

Abstract
Phosphatidylserine (PS) normally localizes to the inner leaflet of cell membranes but becomes exposed in abnormal or apoptotic cells, signaling macrophages to ingest them. Along similar lines, it seemed possible that the removal of red cells from circulation because of normal aging or in hemolytic anemias might be triggered by PS exposure. To investigate the role of PS exposure in normal red cell aging, we used N-hydroxysuccinimide-biotin to tag rabbit red cells in vivo, then used phycoerythrin-streptavidin to label the biotinylated cells, and annexin V-fluorescein isothiocyanate (FITC) to detect the exposed PS. Flow cytometric analysis of these cells drawn at 10-day intervals up to 70 days after biotinylation indicated that older, biotinylated cells expose more PS. Furthermore, our data match a simple model of red cell senescence that assumes both an age-dependent destruction of senescent red cells preceded by several hours of PS exposure and a random destruction of red cells without PS exposure. By using this model, we demonstrated that the exposure of PS parallels the rate at which biotinylated red cells are removed from circulation. On the other hand, using an annexin V-FITC label and flow cytometry demonstrates that exposed PS does not cause the reduced red cell life span of patients with hemolytic anemia, with the possible exception of those with unstable hemoglobins or sickle cell anemia. Thus, in some cases PS exposure on the cell surface may signal the removal of red cells from circulation, but in other cases some other signal must trigger the sequestration of cells.
Phospholipids are asymmetrically distributed in the membranes of normal eukaryotic cells. The outer leaflet contains the cholinephospholipids phosphatidylcholine and sphingomyelin whereas the inner leaflet contains the aminophospholipids phosphatidylserine (PS) and phosphatidylethanolamine. This distribution of the membrane lipids appears to be quite stable, and when the phospholipids are redistributed so that PS or phosphatidylethanolamine are on the outer leaflet, an aminophospholipid translocase (“flippase”) can restore the normal phospholipid distribution (1, 2).
It has been suggested that a “scramblase” enzyme is directly responsible for the loss of asymmetry that occurs when, for example, red cells are treated with a calcium ionophore. The exposure of PS on the cell surface as a result of “scrambling” appears to be an important signal for the ingestion by macrophages of apoptotic nucleated cells (3–5). Erythrocytes cannot undergo apoptosis because they lack a nucleus, but it is attractive to suppose that evolution has seized on the same mechanisms for signaling removal of red cells from the circulation. If this were the case, then the removal of aged red cells, and red cells from patients with hemolytic anemia, might be caused by exposure of PS on the membrane.
Indeed, it has been proposed that red cells with exposed PS are more likely to be ingested by macrophages (6) and that the exposure of PS signals the removal of aged red cells from the circulation (7). There is general agreement that PS is exposed on sickle cells (8–10) and the pathogenesis of some of the features of this disorder have been explained on this basis.
The specific binding of annexin V to PS has greatly facilitated the measurement of the exposure of small amounts of this phospholipid on the outer membrane of cells. Annexin V can be radiolabeled or labeled with a fluorescent compound. The latter makes it possible to use a flow cytometer to estimate the percentage of erythrocytes with PS exposed on their outer surface. Both techniques have been used successfully in quantitating PS on red cell membranes (11, 12).
We now show that increased PS exposure occurs during normal red cell aging, but only in certain patients with hemolytic anemia.
MATERIALS AND METHODS
Reagents.
B-phycoerythrin-streptavidin (PE-streptavidin) was obtained from Molecular Probes, annexin V-fluorescein isothiocyanate (annexin V-FITC) from Nexins Research (Hoeven, The Netherlands), N-hydroxysuccinimide biotin (NHS-biotin), stored desiccated at −20°C, from Calbiochem, N,N-dimethylacetamide, N-ethylmaleimide (NEM), and calcium ionophore A23187 from Sigma. PBS consisted of 146 mM NaCl containing 10 mM Na2HPO4/NaH2PO4, pH 7.4, filtered through no. 2 Whatman paper. Many of the procedures described below have been performed in Hepes buffer instead of PBS with similar results.
Patient Samples.
