

Abstract
X-linked hypophosphatemia is the most prevalent inherited form of rickets. In this disorder, rickets results from hyperphosphaturia and inappropriately normal levels of 1,25(OH)2-vitamin D. Current therapy with oral phosphate and vitamin D improves the rickets, but has significant morbidity and does not significantly affect the short stature and hypophosphatemia. In the present study, we demonstrate that Hyp mice, which have a mutation homologous to that in patients with X-linked hypophosphatemia, have a 2-fold greater urinary prostaglandin E2 (PGE2) excretion than C57/B6 mice. To determine whether PGs were involved in the pathogenesis of this disorder, Hyp and C57/B6 mice received i.p. injections with vehicle or indomethacin (1 mg/kg of body weight twice daily for 4 days) and were studied ≈12 h after the last dose of indomethacin. In the Hyp mice, indomethacin treatment decreased the fractional excretion of phosphate from 13.0 ± 3.2% to 2.2 ± 1.1% (P < 0.05), and increased serum phosphate from 2.9 ± 0.2 mg/dl to 4.1 ± 0.2 mg/dl (P < 0.05). There was no effect of indomethacin in C57/B6 mice. Indomethacin did not affect serum creatinine or inulin clearance, demonstrating that the normalization of urinary phosphate excretion was not caused by changes in glomerular filtration rate. Indomethacin treatment increased renal brush border membrane vesicle NaPi-2 protein abundance in Hyp mice to levels comparable to that of C57/B6 mice, but had no effect in C57/B6 mice. In vitro isolated perfused proximal tubule studies demonstrate directly that 10-6 M bath indomethacin normalized the phosphate transport defect in Hyp mice but had no effect on C57/B6 mice. In conclusion, there is dysregulation of renal PG metabolism in Hyp mice, and indomethacin treatment normalizes the urinary excretion of phosphate by a direct tubular effect.
Keywords: X-linked hypophosphatemia, rickets, sodium/phosphate cotransporter NaPi-2
X-linked hypophosphatemia is the most common inherited cause of rickets (1). Children with this disorder have marked short stature, dental abscesses, and rickets. X-linked hypophosphatemia is inherited as an X-linked dominant disorder characterized by hypophosphatemia, normocalcemia, and a relative deficiency of 1,25(OH)2-vitamin D (1). The hypophosphatemia is caused by impaired proximal tubule phosphate reabsorption (1–6). The vitamin D deficiency is caused by decreased 25(OH)-vitamin D-1α-hydroxylase activity (7–10). Conventional therapy consists of oral administration of pharmacologic doses of vitamin D and phosphate, though hypercalciuria and nephrocalcinosis are frequent complications of such therapy (11–15). Nephrocalcinosis can lead to renal insufficiency and renal tubular acidosis (15). Furthermore, although this therapy is effective for treating rickets and bone pain, these patients have short stature, hypercalciuria, and hypophosphatemia (16).
Much of our understanding of the pathophysiology of X-linked hypophosphatemia has come from studies using the Hyp mouse, which has a mutation homologous to that in patients with this disorder (17–23). Substantial evidence points to a humoral factor mediating the aberrant proximal tubule phosphate transport in this disorder. Transplantation of a normal kidney into a Hyp mouse results in hyperphosphaturia (24), whereas transplantation of a Hyp mouse kidney into a control mouse leads to normal phosphate levels and rates of phosphate excretion (24). In addition, immortalized proximal tubule cells from Hyp mice have normal rates of phosphate transport (25, 26). Thus, there does not appear to be an intrinsic proximal tubule defect in X-linked hypophosphatemia. The circulating factor that inhibits proximal tubule phosphate transport may be liberated from bone, and it has at present been designated as phosphatonin (27). It is possible that the rate of proximal tubule phosphate transport in this disorder can be altered by using techniques that do not directly affect the underlying abnormality.
Prostaglandin E2 (PGE2) has been shown to selectively inhibit phosphate transport in the proximal tubule (28). Thus, PGs could theoretically play a role in the pathogenesis of phosphate wasting in Hyp mice. This study demonstrates that PGs play a pivotal role in mediating the phosphaturia in Hyp mice and that indomethacin corrects the transport defect both in vivo and in vitro.
