column (Amersham Biosciences). Increasing amounts of MOGED were incubated with 0.1 mM DTSSP [3,3′-dithiobis(sulfosuccinimidylpropionate); Pierce] for 30 min at room temperature. The reaction was quenched with 1 M Tris, and the whole reaction was analyzed by SDS/PAGE.
Blue Native PAGE. Blue native PAGE (31) was used to resolve MOGED. Briefly, MOGED was resuspended in Coomassie blue G250 (Serva) loading buffer and resolved on 4–20% native PAGE gels (Bio-Rad) at 15 mA for2hat4°C. The gels were then stained in Coomassie blue R250. Chymotrypsinogen A (≈26 kDa) and ribonuclease A (≈14 kDa) were used as molecular mass standards (2 μg per lane).
Native Gel Electrophoresis. Nondenaturing PAGE was performed by using a discontinuous buffer system. The resolving gel contained 0.37 M Tris, pH 8.8, with 3.8 mM TEMED, 3.0 mM ammonium persulfate, and specified percent acrylamide. A 4% stacking gel was used in all experiments. The gels were run for 30 min at constant voltage of 200 V with SDS-running buffer (0.1 M Tris/0.38 M glycine, pH 8.8). The gels were stained with Coomassie blue.
Biacore Analysis. The biotinylated MOG35-55 and MOG92-106 peptides were obtained from Auspep (Melbourne, Australia) and synthesized by standard fluorenylmethoxycarbonyl (Fmoc) chemistry, and purity (>95%) was determined by mass spectrometry. Biotinylated peptides were immobilized on streptavidin sensor chip and binding of MOGED analyzed by surface plasmon resonance (Biacore, Victoria, Australia). MOGED (10 μg/ml) was passed through the flow cells at a flow rate of 10 μl/min. After completion of each run, the sensor chip was regenerated with a pulse of 100 mM NaOH.
Results
Structure Description. The structure of MOGED has been determined to 1.8-Å resolution (Fig. 1). As predicted from sequence data, the overall structure of MOGED adopts a topology of an Ig-V domain (Fig. 1) with overall dimensions of ≈40 × 35 × 30 Å. The structure adopts a compact, β-sandwich domain with one antiparallel β-sheet (strands A, B, E, and D) packing against a mixed β-sheet (strands A′, G, F, C, C′, and C′′) with the N and C termini at opposite ends of the molecule. In addition to the β-strands, there are four 310 helices located at the periphery of the molecule. The core packing residues of MOGED are similar to what is observed in other Ig-V domains, namely a canonical disulfide bond (Cys-24–Cys-98) that packs against the consensus residue (Trp-39), as well as the conserved and buried salt bridge between residues Arg-68 and Asp-92.
Fig. 1.

Schematic of the MOG monomer. The secondary structure assignment is as follows: βA(4–6); βA′ (12–15); βB (20–27); βC (36–42); βC′ (47–52); βC′′ (55–56); βD (69–72); βE (80–85); βF (94–102); and βG (105–116). The disulfide bond is shown in green. Each β-sheet is color-coded.
The three loops at the membrane-distal end of MOGED that connect the B–C, C′–C′′, and F–G strands have been termed Loops 1, 2, and 3, respectively. These loops are in the homologous location of the complementarity determining regions (CDRs 1, 2, and 3, respectively) of the Ig-V domains. The three loops combine to form a polar and essentially uncharged flat face in which His-103 at the tip of Loop 3 protrudes.
MOGED possesses a significant strip of electronegative charge that runs about half the length of the molecule, terminating at a small band of electropositive charge (Arg-4, Arg-25, Lys-80) (Fig. 2 Upper). This electronegative strip is dominated by residues at the N-terminal region of the B β-strand (Asp-18, Glu-19, Glu-21) but also contains Glu-74 and Glu-116. Surface-exposed hydrophobic patches can also be indicative of regions that mediate protein–protein and/or –ligand interactions. MOGED possesses a number of solvent-accessible hydrophobic residues, including three aromatics: Tyr-10, Phe-44, and Phe-90 (Fig. 2 Lower).
Fig. 2.
