

Abstract
Methionine synthase catalyzes the remethylation of homocysteine to methionine via a reaction in which methylcobalamin serves as an intermediate methyl carrier. Over time, the cob(I)alamin cofactor of methionine synthase becomes oxidized to cob(II)alamin rendering the enzyme inactive. Regeneration of functional enzyme requires reductive methylation via a reaction in which S-adenosylmethionine is utilized as a methyl donor. Patients of the cblE complementation group of disorders of folate/cobalamin metabolism who are defective in reductive activation of methionine synthase exhibit megaloblastic anemia, developmental delay, hyperhomocysteinemia, and hypomethioninemia. Using consensus sequences to predicted binding sites for FMN, FAD, and NADPH, we have cloned a cDNA corresponding to the “methionine synthase reductase” reducing system required for maintenance of the methionine synthase in a functional state. The gene MTRR has been localized to chromosome 5p15.2–15.3. A predominant mRNA of 3.6 kb is detected by Northern blot analysis. The deduced protein is a novel member of the FNR family of electron transferases, containing 698 amino acids with a predicted molecular mass of 77,700. It shares 38% identity with human cytochrome P450 reductase and 43% with the C. elegans putative methionine synthase reductase. The authenticity of the cDNA sequence was confirmed by identification of mutations in cblE patients, including a 4-bp frameshift in two affected siblings and a 3-bp deletion in a third patient. The cloning of the cDNA will permit the diagnostic characterization of cblE patients and investigation of the potential role of polymorphisms of this enzyme as a risk factor in hyperhomocysteinemia-linked vascular disease.
Methionine is an essential amino acid in mammals. It is required for protein synthesis and is a central player in one carbon metabolism. In its activated form, S-adenosylmethionine, it is the methyl donor in hundreds of biological transmethylation reactions and the donor of propylamine in polyamine synthesis. The eventual product of the demethylation of methionine is homocysteine and its remethylation is catalyzed by a cobalamin-dependent enzyme, methionine synthase (1, 2).
The enzyme-bound cobalamin cofactor of methionine synthase plays an essential role in the methyl transfer reaction by acting as an intermediate methyl carrier between methyltetrahydrofolate and homocysteine (Fig. 1A). Cleavage of the methyl-cobalt bond of the methylcob(III)alamin intermediate occurs heterolytically so as to leave the cobalamin in the highly reactive cob(I)alamin oxidation state (3). During the cycling between methylcob(III)alamin and cob(I)alamin, the cofactor may be oxidized to cob(II)alamin with consequent inactivation of the enzyme. The restoration of enzyme activity is dependent on the reductive methylation of cob(II)alamin in a reaction in which S-adenosylmethionine provides the methyl group (4).
Figure 1.
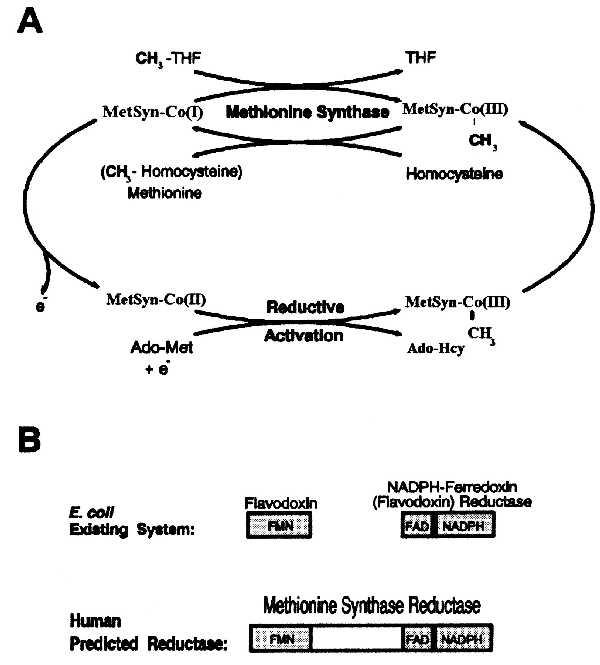
Role and structure of methionine synthase reductase. (A) Transfer of the methyl group of methyltetrahydrofolate (CH3-THF) to homocysteine via methionine synthase-methylcobalamin [MetSyn-CH3-Co(III)] as an intermediate methyl carrier. The reductive methylation in the lower part of the scheme is the mechanism by which S-adenosylmethionine (Ado-Met) together with an electron reactivates the enzyme after oxidative inactivation. Ado-Hcy, S-adenosylhomocysteine. (B) Enzymes involved in the reduction of methionine synthase. (Upper) Scheme shows the two-flavoprotein system involved in the reductive activation of cobalamin-dependent methionine synthase in Escherichia coli. FMN and FAD/NADPH binding sites are in different proteins. (Lower) We propose a “one gene-one enzyme” reduction system for mammalian methionine synthase.
