

Abstract
RPE65 is a protein of unknown function expressed specifically by the retinal pigment epithelium. We examined all 14 exons of this gene in 147 unrelated patients with autosomal recessive retinitis pigmentosa (RP), in 15 patients with isolate RP, and in 45 patients with Leber congenital amaurosis (LCA). Sequence anomalies that were likely to be pathogenic were found in two patients with recessive RP, in one patient with isolate RP recategorized as recessive, and in seven patients with LCA. Cosegregation analysis in each available family showed that all affected individuals were either homozygotes or compound heterozygotes and that all unaffected individuals were either heterozygote carriers or homozygous wild type. In one family, there was one instance of a new mutation not present in either parent of the affected individual. In another family, affected members with recessive RP in three branches (i.e., three distinct pairs of parents) were compound heterozygotes for the same two mutations or homozygous for one of them. Based on our results, mutations in the RPE65 gene appear to account for ≈2% of cases of recessive RP and ≈16% of cases of LCA.
Over 40 loci for human hereditary retinal degenerations involving the photoreceptors and retinal pigment epithelium are known (http://utsph.sph.uth.tmc.edu/www/utsph/RetNet/home.htm). Most of these loci are unidentified genes that have been recognized by linkage studies alone. Our group is engaged in identifying genes responsible for this set of diseases primarily through a candidate gene-based approach (1). In this report, we describe an analysis of the RPE65 gene, which has been assigned to chromosome 1p31 (2). This gene was discovered through its protein product that forms a complex with an antibody raised against human retinal pigment epithelium (3). The cDNA sequence predicts a protein with 533 amino acid residues and a molecular mass of ≈61 kDa (4). The protein is associated with the endoplasmic reticulum of the retinal pigment epithelium in vertebrates (3, 5). Although the biochemical function of the protein product is currently obscure, the tissue-specific expression of the RPE65 gene made it an attractive candidate as a cause for some retinal degenerations because the retinal pigment epithelium has an essential role in maintaining the viability of the neighboring photoreceptor cells. In particular, enzymes in the endoplasmic reticulum are responsible for the recycling of the chromophore used by photoreceptor opsins.
To explore the possible role that defects in RPE65 might play in the etiology of retinal degenerations, we screened a large cohort of unrelated patients with retinitis pigmentosa or Leber congenital amaurosis for mutations. Because RPE65 is expressed specifically in the eye, the study focused on patients with nonsyndromic forms of these diseases. This study was carried out contemporaneously with studies from two other groups that recently have been published (6, 7).
MATERIALS AND METHODS
Ascertainment of Patients.
The index patients in this study were examined and diagnosed by one or more of the authors (G.A.F., A.B.F., or E.L.B.). Most reside in the United States or Canada. All patients had reduced or nondetectable electroretinogram responses. Patients with recessive retinitis pigmentosa (RP) had unaffected parents and at least one affected sibling or were the offspring of a consanguineous mating. If all affected siblings with RP in a sibship were males, the possibility of X-linked disease was evaluated through an ophthalmological examination of the mother including an electroretinogram to search for the carrier state of X-linked RP (8); families with X-linked RP were excluded from this study. Some patients with isolate RP also were included in this study; they had no affected relatives and were not the offspring of a consanguineous mating. Patients with Leber congenital amaurosis (LCA) had absent or severely diminished vision within the first year of life; all had unaffected parents. Control individuals had no known blood relative with a hereditary retinal degeneration and no symptoms of visual malfunction.
After informed consent, we collected between 3 and 50 ml of venous blood from each patient or control individual. Leukocyte nuclei were prepared from the blood samples and stored at −70°C before DNA was purified from them.
Determination of the Intron Sequence Flanking the Exons of the RPE65 Gene.
We synthesized the following oligonucleotide primers based on the published cDNA sequence for the human RPE65 gene: sense, 5′-GTGTAGTTCTGAGTGTGGTG-3′; antisense, 5′-CACAGAGGAAGTATGATTAT-3′. These primers were used to amplify a 369-bp genomic fragment that spanned the end of the coding sequence and the beginning of the 3′ untranslated region. With this set of primers, two human genomic DNA fragments, 50 kb and 65 kb in size and cloned separately in a P1 vector, were isolated (Genome System, St. Louis). To determine flanking intron sequences, the 65-kb genomic fragment was used as a sequencing template for cycle-sequencing (Thermo Sequenase radiolabeled terminator cycle sequencing kit, Amersham) by using primers derived from the published cDNA sequence.
Screening for RPE65 Gene Mutations.