Methods
Clearance Studies. Male C57/B6 and Hyp mice were obtained from The Jackson Laboratory and were of the same genetic background. They were allowed free access to food and water until the time of study. C57/B6 and Hyp mice received i.p. injections with either 1 mg/kg of body weight indomethacin or vehicle. Both groups were injected twice each day for 4 days. Approximately 12 h after the last injection, urine was collected from the mice, they were then anesthetized by using isoflurane, and blood was collected by cutting the tips of their tails. Blood and urine creatinine and phosphate were measured by using a CX9 analyzer (Beckman Coulter). Urinary PGE2 was measured by using ELISA (Amersham Pharmacia). Fractional excretion of phosphate was calculated by using the following equation: FEPO4 = [(urine phosphate/serum phosphate)/(urine creatinine/serum creatinine)] × 100.
For measurement of glomerular filtration rate (GFR), mice were first anesthetized with Nembutal (70 μg/g of body weight; Abbott). The mice were maintained at 37°C by using a heated surgical table. A tracheostomy was performed, the trachea was cannulated with polyethylene (PE) 50 tubing (Intramedic, Clay Adams), and 100% O2 was blown over the tracheostomy. An arterial line was placed in the carotid artery by using stretched PE 10 tubing for blood sampling. A PE 10 i.v. line was placed in the jugular vein for infusion of [3H]methoxyinulin (New England Nuclear), and PE 20 tubing was placed in the bladder for urine collection. All blood removed was replaced with an equal volume of 4% albumin. Inulin was infused at a rate of 25 μl/min per 100 g of body weight. After a 45-min equilibration period, three 20-min urine collections were performed. Midway through each collection period 100 μl of blood was removed for liquid scintillation counting.
In Vitro Microperfusion Flux Studies. Isolated segments of superficial proximal straight tubules were perfused as described (29–31). Briefly, tubules were dissected in Hanks' balanced salt solution containing 137 mM NaCl, 5 mM KCl, 0.8 mM MgSO4, 0.33 mM Na2HPO4, 0.44 mM KH2PO4, 1 mM MgCl2, 10 mM Tris·HCl, 0.25 mM CaCl2, 2 mM glutamine, and 2 mM l-lactate at 4°C. Tubules were transferred to a 1.2-ml temperature-controlled bath chamber. The tubules were perfused by using concentric glass pipettes at 37°C.
Proximal straight tubules were perfused at ≈10 nl/min. The perfusion solution was an ultrafiltrate-like solution containing 115 mM NaCl, 25 mM NaHCO3, 4.0 mM Na2HPO4, 10 mM sodium acetate, 1.8 mM CaCl2, 1 mM MgSO4, 5 mM KCl, 8.3 mM glucose, and 5 mM alanine. The bathing solution contained 6 g/dl BSA. The osmolality of these solutions were adjusted to 295 milliosmoles/kg of H2O. The pH and osmolality of the bathing solution were maintained constant by continuously changing the bath at a rate of 0.5 ml/min. Net volume absorption (JV, in nl·mm-1·min-1) was measured as the difference between the perfusion rate (VO) and collection rates (VL), both in nl/min, normalized per millimeter of tubular length (L). The collection rate was measured with a 50-nl constant-volume pipette. Exhaustively dialyzed [3H]methoxyinulin was added to the perfusate at a concentration of 75 μCi/ml (1 Ci = 37 GBq) so that the perfusion rate, and thus the volume absorption, could be calculated. The length (in mm) was measured with an eyepiece micrometer. Tubules were incubated for at least 20 min before initiation of the control period. Phosphate transport was determined by using the following equation:
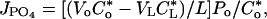 |
where Po is the phosphate concentration in the perfusate in mM and 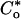 and
and 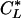 are the phosphate concentrations in the perfusate and collected fluids, respectively, in cpm/nl.
are the phosphate concentrations in the perfusate and collected fluids, respectively, in cpm/nl.
Brush Border Membrane Vesicle (BBMV) Isolation. C57/B6 and Hyp mouse kidneys were removed and placed in an ice-cold isolation buffer containing 300 mM mannitol, 16 mM Hepes, and 5 mM EGTA titrated to pH 7.4 with Tris. The isolation buffer contained aprotinin (2 μg/ml), leupeptin (2 μg/ml), and phenylmethylsulfonyl fluoride (100 μg/ml). The cortex was homogenized with 20 strokes of a Potter-Elvehjem homogenizer at 4°C. BBMV were then isolated by differential centrifugation and magnesium precipitation as described (32). The final BBMV fraction was resuspended in isolation buffer. Protein was assayed by using the bicinchoninic acid (BCA) method (Pierce).