Severe deficiency of methionine synthase activity leads to megaloblastic anemia, developmental delay, hyperhomocysteinemia, and hypomethioninemia (5, 6). Two forms of methionine synthase deficiency are known (5, 7). We (8) and others (9, 10) recently cloned cDNAs encoding human methionine synthase and showed that patients from the cblG complementation group of folate/cobalamin metabolism have mutations in the methionine synthase gene (8, 11). For a second class of patients, belonging to a distinct complementation group, cblE, net methionine synthase activity appears reduced. Their defect is in a reducing system required for maintenance of the enzyme in a functional state (7, 12, 13).
The E. coli methylcobalamin-dependent methionine synthase has been well characterized (14, 15). The reductive activation system required for its maintenance is a two component flavoprotein system (16) consisting of flavodoxin (17) and NADPH-ferredoxin (flavodoxin) oxidoreductase (18), a member of a family of electron transferases termed the “FNR family” (19). Banerjee and colleagues recently documented an NADPH-dependent reducing activity defective in cblE patients (7), suggesting that the reductive-activation system in higher organisms would bear some resemblance to that in E. coli.
Given the absence of flavodoxins in mammalian cells and the occurrence of a single complementation group accounting for defects of the reductive-activation system, we hypothesized that the human counterpart would be a single protein structurally related to the combination of flavodoxin and flavodoxin reductase (Fig. 1B). Significantly, this would imply that FMN, FAD, and NADPH would be bound by a single polypeptide, as is the case for cytochrome P-450 reductase and NO synthase (20). In this study, we present the isolation of cDNA clones corresponding to the reductive activation enzyme defective in the cblE complementation group. The deduced protein is a novel member of the FNR family. We have called this enzyme methionine synthase reductase.
MATERIALS AND METHODS
Cell Lines.
Ten cell lines classified as members of the cblE complementation group were used in this study, of which three were analyzed in detail: WG788 from the original cblE patient (12); WG1146 from his younger brother who had been diagnosed before birth and whose mother was treated with hydroxocobalamin during pregnancy (21); and WG1836 from a patient who had previously been described as having dihydrofolate reductase deficiency (case 1 in ref. 22) and subsequently as having a “new mutation” associated with low methylcobalamin levels and reduced cellular folate uptake (23). We have shown that the fibroblast line from this last patient falls into the cblE complementation group. For mutation analysis, 22 other cell lines were used as normal controls.
Materials.
Radiolabeled compounds were from DuPont. A human multiple tissue Northern blot and a β-actin probe were from CLONTECH. The random-primed DNA labeling kit was from Boehringer Mannheim. The sequenase kit for manual sequencing of crude PCR products was from United States Biochemicals. The oligonucleotides were synthesized by ACGT Corporation (Toronto) or the Sheldon Biotechnology Centre, McGill University. The sequence of oligonucleotides is shown in Fig. 2. A human cDNA library, made in Lambda-ZAP from RNA derived from the human colon carcinoma line Caco-2, was used to obtain 5′ extensions of the cDNA.
Figure 2.