Red cells from 14 patients with hemolytic anemia (including at least 2 who had been subjected to splenectomy), 2 other splenectomized patients (1 with Gaucher disease and the other with lymphoma), and 8 normal controls were studied. Of the patients with hemolytic anemia, 1 had hereditary spherocytosis, 2 were glucose-6-phosphate dehydrogenase deficient, 2 were pyruvate kinase deficient, and 2 had an unstable hemoglobin. No specific diagnosis was established for the other patients with hemolytic anemia.
Blood Filtration.
Blood samples anticoagulated in EDTA were defibrinated by adding CaCl2 to a 6 mM concentration and mixing with glass beads gently for 5–10 min. One milliliter of the red cell suspension was filtered over a 2 ml cellulose column (13) to remove leukocytes and platelets, then washed at 4°C two or three times with 0.9% NaCl and once with PBS.
Annexin Labeling of Erythrocytes.
We measured exposed PS by treating red cells with commercially available FITC-labeled annexin V in the presence of calcium, washing with a calcium-containing buffer, and measuring cell-associated fluorescence in a Becton Dickinson FACScan, essentially following the method of Kuypers et al. (11).
Whole blood was collected in EDTA (≈1 mg/ml) and filtered as described above. The suspension was diluted to a final concentration of 107 red cells/ml in PBS plus 1.2 mM CaCl2. Labeling with annexin was carried out in a system containing 100 μl of the diluted red cell suspension, 9 μl annexin binding buffer, 1 μl annexin-FITC, and sufficient PBS plus 1.2 mM CaCl2 for a final volume of 1.00 ml. The addition of EDTA to a final concentration of 5 mM to chelate the calcium needed for specific annexin binding to PS provided a negative control. The suspensions were incubated for 30 min at room temperature in the dark, then washed once with buffer to remove unbound annexin V. Finally, the cells were resuspended to a volume of 1.00 ml with PBS plus 1.2 mM CaCl2 for analysis on a Becton Dickinson FACScan flow cytometer. Acquisition and data analysis were performed by using lysis ii or cellquest software (Becton Dickinson). Ten thousand to 20,000 events per sample were acquired to minimize sampling errors. Fluorescent channels and light scatter were set at log gain. The FSC setting was at E-1 with a threshold of 36. The SSC PMT voltage was set at 250, and the FL1 PMT voltage at 750. The red cell population was defined by gating a region on the forward and side scatter plots.
Positive controls were incorporated into most of the studies. Normal human blood was drawn into EDTA anticoagulant, filtered as described above, and suspended at a 30% hematocrit in PBS. The suspension was incubated in 10 mM NEM for 30 min at room temperature to inhibit the translocase, then washed twice with PBS. The NEM-treated red cells were diluted to 16% hematocrit with PBS plus 1.2 mM CaCl2 and incubated for 3 min at 37°C. Calcium ionophore A23187 was added to a final concentration of 4 μM, and the suspension was incubated for 1 h at 37°C. The cells were then washed twice with PBS plus 2.5 mM EDTA to remove the Ca2+ and three times more with PBS plus 1% BSA to remove the ionophore. After one additional washing with PBS, the cells were resuspended in PBS plus 1.2 mM CaCl2 and labeled with annexin V.
Rabbit Studies.
Biotinylation. To identify an aging population of erythrocytes, we labeled the red cell membrane proteins of two rabbits with biotin in vivo by using the method described by Dale and Norenberg (14). A preinfusion blood sample was drawn into EDTA anticoagulant. Just prior to injection, 16.6 mg NHS-biotin/kg body weight was dissolved in 0.95 ml N,N-dimethylacetamide and incubated 2 min in the dark at 37°C. The dissolved NHS-biotin was added to 27 ml sterile 0.9% NaCl in a 60-ml syringe and mixed well. The NHS-biotin was then injected into a peripheral ear vein of the rabbit at a rate of 2 ml/min with a Sage model 341 syringe pump (Orion Research, Boston) by using a 23-gauge butterfly needle. Blood samples (2 ml) were drawn into EDTA anticoagulant 2 h postinfusion and every 10 days thereafter.
Detection of biotin and PS on in vivo labeled rabbit erythrocytes.