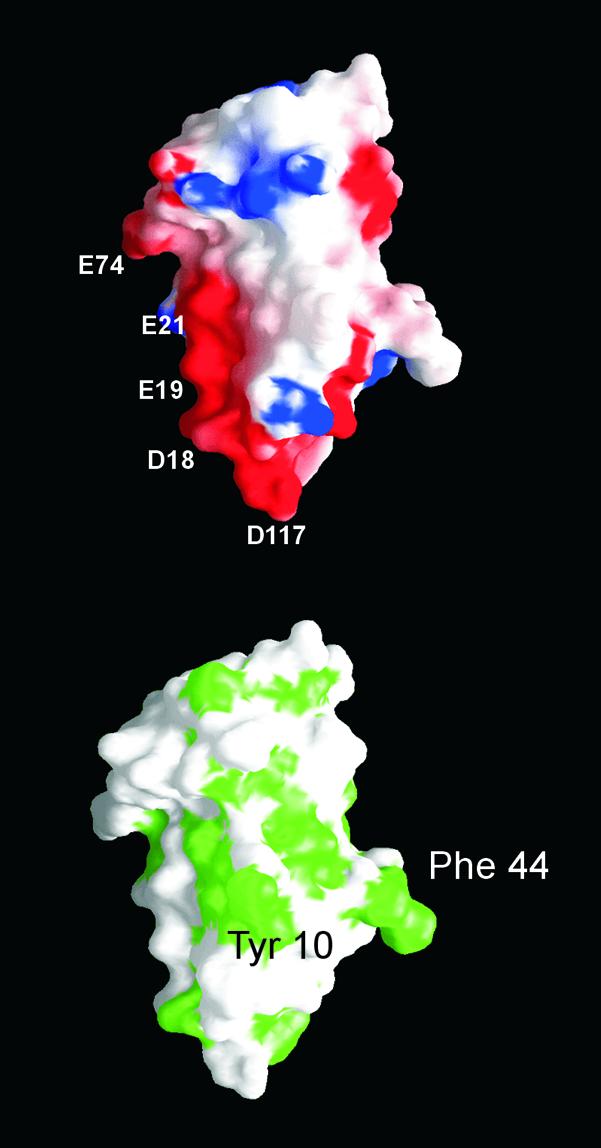
Surface representations of MOG. (Upper) Electrostatic potential, highlighting the electronegative strip. Residues contributing to this feature are labeled. (Lower) The solvent-exposed hydrophobic residues, Tyr-10 and Phe-44, in the same orientation to that in Upper.
Structural Comparisons. Automated structure comparisons (24) against other Ig-V domain structures revealed MOGED to be the most structurally homologous to sialoadhesin (rmsd 1.6 Å for 105 Cα atoms), despite sharing only 24% amino acid sequence identity. Additionally, a number of significant matches were made with other receptors identified in cell adhesion/receptor–ligand interactions, including b7-2 (25), adenovirus receptor D1 domain (CAR) (26), myelin protein zero (P0) (27), and junctional adhesion molecule (JAM) (28). In comparing the structure of MOGED to sialoadhesin (29) and P0, the core secondary structural elements superimpose well, reflecting the conserved core-packing residues (Fig. 3). The major structural differences reside at the A–A′ interconnecting region, Loop 1, the C–C′ loop, part of Loop 2, residues 73–78, and residues within Loop 3.
Fig. 3.

Sequence alignment of the MOG sequences (mMOG, mouse; rMOG, rat; bMOG, bovine; hMOG, human; qMOG, quokka), as well as closely related structural homologues CAR, JAM, and P0. Secondary structure elements relating to the MOG structure are shown above the alignment. Identical residues are boxed and in red, and conservatively substituted residues are in blue. The major encephalitogenic peptides (1–22, 35–55, and 92–106) are shown underneath the MOG sequences. Shaded residues represent regions of sequence differences between the MOG species. Numbering was done according to the MOG sequence.