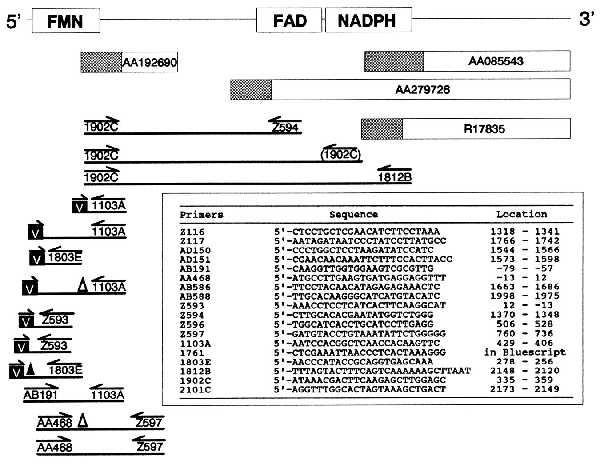
Overlapping clones and PCR fragments analyzed to clone and sequence human methionine synthase reductase. Expressed sequence tag clones are shown as rectangles. The partial sequences that were available from the dbEST database are shown as hatched boxes. PCR fragments are represented as lines. Oligonucleotides are indicated below arrows and are described in the table. The primer in parentheses designates a mispriming outcome that generated valid internal sequence. The letter “V” in black boxes indicates primers annealing to the vector of the cDNA library used as a template for PCR. The presence of a triangle above a segment indicates that it contained a deletion of 154 bp (▵) or 26 bp (▴), due to alternative splicing.
Homology Matches.
Comparisons were made between putative FMN, FAD, and NADPH binding sites and sequences in the National Center for Biotechnology Information (NCBI) databases (dbEST and nr) by using the blast programs (24). The cytochrome P-450 reductase and NO synthase full sequences were also used for homology searching.
PCR Cloning and DNA Sequencing.
RNA isolation, reverse-transcription, PCR reactions, subcloning, and manual sequencing was performed as described (8). Automated sequencing was done by Bio S&T (Montreal) or by the DNA Sequencing Core Facility of the Canadian Genetic Diseases Network.
Northern Blot.
The commercially available multiple tissue Northern blot, prepared from poly(A)+ RNA (2 μg per lane) of the indicated human tissues, was probed with an EcoRI segment of a subclone in pCRII containing an insert spanning positions 335-2148 of the methionine synthase reductase cDNA. Hybridization with human β-actin cDNA served as a control for the quantity and integrity of the RNA in the blot.
Chromosomal Localization.
We performed PCR analysis of DNA isolated from the NIGMS human/rodent somatic cell hybrid mapping panel (no. 2). The oligonucleotide primers, which were specific for the 3′-untranslated region of the gene, amplified a 111-nucleotide product (accession no. G19837 in dbSTS). A P1-derived artificial chromosome clone (104K2) was identified from a total human genomic library (25) by hybridization screening with a methionine synthase reductase cDNA probe (clone 704947, accession no. AA279726 in dbEST) and this genomic clone was then used for fluorescence in situ hybridization mapping (26, 27).
Mutation Analysis.
Total cellular RNA was reverse transcribed by using oligonucleotide 2101C and PCR amplified in nine overlapping segments. PCR products were verified by agarose gel electrophoresis before testing for heteroduplex formation. Heteroduplex analysis was carried out by mixing mutant and control PCR products 1:1, heating the mixture to 95°C for 3 min, cooling to room temperature, and subjecting the samples to electrophoresis on an 8% polyacrylamide gel (8). Fragments displaying heteroduplexes were rerun without mixing to determine if the change was homozygous or heterozygous in the patient sample and then subcloned and sequenced, or sequenced directly.
RESULTS
Cloning Human Methionine Synthase Reductase cDNA.
More than 20 overlapping sequences homologous to the FAD and NADPH-binding domains of cytochrome P-450 reductase were identified in an initial survey of the NCBI dbEST database by using tblastn. We obtained (from the I.M.A.G.E. consortium) and sequenced clones 550341, 704947, and 31776 (accession nos. AA085543, AA279726, and R17835, respectively) to confirm the sequence of this part of the cDNA (Fig. 2). Comparison of this FAD/NADPH binding sequence with sequences in the NCBI databases yielded a sequence from Caenorhabditis elegans (accession no. Z35595) containing the binding site motifs for FMN, FAD, and NADPH. We used the latter sequence to search the dbEST database by using tblastn and identified a human sequence (accession no. AA192690, clone 628497) containing a putative FMN binding site similar to the one encoded by Z35595. We sequenced the entire insert in clone 628497 (obtained from the I.M.A.G.E. consortium). Reverse transcription (RT)–PCR of human fibroblast RNA by using oligonucleotide primers corresponding to the FMN and FAD/NADPH binding regions linked the two sets of cDNA clones and confirmed that our cDNA contained all three putative binding sites. The 5′ end of the sequence was obtained by PCR by using a cDNA library as template, with primers bridging the cDNA and vector arms used to amplify the library.