The red cells from the whole blood were diluted to 107 cells/ml with PBS plus 1.2 mM CaCl2. In rabbit 2, the blood was washed three times in PBS before dilution. Diluted red cell suspension (100 μl) was added to a fluorescent labeling reaction mix containing 10 μl 1 mg/ml PE-streptavidin, 9 μl annexin binding buffer, 1 μl annexin-FITC, and sufficient PBS plus 1.2 mM CaCl2 for a final volume of 1.00 ml. Along with each sample we also prepared a negative control containing 5 mM EDTA and 0.5 mM biotin. The reaction mixtures were incubated for 30 min at room temperature in the dark, washed once with PBS plus 1.2 mM CaCl2, and resuspended to 1 ml with PBS plus 1.2 mM CaCl2. The FACScan analysis was carried out as indicated above. The SSC PMT voltage was set at 250, FL1 PMT voltage at 750, and FL2 PMT voltage at 525. Compensation was adjusted for FL1-FL2 at about 8.0% and for FL2-FL1 at 2.9%.
RESULTS
Aging and Senescence.
The age-dependence of exposed PS in rabbit red cells was studied by in vivo biotinylation of all red cells in two rabbits. Biotinylated cells bind streptavidin-PE and such cells are of necessity at least as old as the time elapsed since biotinylation. Fig. 1 shows plots of exposed PS (measured by FITC fluorescence) versus biotin (measured by PE fluorescence) for various times after biotinylation.
Figure 1.
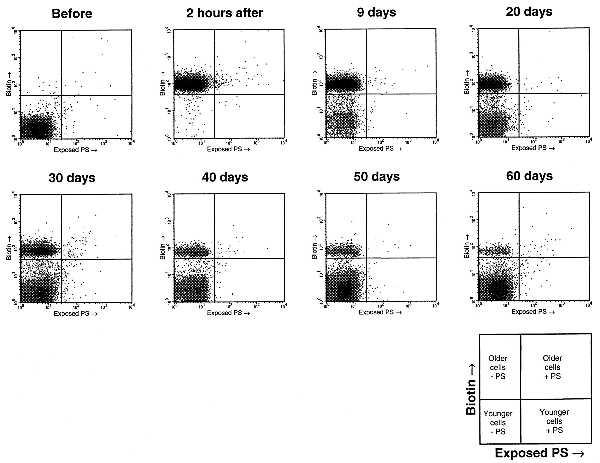
Density plots of exposed PS (measured by FITC fluorescence) vs. biotin (measured by PE fluorescence) in red cells before and after irreversible in vivo biotinylation (rabbit 2). The quadrants indicate cutoffs for determining biotinylation and exposed PS. Notice that biotinylated cells steadily decrease in number after biotinylation, and a greater proportion of them have exposed PS than do nonbiotinylated cells. These experiments were done in duplicate assays (only one shown) for both rabbit 1 and rabbit 2.
Fig. 2 presents the proportion of biotinylated cells with exposed PS as a function of days after biotinylation. The proportion of biotinylated (older) cells with exposed PS rises dramatically with age, as we would expect if exposed PS were the signal for senescent red cell removal.
Figure 2.
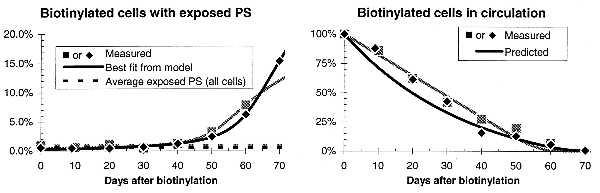
Graphs on the left show that older, biotinylated cells expose more PS in both rabbit 1 (gray points and curves) and rabbit 2 (black points and curves). The parameters for our model of red cell senescence were chosen to provide a least-squares fit to these data on exposed PS in biotinylated cells (r2 > 0.98). Graphs on the right show that the model correctly predicts the rate at which biotinylated cells are removed from circulation (r2 > 0.97). This supports the model, which assumes that the amount of exposed PS is proportional to the rate of senescent death.