The Dimer. MOGED was purified predominantly as a monomer and crystallizes as a monomer in the asymmetric unit. However, the observed contacts within the crystalline lattice was suggestive of a biologically relevant, higher-order oligomer of MOGED, namely a MOG dimer. There are three regions of crystal contacts observed within the lattice of MOG; two of these interactions neither exhibited an extensive interface nor displayed significant shape complementarity. In the third crystal contact region, however, MOGED was observed to lie on a crystallographic twofold, forming an antiparallel, head-to-tail dimer within the lattice. This dimer interface (Fig. 4) is extensive, burying ≈1,800 Å2 of surface area, excluding water molecules. Furthermore, the shape complementarity index at the dimer interface is high (0.7), representing a value that is comparable to that observed in antibody–antigen interactions. Taken together, these global features, namely an extensive interface exhibiting high shape complementarity, suggest that the crystallographic MOGED dimer may represent a biologically relevant MOG dimer.
Fig. 4.

Schematic representation of the crystallographic head-to-tail dimer of MOGED. Mapping of the encephalitogenic peptides onto the dimer: orange, residues 1–22; red, residues 35–55; cyan, residues 92–106. All map onto the face of the β-sheet that mediates dimer contacts. Protomers A and B are colored light gray and dark gray, respectively.
The dimer interface runs along the long axis of the molecule, with interactions predominantly involving the extreme N terminus, the interconnecting loop between A and A′ strands (residues 8–11), the C–C′ loop (residues 40–46), and the F–G strand β-hairpin (residues 95–112) (Figs. 4 and 5). The interface is dominated by van der Waals interactions and hydrogen bonds. There are 10 direct hydrogen bonds per protomer: Gly-9O to Glu′-107OE1; Pro-11N to Glu′-107OE1; Ser-45OG to Ser′-45OG; Thr-97OG1 to Thr′-97OG1; Glu-106NE2 to Glu′-112OE1; Glu-107OE1 to Gly′-9O; Glu-107OE1 to Pro′-11N; Glu-108OE2 to Glu′-112OE1; Glu-112OE1 to Gln′-106NE2; Glu-112OE1 to Gln′-106OE2. Residues from the F–G β-hairpin participate extensively at this interface, with water molecules being largely excluded from this F–G β-hairpin area of the interface (Fig. 5 Upper). The small side chain of Ala-110 interacts with its counterpart. Ala-109 and Ala-110 are flanked by long acidic side chains of Glu-112, Glu-108, and Glu-107 that cluster to form an unusual triad of residues that interact at the interface. The periphery of this interface is dominated by the polar side chains Gln-106 and Tyr-105.
Fig. 5.

Stereoviews of the interactions at the dimer interface. (Upper) Centered around the F–G β-hairpin, shown in the same orientation as Fig. 4. Contacting residues are in ball-and-stick format, and hydrogen bonds are shown as dashed lines. Protomers A and B are colored light gray and dark gray, respectively. (Lower) Close-up of the interactions at C–C′ loop. Residues are shown in ball-and-stick format, water molecule are shown as blue spheres, and hydrogen bonding is shown as a dashed line. Protomers A and B are in yellow and red, respectively.
Adjacent to Ala-109 is a water-filled cavity that mediates interactions with the A–A′ strand loop. This loop is unusual in that it bulges away from the body of the Ig-V fold, toward the dimer interface (Fig. 5 Upper). Within this bulged loop is the prominent aromatic, Tyr-10, which is fully solvent-exposed in the monomer but in the dimeric state stacks between the aliphatic moieties of Gln′-2 and Arg′-4 from the opposing protomer.
The C–C′ loop (residues 40–46) (Figs. 1 and 5 Lower) interacts with each other, in an orientation that is ≈70° to that of the β-hairpin-mediated interaction. This loop contains two aromatics, Phe-44 and Tyr-40, and a cis proline, Pro-43, that packs against Tyr′-40 and Phe′-99. The side chain of Phe-44 is completely solvent-exposed and does not form intersubunit contacts with its side chain. The main chain of Phe-44 does, however, form van der Waals contacts with Ser′-45, a residue that hydrogenbonds onto itself. There are a number of water-mediated hydrogen bonds centered around this loop (Fig. 5 Lower).