The coding sequence of the candidate human methionine synthase reductase contains 2094 bp encoding a polypeptide of 698 amino acids in length (Fig. 3). The predicted molecular mass is 77,700 Da. It shares 38% identity (49% similarity) with human cytochrome P-450 reductase and 43% identity (53% similarity) with the C. elegans putative methionine synthase reductase (Fig. 4). The first in-frame methionine residue is a candidate for the initiation codon as it perfectly aligns with the first methionine of the C. elegans sequence. Further, the presence of a G at positions −3 and −6 places the codon in good context for initiation of translation (28). RT-PCR involving various pairs of primers detected alternatively processed mRNA, including one form with a deletion of 154 bp (nucleotides 130–283) and another lacking a 26-bp segment (−52 to −27), accounting for <20% and 40% of the message, respectively (data not shown). A polyadenylation signal is present at positions 3135–3140. The poly(A) tail is added after position 3165, although we observed some clones with polyadenylation after residue 3157 (data not shown).
Figure 3.

Nucleotide and deduced amino acid sequence of human methionine synthase reductase. The nucleotide and amino acid residues are numbered on the left and right margins, respectively. The stop codon is indicated by ∗∗∗.
Figure 4.

Amino acid sequence comparisons among human methionine synthase reductase (HsMTRR, accession no. AF025794), C. elegans putative methionine synthase reductase (CeMTRR, accession no. Z35595), and human cytochrome P-450 reductase (HsCPR, accession no. A60557). Amino acids residues are numbered on the right margin. Conserved and similar (33) residues are shown under the sequence by stars and dots, respectively. Regions proposed to be involved in binding of FMN, FAD, or NADPH are shown above the sequences (18, 19, 31, 34–36).
Northern Blot Analysis.
The 1.8-kb probe hybridized to one predominant RNA species of 3.6 kb in all tissues tested and is particularly abundant in skeletal muscle (Fig. 5). A smaller band of 3.1 kb was also detected in brain, with a minor species at 6 kb.
Figure 5.
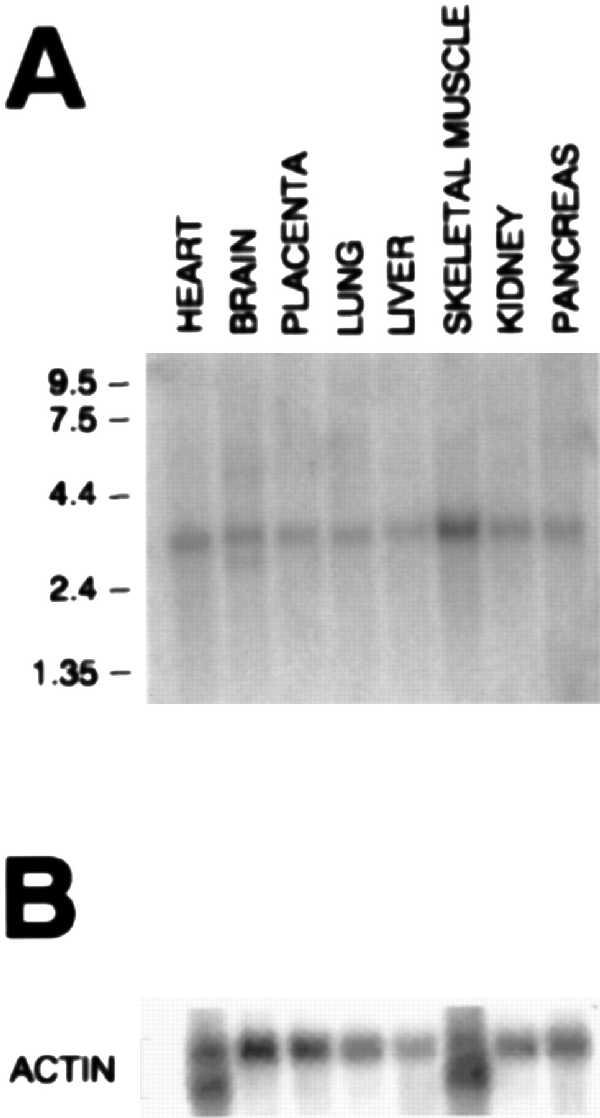
Northern blot analysis of methionine synthase reductase. Northern blot analysis of poly(A)+ RNA from the indicated human tissues. The membranes were hybridized with a [32P]-labeled methionine synthase reductase (A) or β-actin (B) probe. Positions of molecular size (kb) markers are indicated at left.