We developed a simple model to test the correlation between PS exposure and the removal of biotinylated red cells from circulation. This model of red cell aging assumes both a random destruction of red cells and an age-dependent destruction of senescent red cells as discussed previously by Eadie and Brown (15). We assume that the removal of senescent red cells is preceded by a fixed period of PS exposure, and that the age of senescent death follows a gamma distribution (16), a widely used lifetime distribution that successfully models time between inventory restockings, time to recalibration of instruments, and the survival time of irradiated mice. Finally, we assume that red cells are produced and destroyed at a constant rate. This steady-state assumption implies that the percent of all cells with exposed PS should not change over time. However, the measured value for this percentage did fluctuate modestly, suggesting that technical factors influence the number of cells with exposed PS measured on different days. Accordingly, we normalized the data so that the total exposed PS for each individual measurement matched the average value.
The four parameters of the model—rate of random destruction, mean and SD age of senescent death, and duration of PS exposure before the cells are removed from the circulation—were adjusted to provide a least squares fit to the PS exposure data in the left graph of Fig. 2. The model provides an excellent fit to the data (r2 > 0.98). More significantly, the parameters found by using the data on PS exposure in biotinylated cells correctly predict the decline in biotinylated cells over time. The accuracy of the prediction shown in the right graph of Fig. 2 (r2 > 0.98) makes a strong case for the validity of the model, and thus of the connection between PS exposure in red cells and their removal from circulation.
We calculated more accurate estimates of the parameters describing red cell aging by fitting them both to the data on PS exposure and on biotinylation. The parameters thus obtained (Fig. 3 and Table 1) approximately agree with a previously published estimate by Brown and Eadie (17). Between 1% and 1.5% of cells are randomly chosen for destruction each day. Red cells that survive random destruction eventually become senescent and are destroyed at an average age of between 60 and 70 days. The data further indicate that the age of senescence is narrowly distributed with a SD of under 4 days, and that senescent cells expose PS on their surface for several hours before being removed from the circulation. Changing the cutoff fluorescence defining exposed PS affects the calculated PS exposure time but does not significantly change any of the other parameters of the model.
Figure 3.
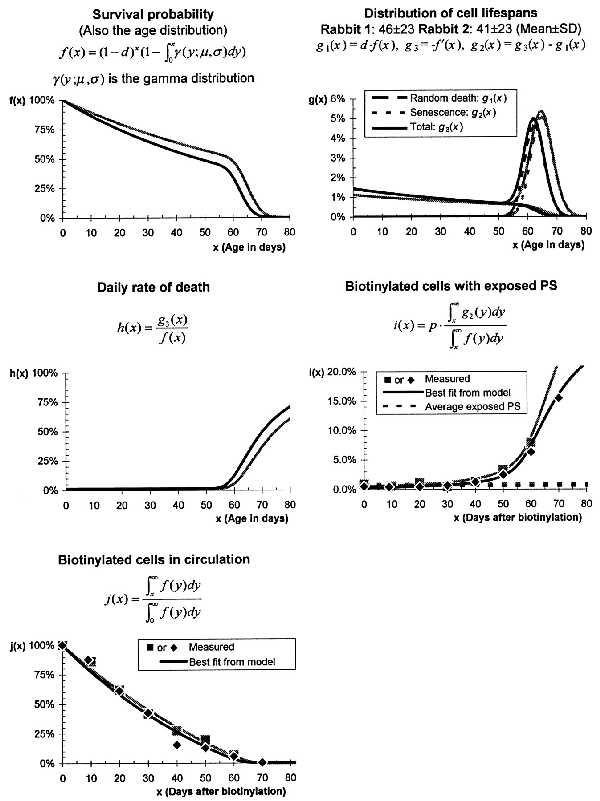
Red cell aging in rabbit 1 (gray points and curves) and rabbit 2 (black points and curves). The parameters shown in Table 1 characterizing these curves were fitted to the experimental data [i(x) and j(x) above] and agree with results from Brown and Eadie (17). The best fit curves for i(x) and j(x) match the experimental data closely (r2 > 0.98).
Table 1.