SDS/PAGE and Immunoblotting. Brush border membrane proteins (25 μg per lane) were denatured and then separated on a 7.5% polyacrylamide gel by using SDS/PAGE as described (32). The proteins were transferred overnight to a poly(vinylidene difluoride) membrane at 120–140 mA at 4°C. The blot was blocked with fresh Blotto (5% nonfat milk/0.1% Tween 20 in PBS, pH 7.4) for 1 h followed by incubation with primary antibody to the sodium/phosphate cotransporter NaPi-2. NaPi-2 antibody, a generous gift from Jurg Biber (University of Zurich), was added at 1:1,000 dilution overnight at 4°C. The blot was then washed extensively with Blotto. The secondary antibody, horseradish peroxidase-conjugated donkey anti-rabbit Ig, was added at 1:10,000 dilution and incubated in room temperature for 1 h. The blot was again washed with Blotto, and enhanced chemiluminescence was used to detect bound antibody (Amersham Pharmacia). The NaPi-2 protein abundance was quantitated by using densitometry. Equal loading of the samples was confirmed by using an antibody to β-actin at a 1:5,000 dilution (Sigma).
Proximal Tubule cAMP Content. To determine whether the effect of indomethacin could be mediated by changes in cAMP, we measured cAMP generation in proximal straight tubules from Hyp and C57/B6 mice. Tubules were dissected, measured with an eyepiece micrometer, and transferred to an Eppendorf tube with 50 μl of Hanks' solution. Tubules were incubated in the presence of IBMX (3-isobutyl-1-methylxanthine; 1 mM) and either indomethacin (10-6 M) or vehicle in a final volume of 180 μl for 30 min at 37°C. Lysis buffer (20 μl, provided in the kit; Amersham Pharmacia cAMP ELISA kit) was added, and the tube was vortexed and allowed to stand at room temperature for 10 min to complete the cell lysis. The supernatant was then assayed (per manufacturer's protocol) for cAMP and expressed as fmol/mm of tubular length per 30 min. The assay was found to be linear at that time. To ensure that the difference in cAMP content was not caused by a difference in protein content between C57/B6 and Hyp mouse proximal tubules, the protein content was measured in seven samples of tubules in both Hyp and C57/B6 mice with 20 mm of tubules in each sample by using the bicinchoninic acid method. The protein content in proximal tubules was 0.40 ± 0.04 μg/mm in C57/B6 mice and 0.45 ± 0.04 μg/mm in Hyp mice (P = not significant).
Statistics. Data are expressed as the mean ± SEM. Data comparing more than two groups were analyzed by using ANOVA unless otherwise designated. Studies comparing two groups were analyzed by using paired or unpaired Student's t test, whichever was appropriate.
Results
Urinary PG Excretion. PGE2 has been shown to inhibit phosphate transport in the proximal tubule (28). We first examined whether there was a difference in urinary PGE2 excretion in C57/B6 and Hyp mice. As shown in Fig. 1, urinary PGE2 excretion was 2-fold higher in Hyp mice than in C57/B6 mice. This finding demonstrates that there is a difference in PGE2 excretion in Hyp mice compared with C57/B6 mice, which may play a role in the pathogenesis of the hyperphosphaturia in Hyp mice.
Fig. 1.

Urinary PGE2/creatine ratio in C57/B6 mice compared with Hyp mice. The same results were obtained when PG was normalized to urine volume. The number of measurements is given in parentheses in this and all figures. *, P < 0.05 vs. C57/B6.
Clearance Studies. To examine whether PGs had an effect in mediating the phosphaturia in Hyp mice, we studied mice treated with indomethacin. The effects of indomethacin in C57/B6 and Hyp mice are shown in Table 1 and Figs. 2 and 3. Indomethacin treatment resulted in a significant decrease in the FEPO4 to levels comparable to that seen in C57/B6 mice and a significant increase in serum phosphate compared with vehicle-treated Hyp mice. Indomethacin treatment did not affect the serum creatinine in either C57/B6 or Hyp mice.