Chromosomal Location.
The methionine synthase reductase gene was localized to human chromosome 5 because the gene-specific primer pair amplified a PCR product of the expected size only from the GM10114 hybrid that contains chromosome 5 as its only human material (data not shown). Moreover, the DNA sequence we determined for the methionine synthase reductase gene matched markers AA002A03 and STSG444 that were also mapped by the NCBI consortium to chromosome 5 between markers D5S406-D5S478 and D5S406-D5S635, respectively (29). To determine the cytogenetic position of the gene on chromosome 5, we mapped a genomic P1-derived artificial chromosome clone encompassing the gene by using fluorescence in situ hybridization. The hybridization efficiency was 100% and among 100 mitotic figures examined each result indicated the gene was localized on chromosome 5p15.2-p15.3 (Fig. 6). We propose MTRR as the gene name for methionine synthase reductase, because the methionine synthase gene has been named MTR.
Figure 6.

Localization of the methionine synthase gene to chromosome 5p15.2–p15.3. The summary of the fluorescence in situ hybridization mapping results is shown with each dot representing a signal detected on human chromosome 5.
Mutations in the cblE Complementation Group.
Patient cell lines from the cblE complementation group were analyzed by RT-PCR-dependent heteroduplex analysis. Three cell lines showed heteroduplexes, one observed in two overlapping RT-PCR segments (Fig. 7 A and B). The heteroduplex-containing samples were subcloned and sequenced and two mutations were identified. One mutation, confirmed in heterozygous fashion in fibroblast line WG788, is a 4-bp deletion, 1675del4, resulting in a frameshift and nearby stop codon. The same mutation was observed in cell line WG1146 from the brother of patient WG788. The second mutation, also heterozygous in a different cell line, WG1836, is an in-frame deletion of 3 bp, 1726delTTG. It results in the loss of a highly conserved leucine at position 576 of the amino acid sequence (Fig. 7C). On direct sequencing of the PCR products, only a very faint background contributed by the normal sequence was observed, suggesting that the other, unidentified mutation in WG1836 was associated with a very low level of steady state mRNA (data not shown).
Figure 7.
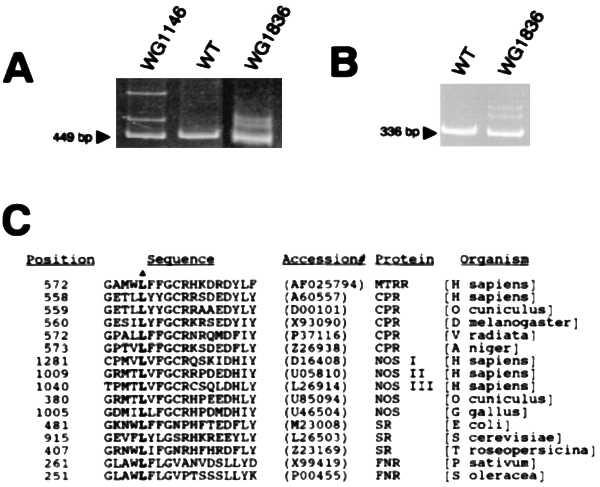
Mutation analysis of cblE patient cell lines. (A) PCR products obtained with primers Z116/Z117 from RT reactions with control sample (WT) and two cblE cell lines. For cell lines WG1146 and WG1836, the bands above the 449 bp amplification product are due to heteroduplexes formed between DNA strands bearing different allelic sequences. The pattern for WG1146 was also seen with WG788 (data not shown). (B) RT-PCR products amplified with primers AB586/AB588 from a control sample and WG1836. Heteroduplexes are observed above the 336-bp segment for WG1836. (C) Sequence comparison among proteins of the FNR family in a part of the NADPH binding region in the vicinity of the leucine residue that is deleted in a cblE patient (denoted by a triangle). Representative examples are shown [MTRR, methionine synthase reductase; CPR, cytochrome P-450 reductase; NOS, NO synthase, SR, sulfite reductase; and FNR, NADPH-ferredoxin (flavodoxin) reductase].