Native MOG has been repeatedly reported to exist as a mixture of monomeric and dimeric species (3, 15, 22). We also observed that, in Western analysis, the MOG-specific monoclonal antibody, 8-18C5, recognizes proteins with relative mass of 26–28 kDa (monomeric MOG) and 54 kDa (dimeric MOG) in CNS myelin from mouse and human brain as well as native MOG purified from human brain (Fig. 6A).
Fig. 6.

Biochemical evidence of MOG self-association. (A) Myelin purified from brain samples (lane 1, wild-type mouse; lane 2, MOG–/– mouse; lane 3, human) was separated by SDS/PAGE and immunoblotted with MOG-specific mAb 8-18C5. Purified native MOG from different human brain samples (lanes 4–7) was separated by SDS/PAGE and immunoblotted with MOG-specific mAb 8-18C5. Each of the three panels represents a separate experiment. (B) Recombinant MOGED was incubated with the cross-linker DTSSP, and the reaction was separated by SDS/PAGE. Dimer formation was observed only in the presence of the cross-linker. (C) Increasing amounts of recombinant MOGED were analyzed by nondenaturing PAGE, resulting in two species. (D) Blue native PAGE. Increasing amounts of MOGED were separated on a 4–20% native gradient PAGE gel and stained with Coomassie blue, revealing monomer dimer species. CT, chymotrypsinogen A; RA, ribonuclease A. (E) Biacore data. MOGED (10 μg/ml) was passed over biotinylated MOG35-55 (blue curve) or MOG92-106 (red curve) immobilized on a streptavidin sensor chip. Binding was analyzed by surface plasmon resonance and shows specific binding of MOGED to the 35–55 peptide. A control protein displayed no binding to either peptide (data not shown). M, monomer; D, dimer.
To confirm that the observed MOGED dimer was not merely due to crystal packing artifacts, several biochemical techniques were used to ascertain whether MOGED formed a dimer in solution. Firstly, we performed cross-linking studies, using an approach that has proved informative in other studies examining receptor self-association (30). In the presence of the primary amine-coupling agent DTSSP, increasing concentrations of MOGED lead to the formation of a MOGED species with the relative mass of 29 kDa, corresponding to the expected size of a dimer (Fig. 6B).
We next determined whether the absence of denaturing agents such as SDS may affect the relative amounts of the monomeric and dimeric species of MOGED. Whereas SDS/PAGE and analytical gel filtration show that MOGED exists primarily as a monomer, native PAGE revealed the presence of two protein species, which is again consistent with a mixture of dimeric and monomeric forms of MOGED (Fig. 6C). The high level of purity of the MOGED sample was verified by mass spectrometry analysis (data not shown). Given the topology of the dimer, the His-tag is unlikely to influence the observed dimerization. In addition, blue native PAGE analysis allowed us to determine the relative mass of the MOGED species. Two species were resolved as in native PAGE, with their masses corresponding approximately to those of the 26- and 14-kDa markers (Fig. 6D).
Moreover, to confirm the role of MOG35-55 in the formation of the MOGED dimer we immobilized biotinylated 35–55 peptide on a streptavidin sensor chip and analyzed binding by Biacore. MOGED (10 μg/ml) specifically bound the immobilized MOG35-55 but displayed no binding to the MOG92-106 (Fig. 6E), confirming that this region is important for dimer formation. A control protein displayed no binding to the peptide (data not shown).
Discussion
The three-dimensional structure determination of MOGED (Fig. 1) represents a detailed structural analysis of an MS-related autoantigen. We have demonstrated that MOGED adopts an Ig-V fold and forms a head-to-tail dimer (Fig. 4), and because all of the residues mediating these dimeric contacts are conserved among species (Fig. 3), this probably represents a common mode of dimerization. The observed dimer interface was extensive and exhibited significant shape complementarity, features that argue against the observed MOGED dimer representing merely a crystallographic artifact.