Parameters describing red cell aging, calculated from data shown in Fig. 3
| Rabbit 1 | Rabbit 2 | Brown and Eadie (17) | |
|---|---|---|---|
| Random death (d) | 1.1%/day | 1.4%/day | 1.35%/day |
| Senescence | |||
| Mean (μ) | 66 days | 63 days | 68 days |
| SD (σ) | 4 days | 3 days | — |
| PS exposure time (p) | 0.51 days (12 h) | 0.30 days (7 h) | — |
Patients with Hemolytic Anemia.
The results of the measurement of exposed PS in patients with hemolytic anemia and normal controls is summarized in Table 2. The red cells of most patients with hemolytic anemia have normal levels of PS exposure, so that some other signal must be responsible for their reduced red cell life span. However, red cells from patients with unstable hemoglobin do have elevated levels of PS on the outer leaflets of their membranes, perhaps accounting for their shortened circulating lifetime.
Table 2.
Exposure of PS on red cells from normal controls and patients
| Patient | Diagnosis | % of cells with exposed PS |
|---|---|---|
| 1 | Hemolytic anemia | 0.00 |
| 2 | Hemolytic anemia | 0.00 |
| Ctrl1 | Normal control | 0.10 |
| 3 | Hemolytic anemia (G6PD deficiency) | 0.11 |
| 4 | Hemolytic anemia | 0.13 |
| 5 | Hemolytic anemia | 0.13 |
| 6 | Hemolytic anemia (G6PD deficiency: A-) | 0.15 |
| 7 | Hemolytic anemia (PK deficiency) | 0.17 |
| Ctrl2 | Normal control | 0.18 |
| 8 | Hemolytic anemia (Hereditary spherocytosis) | 0.23 |
| Ctrl3 | Normal control | 0.29 |
| Ctrl4 | Normal control | 0.42 |
| Ctrl5 | Normal control | 0.42 |
| Ctrl6 | Normal control | 0.43 |
| 9 | Hemolytic anemia | 0.53 |
| Ctrl7 | Normal control | 0.53 |
| 10 | Hemolytic anemia | 0.56 |
| 11 | Hemolytic anemia | 0.56 |
| 12 | Lymphoma* | 0.69 |
| Ctrl8 | Normal control | 0.83 |
| 13 | Gaucher disease* | 1.16 |
| 14 | Hemolytic anemia (PK deficiency) | 1.51 |
| 15 | Hemolytic anemia (unstable hemoglobin)* | 3.72 |
| 16 | Hemolytic anemia (unstable hemoglobin)* | 6.78 |
Patients shown in boldface type have an elevated (>1%) percentage of cells exposing PS.
Patient had a splenectomy.
A possible complicating factor in the analysis of red cells from patients with hemolytic anemia is that some of them have been splenectomized. Splenectomy might increase the fraction of cells with exposed PS by allowing senescent red cells to remain in the circulation longer or by allowing more cells to survive to an age when they will senesce and expose PS. We therefore measured exposed PS on the red cell membranes of two splenectomized patients who did not have hemolytic anemia (patients 12 and 13). This small sample suggests that splenectomized patients may have slightly elevated levels of exposed PS. Much more PS is exposed in the two splenectomized patients with unstable hemoglobins. This elevated level of PS exposure might be caused by either the unstable hemoglobin alone or the combination of splenectomy and unstable hemoglobin. It is notable, in this regards, that sickle cell hemoglobin, which is to some extent unstable (18), is also associated with abnormally high levels of exposed PS (8–10).
DISCUSSION
Many signals for the removal of senescent red cells from the circulation have been proposed, including binding of antibodies to clustered band 3 (19), exposure of a “senescent antigen” that binds circulating antibodies (20), and loss of ATP (21–23). Most of these theories have been supported chiefly by studies based on separation of cells by density. Recently, for instance, it was shown that denser and supposedly older mouse red cells had an impaired ability to translocate PS from the outer to the inner leaflets and exposed more PS as indirectly measured by a prothrombinase assay (7). Autologous monocytes bound denser cells with exposed PS, and some evidence was presented that the denser cells were older.