DISCUSSION
We conclude that the cDNA that we have identified corresponds to human methionine synthase reductase. It is homologous to reductases of the FNR family, consistent with its role in the reductive activation of methionine synthase. In addition, the corresponding gene contains mutations in cblE patients with a functional deficiency of methionine synthase. Our key finding confirming the identification of the cDNA was the 4-bp frameshift mutation in the two affected siblings. The occurrence of a functionally null mutation in a candidate gene forms compelling evidence that the mutation is causative of disease in the affected patients. Similarly, the 3-bp deletion in a third patient is also highly likely to cause an enzyme defect.
The survey of the NCBI databases for homology to the human methionine synthase reductase by using blastp or tblastn yielded the putative methionine synthase reductase of C. elegans (P = 9 × 10−92). Such a reductase in C. elegans was expected, because its putative cobalamin-dependent methionine synthase is highly homologous to the human enzyme (8). Proteins of the FNR family were also found by blast programs. The strongest homology was found with cytochrome P-450 reductase (P > 3 × 10−68), followed by NO synthase (three isoforms, P > 4 × 10−52), and sulfite reductase (P > 6 × 10−39). Lower, but still significant homology was found with flavodoxin reductase (P > 2 × 10−9) and flavodoxin (P > 3 × 10−2), the two E. coli proteins that gave impetus to our analysis.
Our finding suggests a convergent evolution of the two genes specifying flavodoxin/flavodoxin reductase to a single gene encoding a fused version of the two proteins in humans, as proposed previously in relation to the structure of cytochrome P-450 reductase (30). Nevertheless, a two component system has been proposed for human cells based on fractionation of the reducing system by DEAE chromatography (7). It remains to be determined if another intermediate electron carrier might be involved.
The N terminus of cytochrome P-450 reductase is hydrophobic and compatible with an anchor to the endoplasmic reticulum. Interestingly, the C. elegans genome encodes an enzyme (accession no. U21322) similar to human cytochrome P-450 reductase that also contains a hydrophobic N terminus as expected. In contrast, the N terminus of methionine synthase reductase would be expected to yield a cytosolic protein because its target, methionine synthase, is a cytosolic enzyme. The sequence of the proposed N terminus of human methionine synthase reductase is consistent with cytosolic targeting. Similarly, the sequence of the putative N terminus of methionine synthase reductase from C. elegans (accession no. Z35595) is also consistent with cytosolic targeting.
The two mutations we have shown to be associated with cblE disease are located in the vicinity of the NADPH binding domain by comparison with proteins of the FNR family. The 4-bp deletion yields a truncated protein that is expected to be deficient in NADPH binding and possibly in FAD binding, because the C terminus of the enzyme may be involved in both (31). Leu-576, which is removed by the 3-bp deletion, is located between two nearby sequences proposed to be involved in NADPH binding. Leu-576 is well conserved among many reductases that are similar to the methionine synthase reductase (Fig. 7C). This supports the idea that 1726delTTG results in an enzymatic defect, although confirmation will require expression of the mutant protein. This residue is also conserved in the flavodoxin reductase enzymes of several organisms, although the homology with this portion of the protein is low in some cases. It is possible that the deletion alters the relationship of the two local NADPH-binding motif components.