Table 1. Effect of indomethacin on serum phosphate and urinary phosphate excretion in C57/B6 and Hyp mice.
| Mice | Treatment | Serum creatinine, mg/dl | Serum phosphate, mg/dl | Urine creatinine, mg/dl | Urine phosphate, mg/dl | FEPO4, % |
|---|---|---|---|---|---|---|
| C57/B6 | Vehicle | 0.2 ± 0.1 | 7.2 ± 0.4 | 30 ± 5 | 25.7 ± 12.0 | 2.7 ± 1.2 |
| C57/B6 | Indomethacin | 0.3 ± 0.1 | 8.0 ± 0.4 | 40 ± 3 | 15.1 ± 5.7 | 2.6 ± 0.6 |
| Hyp | Vehicle | 0.2 ± 0.1 | 2.9 ± 0.2* | 47 ± 6 | 80.3 ± 32.5 | 13.0 ± 3.2* |
| Hyp | Indomethacin | 0.2 ± 0.2 | 4.1 ± 0.2† | 47 ± 6 | 22.5 ± 11.4 | 2.2 ± 1.1 |
In all cases, n ≥ 6
, P < 0.05 vs. other groups
, P < 0.05 vs. C57/B6, C57/B6 indomethacin, and Hyp vehicle
Fig. 2.

Effect of indomethacin or vehicle treatment on FEPO4 in C57/B6 and Hyp mice. Indomethacin treatment normalized the urinary phosphate excretion in Hyp mice but had no effect on C57/B6 mice. Urinary phosphate and creatinine were determined ≈12 h after the last dose of indomethacin. *, P < 0.05 vs. other groups.
Fig. 3.

Effect of indomethacin treatment on serum phosphate in C57/B6 and Hyp mice. Indomethacin treatment increased the serum phosphate levels in Hyp mice but had no significant effect on C57/B6 mice. Levels were determined ≈12 h after the last dose of indomethacin. *, P < 0.05 vs. other groups; +, P < 0.05 vs. Hyp.
Although there was no change in serum creatinine (Table 1), we wanted to examine more precisely whether this dose of indomethacin affected the glomerular filtration rate. Inulin clearance in C57/B6 mice and Hyp mice treated with vehicle and indomethacin is shown in Fig. 4. As can be seen, indomethacin treatment had no effect on inulin clearance in Hyp or C57/B6 mice.
Fig. 4.

Effect of indomethacin (INDO) treatment on glomerular filtration rate measured as inulin clearance in C57/B6 and Hyp mice.
Effect of Indomethacin on Renal BBMV NaPi-2 Abundance. In the next series of experiments, we examined the effect of indomethacin on renal brush border membrane NaPi-2 protein abundance in C57/B6 and Hyp mice. C57/B6 and Hyp mice received i.p. injections using the same protocol described for our clearance studies. As is shown in Fig. 5, BBMV from Hyp mice that received vehicle had significantly less NaPi-2 protein abundance than C57/B6 mice. Treatment with indomethacin had no effect on BBMV NaPi-2 abundance in C57/B6 mice, but increased NaPi-2 abundance in Hyp mice.
Fig. 5.

(A) The immunoblot demonstrates the effect of vehicle or i.p. indomethacin (INDO) on NaPi-2 brush border membrane abundance in C57/B6 mice and Hyp mice. The last dose of indomethacin was administered ≈12 h before sacrifice. As is shown, there is no difference in brush border membrane NaPi-2 abundance in C57/B6 mice with indomethacin treatment, whereas there is an increase in brush border membrane NaPi-2 abundance in Hyp mice. (B) As is shown, C57/B6 male mice had significantly greater BBMV NaPi-2/β-actin abundance compared with Hyp mice treated with vehicle, but this difference was not present after indomethacin treatment. *, P < 0.05 vs. other groups.
Microperfusion Studies. In the first set of studies, we examined whether the rate of volume absorption and phosphate transport was stable in C57/B6 mouse proximal tubules perfused in vitro. Measurements of phosphate transport and volume absorption were performed after 15 min of incubation and repeated after at least 1 h of incubation. The rate of phosphate transport was 3.3 ± 0.53 and 2.9 ± 0.35 pmol/mm·min in the two periods, respectively (n = 7, P = not significant). Volume absorption was 0.44 ± 0.09 and 0.40 ± 0.11 nl/mm·min in the two periods, respectively (n = 7, P = not significant). Thus volume absorption and phosphate transport is stable in proximal tubules from C57/B6 mice.