Unfortunately, density is not a good criterion for identifying old erythrocytes (24, 25). The elegant technique of Dale and his coworkers (26) for irreversibly biotinylating red cells without affecting their function provides the best way to isolate aged cells; this technique confirmed that density centrifugation results in little enrichment with old cells (14). When the biotinylation technique was used to isolate old red cells, they did not bind more Ig, they were not ATP depleted, and their density and deformity were only modestly increased (27, 28). Linderkamp et al. (29) confirmed the lack of importance of the latter changes by showing that the red cells of children with transient erythroblastopenia of childhood exhibited no changes in density or deformability. We noticed in our experiments (data not shown) that older, biotinylated rabbit cells have the same forward scatter/side scatter distribution as other cells, suggesting that the physical properties of red cells change little with age. Similarly, normal human red cells with exposed PS have the same forward scatter/side scatter distribution as cells without exposed PS. However, the exposure of PS does significantly increase as cells age, and this exposure parallels the rate at which biotinylated red cells are removed from circulation. Thus, aged red cells, like sickle cell anemia red cells and apoptotic lymphocytes, expose their PS, a change that could signal macrophages to remove them from the bloodstream.
Abnormal exposure of PS on sickle cells was reported in 1981 (8). The methodology available at that time was cumbersome, and although this finding was initially challenged (30), it has now been confirmed by using more facile modern methods (11, 12, 31). In the case of enzyme-deficient hemolytic anemias, the proximate cause for red cell removal is unknown or at best controversial (32, 33). Our studies show that exposure of PS is not the death signal in most forms of hemolytic anemia. We examined the red cells both of patients with known red cell enzyme deficiencies and of those in whom no specific defect had been identified. Only two patients with unstable hemoglobin and one patient with pyruvate kinase deficiency exposed excess amounts of PS on the outer leaflet of the red cell membrane.
By using the model of red cell senescence we developed earlier in this paper, the proportion of cells with exposed PS is given by p/μ in the absence of random destruction, where p is the length of time that cells expose PS before being removed from the circulation and μ is the average age of PS exposure. Thus, patients might have a greater proportion of cells with exposed PS if (i) random destruction decreases; (ii) cells with exposed PS remain in the circulation longer, as might be the case with splenectomized patients; or (iii) the average age at which PS is exposed decreases. Any of these three reasons might account for the increased PS exposure in the two splenectomized patients with unstable hemoglobins.
We conclude that PS exposure occurs during red cell aging, but does not play a detectable role in most cases of hemolytic anemia. It seems likely that PS exposure could, indeed, be the long-sought signal for red cell senescence, but the search for the signals that target macrophages to remove red cells in most forms of hemolytic anemia will have to continue.
Acknowledgments
This is manuscript 10997-MEM from The Scripps Research Institute. This work was supported by National Institutes of Health Grants HL25552 and RR00833 and the Stein Endowment Fund.
ABBREVIATIONS
- PS
phosphatidylserine
- FITC
fluorescein isothiocyanate
- PE
phycoerythrin
- NHS
N-hydroxysuccinimide
- NEM
N-ethylmaleimide
References
- 1.Diaz C, Schroit A J. J Membr Biol. 1996;151:1–9. doi: 10.1007/s002329900051. [DOI] [PubMed] [Google Scholar]