With the cloning of the cDNA for human methionine synthase reductase, it will now be possible to characterize the human enzyme involved in the reductive activation of methionine synthase, to identify other mutations in patients with severe deficiency of the enzyme activity and to judge if this reductase can act on other substrates than methionine synthase. It will also be possible to determine if common amino acid polymorphisms exist that lead to mildly elevated homocysteine and that may contribute as a risk factor in cardiovascular disease or neural tube defects, as discovered for methylenetetrahydrofolate reductase (32).
Acknowledgments
We thank N. V. Matiaszuk for the complementation studies and G. Dunbar and P. Zhao for growing cell cultures. We are grateful to the physicians who provided the cell lines used in this study and the I.M.A.G.E. consortium (Livermore, CA) for providing human cDNA clones. We thank Rowena Matthews for helpful discussion and the Canadian Genetic Diseases Network for assistance with some of the sequence analysis. These studies were supported by grants from the Medical Research Council of Canada Group in Medical Genetics (GR-13297), the National Heart, Blood and Lung Institute (HL58955–01), and the Canadian Genome Analysis and Technology Program.
ABBREVIATIONS
- NCBI
National Center for Biotechnology Information
- RT
reverse transcription
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF025794).
References
- 1.Rosenblatt D S. In: The Metabolic and Molecular Bases of Inherited Disease. 7th Ed. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw-Hill; 1995. pp. 3111–3128. [Google Scholar]
- 2.Fenton W A, Rosenberg L E. In: The Metabolic and Molecular Bases of Inherited Disease. 7th Ed. Scriver C R, Beaudet A L, Sly W, S, Valle D, editors. New York: McGraw-Hill; 1995. pp. 3129–3149. [Google Scholar]
- 3.Banerjee R. Chem Biol. 1997;4:175–186. doi: 10.1016/s1074-5521(97)90286-6. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig M L, Matthews R G. Annu Rev Biochem. 1997;66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 5.Watkins D, Rosenblatt D S. Am J Med Genet. 1989;34:427–434. doi: 10.1002/ajmg.1320340320. [DOI] [PubMed] [Google Scholar]
- 6.Harding C O, Arnold G, Barness L A, Wolff J A, Rosenblatt D S. Am J Med Genet. 1997;71:384–390. doi: 10.1002/(sici)1096-8628(19970905)71:4<384::aid-ajmg3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 7.Gulati S, Chen Z Q, Brody L C, Rosenblatt D S, Banerjee R. J Biol Chem. 1997;272:19171–19175. doi: 10.1074/jbc.272.31.19171. [DOI] [PubMed] [Google Scholar]
- 8.Leclerc D, Campeau E, Goyette P, Adjalla C E, Christensen B, Ross M, Eydoux P, Rosenblatt D S, Rozen R, Gravel R A. Hum Mol Genet. 1996;5:1867–1874. doi: 10.1093/hmg/5.12.1867. [DOI] [PubMed] [Google Scholar]
- 9.Li Y N, Gulati S, Baker P J, Brody L C, Banerjee R, Kruger W D. Hum Mol Genet. 1996;5:1851–1858. doi: 10.1093/hmg/5.12.1851. [DOI] [PubMed] [Google Scholar]
- 10.Chen L H, Liu M L, Hwang H Y, Chen L S, Korenberg J, Shane B. J Biol Chem. 1997;272:3628–3634. [PubMed] [Google Scholar]
- 11.Gulati S, Baker P, Li Y N, Fowler B, Kruger W, Brody L C, Banerjee R. Hum Mol Genet. 1996;5:1859–1865. doi: 10.1093/hmg/5.12.1859. [DOI] [PubMed] [Google Scholar]
- 12.Schuh S, Rosenblatt D S, Cooper B A, Schroeder M L, Bishop A J, Seargeant L E, Haworth J C. N Engl J Med. 1984;310:686–690. doi: 10.1056/NEJM198403153101104. [DOI] [PubMed] [Google Scholar]