The single-strand conformation polymorphism technique (9) was used to screen for point mutations and other small-scale sequence changes. Genomic DNA was amplified by using PCR. PCRs were performed in the wells of 96-well microtiter plates. In each well was 20 ng of leukocyte DNA in 20 μl of a buffer containing 20 mM Tris⋅HCl (pH 8.4 or 8.6), 0.25–1.5 mM MgCl2, 50 mM KCl, 0.02 mM dATP, 0.02 mM dTTP, 0.02 mM dGTP, 0.002 mM dCTP, 0.6 μCi [α-32P]dCTP (3000 Ci/mmol), 0.1 mg/ml BSA, 0 or 10% dimethyl sulfoxide, and 0.25 units of Taq polymerase. The pH, Mg+2 concentration, and presence or absence of 10% dimethyl sulfoxide were tailored to each primer pair to yield optimal amplification. After initial denaturation (94°C for 5 min), 35 cycles of PCR amplification were performed. Each cycle consisted of denaturation (94°C for 30 s), primer annealing (54°C for 30 s), and extension (71°C for 30 s). The final extension was at 71°C for 5 min. The amplified DNA fragments were heat-denatured, and aliquots of the single-stranded fragments were separated through two sets of 6% polyacrylamide gels, one set with and one without 10% glycerol. Gels were run at 8–12 W for 5–20 h before drying and autoradiography.
Primer pairs are as follows (sense/antisense): exon 1, sense, CCCCTGGCTGAGAACTTCCT, antisense, GGAGCCCTTGAAATAGCACA; exon 2, sense, GGGCTTCTTCCTTATTCTTC, antisense, ACTGACATAAAAGAGGATGG; exon 3, sense, GGCAGGGATAAGAAGCAATG, antisense, CTTTGAGGAGGAGGAGTGGC; exon 4, sense, CTCTTCCCTATGTTTCAATG, antisense, GTAACATTCAGTTTGGGTTC; exon 5, sense, GCTTGAAAATTACTGGACTG, antisense, CTAAATTCCTGAACATCACC; exon 6, sense, CTCTCAACTGGAGGACATTC, antisense, TCAGAAGAGGACAGATTGGT; exon 7, sense, GCTCTTCCAAAGCCTTTTAA, antisense, AAAGGTAGGCAAAGCAAATC; exon 8, sense, CAGCCCTTTCATTCACAAGC, antisense, TCTTCAGAATCACAAACTTG; exon 9, sense, GTGATTCTGAAGAAGATGTG, antisense, CAGGAACAATGGGAGGTGTC; exon 10, sense, TTGTCATTGCCTGTGCTCAT, antisense, CTGAAATCTACAGAGAAGCA; exon 11, sense, AATTCTTTCCTGCTCACTGA, antisense, GTTACCTCCCGTGTGAAGTT; exon 12, sense, GAGTTTTCCTAAGCATGTGC, antisense, AGCATATACTCAAAGCACTG; exon 13, sense, GCATATTGACTGATTGCTTG, antisense, GCAGTAAGAAGAGTATTCAG; exon 14a, sense, CAGAAAAAGAAGTCAGGTCA, antisense, GCTTTTGTCCTGCTCCTGGG; and exon 14b, sense, GTGTAGTTCTGAGTGTGGTG, antisense, GACCTGAAGCTGATTTTCTC. Variant bands were evaluated by sequencing of corresponding PCR-amplified DNA segments by using a cycle-sequencing protocol (Thermo Sequenase radiolabeled terminator cycle sequencing kit). Sequence variations expected to affect protein sequence or expression were evaluated further by recruiting the relatives of the index patients to participate in a study to determine whether the variant alleles cosegregated with the disease. For this purpose, leukocyte DNA samples from the relatives were analyzed by single-strand conformation polymorphism or direct genomic sequencing for the presence of the variant sequences.
RESULTS
To evaluate RPE65 as a possible cause for retinal degeneration, we first confirmed the reported intron/exon arrangement of the corresponding genomic locus (5) and sequenced intron DNA flanking each of the 14 exons. The resulting genomic sequences have been deposited in The Genome Database (accession nos. AF039855–AF039868). Based on the 5′ untranslated, 3′ untranslated, and flanking intron sequences, we developed assays by using the single-strand conformation polymorphism technique to screen for mutations in the entire coding sequence and the intron sequences immediately flanking each exon. With these assays, we evaluated 147 patients from separate families with autosomal recessive RP, 15 patients with isolate RP, and 45 unrelated patients with LCA.
The following pieces of information were considered to determine whether each discovered sequence anomaly (Table 1) was pathogenic: its predicted effect on the protein product, its relative frequency among patients vs. controls, and its segregation in affected families. Two sequence anomalies, silent changes within codon 352 and intron 12 (Table 1), were interpreted as nonpathogenic polymorphisms because they were at approximately equal frequency in patients and control individuals. Six anomalies were considered likely to be rare nonpathogenic variants because they had an allele frequency <1% and were predicted to have no effect on the encoded protein (i.e., the silent changes affecting codons 16, 133, 144, 326, 385, and 434). One additional rare variant (Ala434Val) was interpreted as nonpathogenic because segregation analysis showed that the only index case with this change carried it heterozygously while an unaffected sibling carried it homozygously. The pathogenicity of three missense anomalies was uncertain. Two of these, Lys294Thr and Val407Ala, were found in only one index patient each, and, in both instances, the index case had no sequence anomaly of the other allele found through single-strand conformation polymorphism and direct sequencing. The third, Asn205Ser, was not found in any affected patient but only in a control individual, who was heterozygous.
Table 1.