The absence of an effect of indomethacin to decrease the glomerular filtration rate, and thus decrease the filtered load of phosphate, left the possibility that indomethacin was acting to increase the rate of phosphate transport by a direct tubular mechanism. To directly examine this, we perfused proximal tubules from C57/B6 mice and Hyp mice in vitro. The rates of volume absorption were 0.41 ± 0.09 and 0.27 ± 0.07 nl/mm·min in the C57/B6 mice and Hyp mice, respectively, and 0.25 ± 0.04 and 0.14 ± 0.06 nl/mm·min in the C57/B6 and Hyp mouse proximal tubules incubated with 10-6 M bath indomethacin (n ≥ 7 in each group). There was no difference in the rate of volume absorption by ANOVA or Student's t test. The effect of 10-6 M bath indomethacin or vehicle on phosphate transport is shown in Fig. 6. As is apparent, addition of indomethacin to the bathing solution resulted in a complete normalization of the rate of phosphate transport in Hyp mice to that seen in C57/B6 mice.
Fig. 6.

Effect of 10-6 M bath indomethacin or vehicle in isolated perfused proximal tubules from C57/B6 and Hyp mice. As can be seen, Hyp mice have a lower rate of phosphate transport than C57/B6 mice. Hyp proximal tubules with indomethacin in the bathing solution had rates of phosphate transport comparable to C57/B6 mice. *, P < 0.05 vs. other groups.
In the final series of experiments, we compared the rate of cAMP generation in dissected proximal tubules from C57/B6 and Hyp mice incubated with vehicle or 10-6 M indomethacin. The results are shown in Table 2. There was no difference in the rate of cAMP generation between proximal tubules from C57/B6 and Hyp proximal tubules. Indomethacin resulted in ≈10–20% reduction in cAMP generation in both Hyp and C57/B6 mouse proximal tubules, but this small change was not statistically significant. Thus the increase in phosphate transport with indomethacin in Hyp mice was not mediated by cAMP.
Table 2. Effect of 10-6 M indomethacin on cAMP production in C57/B6 and Hyp proximal tubules in vitro.
| cAMP, fmol/mm per 30 min
|
||
|---|---|---|
| Proximal tubules | Vehicle | Indomethacin |
| C57/B6 | 943 ± 84 | 755 ± 43 |
| ÿ ß∞ | 819 ± 68 | 717 ± 48 |
For all groups, ≤ = 16
Discussion
X-linked hypophosphatemia is caused by a mutation in the PHEX gene (18–23). The PHEX gene is a phosphate-regulating gene encoding a protein with endopeptidase activity, which is located on the X chromosome. It is postulated that inactivation of PHEX in Hyp mice and patients with X-linked hypophosphatemia somehow increases the serum concentration of a putative phosphaturic hormone, phosphatonin (33). Both fibroblast growth factor 23 (FGF 23) and frizzled-related protein 4 (FRP 4) have been identified as potential candidates for phosphatonins (34, 35).
The kidney produces PGs, which have a myriad of effects on renal function (36, 37). The major PG produced is PGE2 (38). The distal nephron production of PGs far exceeds proximal tubule production (38–40). However, studies have demonstrated that the glomerulus and all nephron segments produce PGs to some extent (38–40).
PGs oppose vasoconstrictor hormones that reduce the glomerular filtration rate (36, 37, 41–43). Thus, indomethacin could potentially reduce phosphate excretion by reducing the filtered load of phosphate delivered to the proximal tubule. However, at the doses used in this study, there was no change in the serum creatinine or inulin clearance. Thus, the reduction in phosphate excretion was not caused by a decrement in glomerular filtration rate. PGs produced by the kidney can affect tubular transport in an autocrine or paracrine fashion (44). Of importance to this study, PGE2 has been shown to inhibit phosphate transport when added to the bathing solution of rabbit proximal straight tubules perfused in vitro (28). Consistent with a tubular mechanism to explain the reduction in phosphate excretion was the dramatic fall in FEPO4 with indomethacin treatment in Hyp mice. In addition, as shown in Fig. 5, indomethacin treatment resulted in an increase in BBMV NaPi-2 protein abundance in Hyp mice, but had no effect on C57/B6 mice. Thus, the effect of indomethacin to reduce the phosphaturia in Hyp mice was paralleled by an increase in renal BBMV NaPi-2 transporter abundance in Hyp mice.
This study examined proximal tubular transport directly in Hyp mice compared with C57/B6 mice. We show that the rate of phosphate transport in Hyp mice is far less than the rate in C57/B6 mice. This directly demonstrates that Hyp tubules have a phosphate transport defect that is maintained when the tubules are perfused in vitro. This apparent discrepancy between studies that have shown that there is not an inherent proximal tubule transport defect is likely caused by a well described memory effect where the tubule maintains its transport characteristics for a time after it is removed from the in vivo environment (45–48). This may be caused by the phosphatonin adhering to the receptor or maintenance of the signal transduction activation by a phosphatonin for a time after removal of the tubule.