- 13.Rosenblatt D S, Cooper B A, Pottier A, Lue-Shing H, Matiaszuk N, Grauer K. J Clin Invest. 1984;74:2149–2156. doi: 10.1172/JCI111641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drennan C L, Huang S, Drummond J T, Matthews R G, Lidwig M L. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 15.Dixon M M, Huang S, Matthews R G, Ludwig M. Structure (London) 1996;4:1263–1275. doi: 10.1016/s0969-2126(96)00135-9. [DOI] [PubMed] [Google Scholar]
- 16.Fujii K, Huennekens F M. J Biol Chem. 1974;249:6745–6753. [PubMed] [Google Scholar]
- 17.Osborne C, Chen L M, Matthews R G. J Bacteriol. 1991;173:1729–1737. doi: 10.1128/jb.173.5.1729-1737.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi V, Reichard P, Eliasson R, Pontis E, Krook M, Jornvall H, Haggard-Ljungquist E. J Bacteriol. 1993;175:1590–1595. doi: 10.1128/jb.175.6.1590-1595.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Correll C C, Ludwig M L, Bruns C M, Karplus P A. Prot Sci. 1993;2:2112–2133. doi: 10.1002/pro.5560021212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bredt D S, Hwang P M, Glatt C E, Lowenstein C, Reed R R, Snyder S H. Nature (London) 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 21.Rosenblatt D S, Cooper B A, Schmutz S M, Zaleski W A, Casey R E. Lancet. 1985;1:1127–1129. doi: 10.1016/s0140-6736(85)92433-x. [DOI] [PubMed] [Google Scholar]
- 22.Tauro G P, Danks D M, Rowe P B, Van der Weyden M B, Schwartz M A, Collins V L, Neal B W. N Engl J Med. 1976;294:466. doi: 10.1056/NEJM197602262940903. [DOI] [PubMed] [Google Scholar]
- 23.Brasch, J., Sawyer, M., Hayman, R. & Van der Weyden, M. B. (1988) Aust. N.Z. J. Med. 18 Suppl., 434.
- 24.Altschul S F, Boguski M S, Gish W, Wootton J C. Nat Genet. 1994;6:119–129. doi: 10.1038/ng0294-119. [DOI] [PubMed] [Google Scholar]
- 25.Ioannou P A, Amemiya C T, Garnes J, Kroisel P M, Shizuya H, Chen C, Batzer M A, de Jong P J. Nat Genet. 1994;6:84–89. doi: 10.1038/ng0194-84. [DOI] [PubMed] [Google Scholar]
- 26.Heng H H, Squire J, Tsui L C. Proc Natl Acad Sci USA. 1992;89:9509–9513. doi: 10.1073/pnas.89.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heng H H, Tsui L C. Chromosoma. 1993;102:325–332. doi: 10.1007/BF00661275. [DOI] [PubMed] [Google Scholar]
- 28.Kozak M. J Biol Chem. 1991;266:19867–19870. [PubMed] [Google Scholar]
- 29.Hudson T J, Stein L D, Gerety S S, Ma J, Castle A B, Silva J, Slonim D K, Baptista R, Kruglyak L, Xu S H, et al. Science. 1995;270:1945–1954. doi: 10.1126/science.270.5244.1945. [DOI] [PubMed] [Google Scholar]
- 30.Porter T D, Kasper C B. Biochemistry. 1986;25:1682–1687. doi: 10.1021/bi00355a036. [DOI] [PubMed] [Google Scholar]
- 31.Wang M, Roberts D L, Paschke R, Shea T M, Masters B S S, Kim J J P. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rozen R. Clin Invest Med. 1996;19:171–178. [PubMed] [Google Scholar]
- 33.Bordo D, Argos P. J Mol Biol. 1991;217:721–729. doi: 10.1016/0022-2836(91)90528-e. [DOI] [PubMed] [Google Scholar]
- 34.Miles J S. Biochem J. 1992;287:195–200. doi: 10.1042/bj2870195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenkins C M, Waterman M R. J Biol Chem. 1994;269:27401–27408. [PubMed] [Google Scholar]
- 36.Neidle E L, Hartnett C, Ornston L N, Bairoch A, Rekik M, Harayama S. J Bacteriol. 1991;173:5385–5395. doi: 10.1128/jb.173.17.5385-5395.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]