Importantly, a dimeric species of MOG was also observed after isolation from the CNS, as well as in situ. Moreover, we have demonstrated (Fig. 6) that MOGED has a propensity to specifically dimerize in solution. The affinity for the dimer must be weak, however, because the predominant species isolated from gel filtration was monomeric. Many receptors containing this fold, for example P0, have additionally been observed to display weak affinities (mM range) of self-association. Unfortunately, the concentration-dependent oligomerization of MOGED could not be ascertained by using analytical ultracentrifugation, because the maximal concentration of MOGED that could be achieved was only 1.3 mg/ml. In addition, the dimerization was not observed to be salt- or pH-dependent. Mutation of a number of key residues at the dimer interface (Fig. 5) would be predicted to disrupt the level of observed dimerization.
We propose that the observed dimer represents not only the dimeric form of MOG that is obtained after isolation from the CNS, but also the dimeric form that has been observed in vivo. Although we consider it unlikely, we cannot eliminate the possibility that the dimeric species observed in vivo (15, 22) is mediated via a cis interaction or that the dimerization domain may additionally reside in the transmembrane/cytoplasmic domain.
Supporting the evidence that this dimeric form of MOGED exists in vivo, receptors that are structurally related to MOGED, such as P0 (27), JAM (28), and CAR (26), likewise self-associate similarly to mediate adhesion (in trans) between opposing membranes. For example, P0, the major protein of myelin of the peripheral nervous system (PNS), functions to mediate membrane adhesion within the myelin sheath, also forms a head-to-tail dimer (27). Moreover, the contact regions involved correspond to those that mediate intersubunit contacts within the MOGED dimer, namely the extreme N terminus (residues 1–7) and residues from the C′ strand (27).
As stated previously, several features favor the role of MOG as an adhesin in the completion and/or compaction of the myelin sheath (3). Our hypothesis is that MOG acts as an adhesin in trans, possibly functioning to abut opposing membranes and thereby acting as an adhesive glue between neighboring myelinated fibers. This hypothesis is consistent with the observation that in the PNS, where MOG is not expressed, myelin sheaths do not appear to be in contact with each other (22).
Other proteins contain self-associating Ig-V domains and additionally interact with heterologous receptors (31). Thus, as well as the observed homophilic association, MOGED may interact heterologously, and certain observations support this view. For example, MOG possesses three loops that are analogous to the CDR loops of IGV domains, a significant electronegative strip, and an exposed aromatic residue (Phe-44 of the C–C′ loop) (Fig. 2), features that may represent a potential binding site (32). Interestingly, Tyr-44 of sialoadhesin, the structural equivalent of MOGED Phe-44, interacts with the ligand, sialic acid (29). One candidate ligand for MOG is the complement component C1q (33), which was shown to bind specifically in vitro to MOG.
Several MOG-derived peptides are encephalitogenic, but the most promiscuous among these is MOG35-55, for which immunization induces a severe demyelinating disease in several animal models (23). Also, MOG35-55 has been identified as an immunodominant epitope for T cell responses in MS (34). Our finding that MOG35-55 forms an integral part of the MOGED dimer (Fig. 4) interface leads us to speculate that the spatial location of this peptide and its participation in the dimeric structure are critical aspects in loss of immune tolerance to this particular sequence. Of interest, the two other regions of MOG that map to the dimer interface (Fig. 4) (MOG1-22 and MOG92-106) are also encephalitogenic in several animal models (4, 6), further supporting a relationship between localization of peptides within the MOG dimer interface and induction of demyelinating disease.
Peptides that lay deeply buried within MOG may not be easily released during antigen processing and therefore do not induce tolerance during development. Introduction of the peptide later in life, either experimentally or as a result of myelin damage, could cause disruption of the MOG dimer and so may then stimulate pathogenic immune responses given a suitable MHC background. This hypothesis is supported by the observation that in contrast to MOG35-55, induction of disease by immunization with MOGED requires processing by antigen-presenting cells (35, 36).
Aside from T cells, MOG is also a known B cell autoantigen in both EAE and MS (10–12). Various MOG-specific autoantibodies cannot be mapped to any linear sequence in the MOGED and hence appear to recognize a conformational epitope (6). Not unexpectedly, conformation-dependent antibodies seem more efficient at inducing demyelination than those recognizing linear epitopes (12).