The regulation in phosphate transport in Hyp mice by a circulating phosphatonin may be analogous to parathyroid hormone (PTH) regulation of the NaPi-2 cotransporter (49–51). Administration of PTH for as little as 2 h to parathyroidectomized rats results in a reduction in NaPi-2 mRNA abundance, demonstrating transcriptional regulation (49). PTH also affects phosphate transport by regulating NaPi-2 cotransporter trafficking (50, 51). As speculation, the circulating phosphatonin in Hyp mice might similarly have an effect on posttranscriptional regulation of the NaPi-2 transporter that could be affected by indomethacin to explain the acute increase in phosphate transport in Hyp proximal tubules.
Previous studies have examined the role of cAMP and protein kinase A in the pathogenesis of the hyperphosphaturia in Hyp mice (52–54). Although basal urinary cAMP levels are slightly but significantly higher in Hyp mice (52), there is no significant difference in cAMP production in cortical slices between Hyp and C57/B6 mice (53). Furthermore, protein kinase A activity is comparable in the two strains, as is the apparent Km for cAMP (54). The present study compared cAMP production in dissected proximal tubules from Hyp and C57/B6 mice. If cAMP were mediating the lower rates of phosphate transport in Hyp mice proximal tubules, we would have expected a greater rate of production in Hyp mouse proximal tubules than in C57/B6 mice, and a significant reduction in cAMP production in Hyp proximal tubules incubated with indomethacin. However, our data are in concordance with studies showing that higher rates of cAMP production do not explain the phosphate transport defect in Hyp mice. Furthermore, we found no significant effect of indomethacin on cAMP production in either Hyp or C57/B6 tubules. Thus, the increase in proximal tubule phosphate transport with indomethacin is not mediated by cAMP.
In the present study, there was a dramatic fall in FEPO4 measured ≈12 h after the last dose of indomethacin in Hyp mice to levels seen in C57/B6 mice. Furthermore, there was a significant increase in serum phosphate, although not to levels of C57/B6 mice. The question remains as to why indomethacin treatment resulted in a decrease in the FEPO4 and an increase in serum phosphate in Hyp mice, while having no effect in C57/B6 mice. There are several possible explanations: First, FEPO4 in C57/B6 mice was extremely low, and tubular transport may have been at a maximum. Thus, indomethacin may not be able to increase phosphate transport further, whereas an augmentation was possible with indomethacin therapy in Hyp mice that had low rates of phosphate reabsorption. It is also possible that the rate of renal PG production was higher in Hyp mice or that Hyp mice respond to the normally produced PGs in an augmented fashion compared with C57/B6 mice. Although there was a significant increase in serum phosphate levels in Hyp mice with indomethacin treatment, it is also unclear, at present, why the serum phosphate did not normalize to levels comparable to C57/B6 mice, despite the marked improvement in phosphate excretion. As speculation, it is likely that much of the phosphate reabsorbed went into phosphate-depleted bones of the Hyp mice.
In summary, the present study shows that indomethacin can reduce phosphate excretion and increase serum phosphate in Hyp mice. The data are consistent with indomethacin having a direct tubular action to increase phosphate transport in Hyp mice. These studies suggest that indomethacin may be an effective form of therapy in humans with X-linked hypophosphatemia.
Acknowledgments
We are grateful for the technical assistance of Sumana Chakravarty. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-41612 (to M.B.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PG, prostaglandin; BBMV, brush border membrane vesicle.
References
- 1.Tenenhouse, H. S. & Econs, M. J. (2001) in The Metabolic Basis of Inherited Disease, eds. Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D. (McGraw–Hill, New York), pp. 5039-5067.