- 2.Yaffe M P, Kennedy E P. Biochemistry. 1983;22:1497–1507. doi: 10.1021/bi00275a026. [DOI] [PubMed] [Google Scholar]
- 3.Savill J, Fadok V, Henson P, Haslett C. Immunol Today. 1993;3:131–136. doi: 10.1016/0167-5699(93)90215-7. [DOI] [PubMed] [Google Scholar]
- 4.Fadok V A, Laszlo D J, Noble P W, Weinstein L, Riches D W, Henson P M. J Immunol. 1993;151:4274–4285. [PubMed] [Google Scholar]
- 5.Fadok V A, Voelker D R, Campbell P A, Cohen J J, Bratton D L, Henson P M. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 6.McEvoy L, Williamson P, Schlegel R A. Proc Natl Acad Sci USA. 1986;83:3311–3315. doi: 10.1073/pnas.83.10.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor J, Pak C C, Schroit A J. J Biol Chem. 1994;269:2399–2404. [PubMed] [Google Scholar]
- 8.Chiu D, Lubin B, Roelofsen B, van Deenen L L. Blood. 1981;58:398–401. [PubMed] [Google Scholar]
- 9.Wagner G M, Schwartz R S, Chiu D T, Lubin B H. Clin Haematol. 1985;14:183–200. [PubMed] [Google Scholar]
- 10.Wang R H, Phillips G, Jr, Medof M E, Mold C. J Clin Invest. 1993;92:1326–1335. doi: 10.1172/JCI116706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuypers F A, Lewis R A, Hua M, Schott M A, Discher D, Ernst J D, Lubin B H. Blood. 1996;87:1179–1187. [PubMed] [Google Scholar]
- 12.Tait J F, Gibson D. J Lab Clin Med. 1994;123:741–748. [PubMed] [Google Scholar]
- 13.Beutler E, West C, Blume K G. J Lab Clin Med. 1976;88:328–333. [PubMed] [Google Scholar]
- 14.Dale G L, Norenberg S L. Biochim Biophys Acta. 1990;1036:183–187. doi: 10.1016/0304-4165(90)90032-r. [DOI] [PubMed] [Google Scholar]
- 15.Eadie G S, Brown I W., Jr Blood. 1953;8:1110–1136. [PubMed] [Google Scholar]
- 16.Gross A J, Clark V. Survival Distributions: Reliability Applications in the Biomedical Services. New York: Wiley; 1975. [Google Scholar]
- 17.Brown I W, Jr, Eadie G S. J Gen Physiol. 1953;36:327–343. doi: 10.1085/jgp.36.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bensinger T A, Beutler E. Am J Clin Pathol. 1977;67:180–183. doi: 10.1093/ajcp/67.2.180. [DOI] [PubMed] [Google Scholar]
- 19.Low P S, Waugh S M, Zinke K, Drenckhahn D. Science. 1985;227:531–533. doi: 10.1126/science.2578228. [DOI] [PubMed] [Google Scholar]
- 20.Kay M M B, Marchalonis J J, Schluter S F, Bosman G. Transfus Med Rev. 1991;5:173–195. doi: 10.1016/s0887-7963(91)70207-1. [DOI] [PubMed] [Google Scholar]
- 21.Brok F, Ramot B, Zwang E, Danon D. Isr J Med Sci. 1966;2:291–296. [PubMed] [Google Scholar]
- 22.Bartosz G, Grzelinska E, Wagner J. Experientia. 1982;38:575. doi: 10.1007/BF02327057. [DOI] [PubMed] [Google Scholar]
- 23.Cohen N S, Ekholm J E, Luthra M G, Hanahan D J. Biochim Biophys Acta. 1976;419:229–242. doi: 10.1016/0005-2736(76)90349-7. [DOI] [PubMed] [Google Scholar]
- 24.Beutler E. Br J Haematol. 1986;64:408–409. [Google Scholar]
- 25.Beutler E. Blood Cells. 1988;14:1–5. [PubMed] [Google Scholar]
- 26.Suzuki T, Dale G L. Blood. 1987;70:44A. [PubMed] [Google Scholar]
- 27.Waugh R E, Narla M, Jackson C W, Mueller T J, Suzuki T, Dale G L. Blood. 1992;79:1351–1358. [PubMed] [Google Scholar]
- 28.Dale G L, Daniels R B, Beckman J, Norenberg S L. Adv Exp Med Biol. 1991;307:93–103. doi: 10.1007/978-1-4684-5985-2_9. [DOI] [PubMed] [Google Scholar]
- 29.Linderkamp O, Friederichs E, Boehler T, Ludwig A. Br J Haematol. 1993;83:125–129. doi: 10.1111/j.1365-2141.1993.tb04642.x. [DOI] [PubMed] [Google Scholar]
- 30.Raval P J, Allan D. Biochem J. 1984;223:555–557. doi: 10.1042/bj2230555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wood B L, Gibson D F, Tait J F. Blood. 1996;88:1873–1880. [PubMed] [Google Scholar]
- 32.Valentine W N, Paglia D E. Blood Cells. 1980;6:819–825. [PubMed] [Google Scholar]
- 33.Beutler E. Blood Cells. 1980;6:827–829. [PubMed] [Google Scholar]