In Lewis rats immunized with MOG35-55, antibodies to the peptide are associated with severe EAE, and fine epitope mapping of these antibodies reveals MOG37-46 as an immunodominant region (7). This region is located at the dimer interface, an observation that raises two considerations. First, these MOG37-46 autoantibodies could preferentially recognize the dimeric form of MOG, and this ability, which for example might mimic the natural ligand, may be related to their demyelinating activity. Second, given that the epitope is partly buried at the dimer interface, some autoantibodies may only recognize MOG after the dimer is disrupted. Further experimental evidence is required to clarify these considerations.
The MOGED structure suggests a link between the physiological function of MOG and its immunopathological features in MS. This work could eventually lead to the development of therapeutics designed to interfere with the anti-MOG T and B cell response in patients with MS.
Acknowledgments
We thank I. Mackay, T. Backstrom, and J. Villadangos for critical reading of the manuscript, and S. Bottomley for useful discussions. J.R. is supported by a Wellcome Trust Senior Research Fellowship in Biomedical Science in Australia. This work was supported partly by grants from the National Health and Medical Research Council, MS Australia, the J. B. Were and Son Trust, and Toward a Cure.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DTSSP, 3,3′-dithiobis(sulfosuccinimidylpropionate); EAE, experimental autoimmune encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; MOGED, MOG extracellular domain; MS, multiple sclerosis; P0, myelin protein zero.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1PY9).
References
- 1.Noseworthy, J. H., Lucchinetti, C., Rodriguez, M. & Weinshenker, B. G. (2000) N. Engl. J. Med. 343, 938–952. [DOI] [PubMed] [Google Scholar]
- 2.Bernard, C. C., Johns, T. G., Slavin, A., Ichikawa, M., Ewing, C., Liu, J. & Bettadapura, J. (1997) J. Mol. Med. 75, 77–88. [DOI] [PubMed] [Google Scholar]
- 3.Johns, T. G. & Bernard, C. C. (1999) J. Neurochem. 72, 1–9. [DOI] [PubMed] [Google Scholar]
- 4.Linington, C., Berger, T., Perry, L., Weerth, S., Hinze-Selch, D., Zhang, Y., Lu, H. C., Lassmann, H. & Wekerle, H. (1993) Eur. J. Immunol. 23, 1364–1372. [DOI] [PubMed] [Google Scholar]
- 5.Slavin, A., Ewing, C., Liu, J., Ichikawa, M., Slavin, J. & Bernard, C. C. (1998) Autoimmunity 28, 109–120. [DOI] [PubMed] [Google Scholar]
- 6.Amor, S., Groome, N., Linington, C., Morris, M. M., Dornmair, K., Gardinier, M. V., Matthieu, J. M. & Baker, D. (1994) J. Immunol. 153, 4349–4356. [PubMed] [Google Scholar]
- 7.Ichikawa, M., Johns, T. G., Liu, J. & Bernard, C. C. (1996) J. Immunol. 157, 919–926. [PubMed] [Google Scholar]
- 8.Genain, C. P. & Hauser, S. L. (1996) Methods 10, 420–434. [DOI] [PubMed] [Google Scholar]
- 9.Ichikawa, M., Johns, T. G., Adelmann, M. & Bernard, C. C. (1996) Int. Immunol. 8, 1667–1674. [DOI] [PubMed] [Google Scholar]
- 10.Genain, C. P., Cannella, B., Hauser, S. L. & Raine, C. S. (1999) Nat. Med. 5, 170–175. [DOI] [PubMed] [Google Scholar]
- 11.Brehm, U., Piddlesden, S. J., Gardinier, M. V. & Linington, C. (1999) J. Neuroimmunol. 97, 9–15. [DOI] [PubMed] [Google Scholar]