- 2.Insogna, K. I., Broadus, A. E. & Gertner, J. M. (1983) J. Clin. Invest. 71, 1562-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giasson, S. D., Brunette, M. G., Danan, G., Vigneault, N. & Carriere, S. (1977) Pflügers Arch. 371, 33-38. [DOI] [PubMed] [Google Scholar]
- 4.Cowgill, L. D., Goldfarb, S., Lau, K., Slatopolsky, E. & Agus, Z. S. (1979) J. Clin. Invest. 63, 1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiebzak, G. M., Meyer, R. A. & Mish, P. M. (1981) Miner. Electrolyte Metab. 6, 153-164. [Google Scholar]
- 6.Tenenhouse, H. S., Werner, A., Biber, J., Ma, S., Martel, J., Roy, S. & Murer, H. (1994) J. Clin. Invest. 93, 671-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scriver, C. R., Reade, T. M., DeLuca, H. F. & Hamstra, A. J. (1978) N. Engl. J. Med. 299, 976-979. [DOI] [PubMed] [Google Scholar]
- 8.Drezner, M. K., Lyles, K. W., Haussler, M. R. & Harrelson, J. M. (1980) J. Clin. Invest. 66, 1020-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenenhouse, H. S. & Jones, G. (1990) J. Clin. Invest. 85, 1450-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukase, M., Avioli, L. V., Birge, S. J. & Chase, L. R. (1984) Endocrinology 114, 1203-1207. [DOI] [PubMed] [Google Scholar]
- 11.Verge, C. F., Lam, A., Simpson, J. M., Cowell, C. T., Howard, N. J. & Silink, M. (1991) N. Engl. J. Med. 325, 1843-1848. [DOI] [PubMed] [Google Scholar]
- 12.Goodyer, P. R., Kronick, J. B., Jequier, S., Reade, T. M. & Scriver, C. R. (1987) J. Pediatr. (Berlin) 111, 700-704. [DOI] [PubMed] [Google Scholar]
- 13.Alon, U., Donaldson, D. L., Hellerstein, S., Warady, B. A. & Harris, D. J. (1992) J. Pediatr. (Berlin) 120, 899-905. [DOI] [PubMed] [Google Scholar]
- 14.Reusz, G. D., Hoyer, P. F., Lucas, M., Krohn, P., Ehrich, H. H. & Brodehl, J. (1990) Lancet 335, 1240-1243. [DOI] [PubMed] [Google Scholar]
- 15.Seikaly, M. G., Browne, R. & Baum, M. (1996) Pediatrics 97, 91-93. [PubMed] [Google Scholar]
- 16.Seikaly, M. G., Browne, R. H. & Baum, M. (1994) Pediatrics 94, 478-481. [PubMed] [Google Scholar]
- 17.Eicher, E. M., Southard, J. L., Scriver, C. & Glorieux, F. H. (1976) Proc. Natl. Acad. Sci. USA 73, 4667-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowe, P. S., Oudet, C. L., Francis, F., Sinding, C., Pannetier, S., Econs, M. J., Strom, T. M., Meitinger, T., Garabedian, M., David, A., et al. (1997) Hum. Mol. Genet. 6, 539-549. [DOI] [PubMed] [Google Scholar]
- 19.The HYP Consortium (1995) Nat. Genet. 11, 130-136. [DOI] [PubMed] [Google Scholar]
- 20.Du, L., Desbarats, M., Viel, J., Glorieux, F. H., Cawthorn, C. & Ecarot, B. (1996) Genomics 36, 22-28. [DOI] [PubMed] [Google Scholar]
- 21.Guo, R. & Quarles, L. D. (1997) J. Bone Miner. Res. 12, 1009-1017. [DOI] [PubMed] [Google Scholar]
- 22.Holm, I. A., Huang, X. & Kunkel, L. M. (1997) Am. J. Hum. Genet. 60, 790-797. [PMC free article] [PubMed] [Google Scholar]
- 23.Beck, L., Soumounou, Y., Martel, J., Krishnamurthy, G., Gauthier, C., Goodyer, C. G. & Tenenhouse, H. S. (1997) J. Clin. Invest. 99, 1200-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nesbitt, T., Coffman, T. M., Griffiths, R. & Drezner, M. K. (1992) J. Clin. Invest. 89, 1453-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nesbitt, T., Econs, M. J., Byun, K. J., Martel, J., Tenenhouse, H. S. & Drezner, M. K. (1995) J. Bone. Miner. Res. 10, 1327-1333. [DOI] [PubMed] [Google Scholar]
- 26.Nesbitt, T., Byun, J. K. & Drezner, M. K. (1996) Endocrinology 137, 943-948. [DOI] [PubMed] [Google Scholar]
- 27.LaJeuness, D., Meyer, R. A., Jr., & Hamel, L. (1996) Kidney Int. 50, 1531-1538. [DOI] [PubMed] [Google Scholar]
- 28.Dominguez, J. H., Pitts, T. O., Brown, T., Puschett, D. B., Schuler, F., Chen, T. C. & Puschett, J. B. (1984) Kidney Int. 26, 404-410. [DOI] [PubMed] [Google Scholar]
- 29.Baum, M. (1987) J. Clin. Invest. 79, 1104-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quigley, R. & Baum, M. (1991) J. Clin. Invest. 88, 368-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quigley, R. & Baum, M. (2002) Am. J. Physiol. 283, F525-F531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta, N., Tariff, S. R., Seikaly, M. & Baum, M. (2001) Kidney Int. 60, 173-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drezner, M. K. (2000) Kidney Int. 57, 9-18. [DOI] [PubMed] [Google Scholar]
- 34.Kronenberg, H. M. (2002) N. Engl. J. Med. 347, 1022-1024. [DOI] [PubMed] [Google Scholar]
- 35.Schiavi, S. C. & Moe, O. W. (2002) Curr. Opin. Nephrol. Hypertens. 11, 423-430. [DOI] [PubMed] [Google Scholar]
- 36.Imic, J. D., Kitiyakara, C. & Wilcox, C. S. (2000) in The Kidney: Physiology and Pathophysiology, eds. Seldin, D. W. & Giebisch, G. (Lippincott, Philadelphia), pp. 873-887.