- 12.von Budingen, H. C., Hauser, S. L., Fuhrmann, A., Nabavi, C. B., Lee, J. I. & Genain, C. P. (2002) Proc. Natl. Acad. Sci. USA 99, 8207–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pham-Dinh, D., Mattei, M. G., Nussbaum, J. L., Roussel, G., Pontarotti, P., Roeckel, N., Mather, I. H., Artzt, K., Lindahl, K. F. & Dautigny, A. (1993) Proc. Natl. Acad. Sci. USA 90, 7990–7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilton, A. A., Slavin, A. J., Hilton, D. J. & Bernard, C. C. (1995) J. Neurochem. 65, 309–318. [DOI] [PubMed] [Google Scholar]
- 15.Slavin, A. J., Johns, T. G., Orian, J. M. & Bernard, C. C. (1997) Dev. Neurosci. 19, 69–78. [DOI] [PubMed] [Google Scholar]
- 16.Vanguri, P. & Shin, M. L. (1986) J. Neurochem. 46, 1535–1541. [DOI] [PubMed] [Google Scholar]
- 17.Pflugrath, J. W. (1999) Acta Crystallogr. D 55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 19.Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 20.Perrakis, A., Harkiolaki, M., Wilson, K. S.. & Lamzin, V. S. (2001) Acta Crystallogr. D 57, 1445–1450. [DOI] [PubMed] [Google Scholar]
- 21.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 22.Linington, C., Webb, M. & Woodhams, P. L. (1984) J. Neuroimmunol. 6, 387–396. [DOI] [PubMed] [Google Scholar]
- 23.Iglesias, A., Bauer, J., Litzenburger, T., Schubart, A. & Linington, C. (2001) Glia 36, 220–234. [DOI] [PubMed] [Google Scholar]
- 24.Holm, L. & Sander, C. (1995) Trends Biochem. Sci. 20, 478–480. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz, J. C., Zhang, X., Fedorov, A. A., Nathenson, S. G. & Almo, S. C. (2001) Nature 410, 604–608. [DOI] [PubMed] [Google Scholar]
- 26.van Raaij, M. J., Chouin, E., van der Zandt, H., Bergelson, J. M. & Cusack, S. (2000) Struct. Folding Des. 15, 1147–1155. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro, L., Doyle, J. P., Hensley, P., Colman, D. R. & Hendrickson, W. A. (1996) Neuron 17, 435–449. [DOI] [PubMed] [Google Scholar]
- 28.Kostrewa, D., Brockhaus, M., D'Arcy, A., Dale, G. E., Nelboeck, P., Schmid, G., Mueller, F., Bazzoni, G., Dejana, E., Bartfai, T., et al. (2001) EMBO J. 20, 4391–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.May, A. P., Robinson, R. C., Vinson, M., Crocker, P. R. & Jones, E. Y. (1998) Mol. Cell 1, 719–728. [DOI] [PubMed] [Google Scholar]
- 30.Yang, J., Dokurno, P., Tonks, N. K. & Barford, D. (2001) EMBO J. 20, 3645–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schagger, H. & von Jagow, G. (1991) Anal. Biochem. 199, 223–231. [DOI] [PubMed] [Google Scholar]
- 32.Rossjohn, J., McKinstry, W. J., Woodcock, J. M., McClure, B. J., Hercus, T. R., Parker, M. W., Lopez, A. F. & Bagley, C. J. (2000) Blood 95, 2491–2498. [PubMed] [Google Scholar]
- 33.Johns, T. G. & Bernard, C. C. (1997) Mol. Immunol. 34, 33–38. [DOI] [PubMed] [Google Scholar]
- 34.Kerlero de Rosbo, N., Hoffman, M., Mendel, I., Yust, I., Kaye, J., Bakimer, R., Flechter, S., Abramsky, O., Milo, R., Karni, A. & Ben-Nun, A. (1997) Eur. J. Immunol. 27, 3059–3069. [DOI] [PubMed] [Google Scholar]
- 35.Slavin, A. J., Soos, J. M., Stuve, O., Patarroyo, J. C., Weiner, H. L., Fontana, A., Bikoff, E. K. & Zamvil, S. S. (2001) J. Clin. Invest. 108, 1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tompkins, S. M., Padilla, J., Dal Canto, M. C., Ting, J. P., Van Kaer, L. & Miller, S. D. (2002) J. Immunol. 168, 4173–4183. [DOI] [PubMed] [Google Scholar]