- 37.Dunn, M. J. & Hood, V. L. (1977) Am. J. Physiol. 233, F169-F174. [Google Scholar]
- 38.Farman, N., Pradelles, P. & Bonvalet, J. P. (1987) Am. J. Physiol. 252, F53-F59. [DOI] [PubMed] [Google Scholar]
- 39.Farman, N., Pradelles, P. & Bonvalet, J. P. (1986) Am. J. Physiol. 251, F238-244. [DOI] [PubMed] [Google Scholar]
- 40.Imbert-Teboul, M., Siaume, S. & Morel, F. (1986) Mol. Cell. Endocrinol. 45, 1-10. [DOI] [PubMed] [Google Scholar]
- 41.Baylis, C. & Brenner, B. M. (1978) Circ. Res. 43, 889-898. [DOI] [PubMed] [Google Scholar]
- 42.Navar, L. G., Inscho, E. W., Majid, S. A., Imig, J. D., Harrison-Bernard, L. M. & Mitchell, K. D. (1996) Physiol. Rev. 76, 425-536. [DOI] [PubMed] [Google Scholar]
- 43.Aiken, J. W. & Vane, J. R. (1973) J. Pharmacol. Exp. Ther. 184, 678-685. [PubMed] [Google Scholar]
- 44.Schlondorff, D., Satriano, J. A. & Schwartz, G. J. (1985) Am. J. Physiol. 248, F134-F144. [DOI] [PubMed] [Google Scholar]
- 45.McKinney, T. D. & Burg, M. B. (1977) J. Clin. Invest. 60, 766-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinney, T. D. & Davidson, K. K. (1987) Am. J. Physiol. 252, F509-F516. [DOI] [PubMed] [Google Scholar]
- 47.Alpern, R. J., Horic, S., Moe, O., Tejedor, A., Miller, R. T. & Preisig, P. A. (1991) Kidney Int. Suppl. 33, S29-S32. [PubMed] [Google Scholar]
- 48.Shah, M., Quigley, R. & Baum, M. (2000) Am. J. Physiol. 278, F596-F602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kempson, S. A., Lotscher, M., Kaissling, B., Biber, J., Murer, H. & Levi, M. (1995) Am. J. Physiol. 268, F784-F791. [DOI] [PubMed] [Google Scholar]
- 50.Traebert, M., Roth, J., Biber, J., Murer, H. & Kaissling, B. (2000) Am. J. Physiol. 278, F148-F154. [DOI] [PubMed] [Google Scholar]
- 51.Lötscher, M., Scarpetta, Y., Levi, M., Halaihel, N., Wang, H., Zajicek, H. K., Biber, J., Murer, H. & Kaissling, B. (1999) J. Clin. Invest. 104, 483-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tenenhouse, H. S. (1985) Comp. Biochem. Physiol. A Physiol. 81, 167-171. [DOI] [PubMed] [Google Scholar]
- 53.Tenenhouse, H. S. & Veksler, A. (1986) Endocrinology 118, 1047-1053. [DOI] [PubMed] [Google Scholar]
- 54.Tenenhouse, H. S. & Henry, H. L. (1985) Endocrinology 117, 1719-1726. [DOI] [PubMed] [Google Scholar]