Polymorphisms, rare silent variants, and rare variants of uncertain pathogenicity
| Amino acid change | Sequence variation | Exon | No. of respective alleles (allele frequency)
|
|
|---|---|---|---|---|
| Patients | Controls | |||
| Phe16Phe | TTT vs. TTC | 2 | 414:0 | 189:1 |
| Leu133Leu | CTT vs. CTC | 5 | 413:1 | 190:0 |
| Tyr144Tyr | TAC vs. TAT | 5 | 413:1 | 190:0 |
| Asn205Ser | AAC vs. AGC | 6 | 414:0 | 189:1 |
| Lys294Thr | AAA vs. ACA | 9 | 413:1 | 192:0 |
| Val326Val | GTG vs. GTT | 9 | 413:1 | 192:0 |
| Glu352Glu | GAG vs. GAA | 10 | 352:62 (0.15) | 163:27 (0.14) |
| Thr385Thr | ACG vs. ACA | 11 | 413:1 | 192:0 |
| Val407Ala | GTT vs. GCT | 11 | 413:1 | 192:0 |
| Ala434Val | GCG vs. GTG | 12 | 413:1 | 192:0 |
| Ala434Ala | GCG vs. GCC | 12 | 413:1 | 192:0 |
| Intron 12 | IVS12+20 A vs. C | IVS12 | 401:13 (0.031) | 181:8 (0.042) |
Those sequence variants that were deemed pathogenic changed the predicted amino acid sequence of the encoded protein, were found in affected patients who were either homozygotes or compound heterozygotes for changes in this category, were not found among normal controls, and cosegregated with disease in the available relatives of the corresponding index patients. Sequence anomalies falling into this category (Fig. 1) were found in two patients with recessive RP, one patient with isolate RP recategorized as recessive, and seven patients with LCA. Mutations found in patients with RP were Arg91Trp (CGG to TGG), Ala132Thr (GCC to ACC), Leu341Ser (TTA to TCA), Glu404(4-bp ins) (GAG to GCTGGAG), and Val452Gly (GTC to GGC). The families of these patients participated in segregation analyses. Only the affected individuals in these families were found to be homozygotes or compound heterozygotes (Fig. 2A). Of special note is family 7828, in which three branches of the family have affected children due to homozygosity or compound heterozygosity for the mutations Leu341Ser and Glu404(4-bp ins). Also noteworthy is that the affected individual in family E070 originally was classified as being an isolate case of RP. In view of her being a compound heterozygote with two missense mutations, she was recategorized as having recessive RP. One of those two mutations, Arg91Trp, also was found in two patients with LCA (see below).
Figure 1.

DNA sequence of mutations found in patients with RP (A) and patients with LCA (B). In each panel, the left sequence with the heading “control” is the wild type and the right sequence is that of an index patient whose identification number is at the top and the mutation at the bottom. All sequences are in the sense direction going from bottom to top, except the sequence showing the frameship mutation E404(4-bp ins) in patient 003–117, which is in the antisense direction from bottom to top. Patients 003–117, 003–119, and 003–179 are the index patients with RP from families 7828, 0356, and E070, respectively. Patients 048–040, 048–017, 048–054, 048–011, 048–033, 048–041, and 048–008 are the index patients with LCA from families 9404, 9148, 7775, 3867, 9190, 0748, and 9145, respectively.
Figure 2.
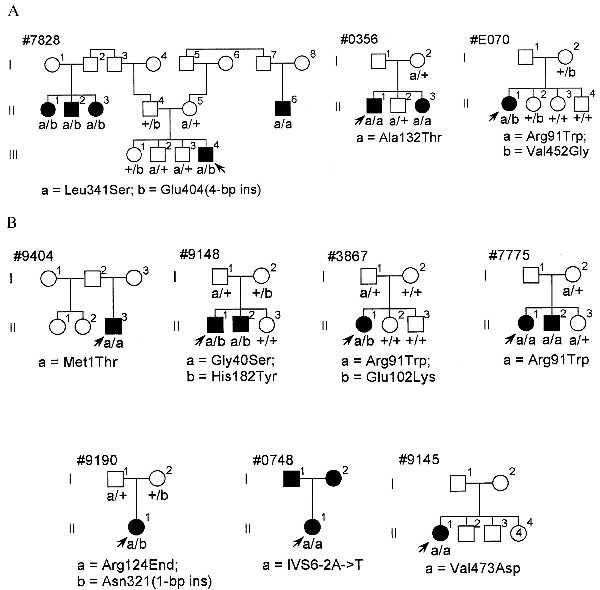
Pedigrees segregating mutations in the families of index patients having RP (A) or LCA (B). Filled symbols are affected, open symbols unaffected. Underneath each family member’s symbol is the RPE65 genotype with small letters indicating the alleles designated at the bottom of each pedigree and “+” indicating the wild-type sequence. Arrows point to index patients. In family 0748, the index patient has LCA and her two parents have RP by history.
Sequence abnormalities interpreted as pathogenic mutations were found in seven patients with LCA. The mutations were Met1Thr (ATG to ACG), Gly40Ser (GGC to AGC), Arg91Trp (CGG to TGG), Glu102Lys (GAA to AAA), Arg124End (CGA to TGA), His182Tyr (CAC to TAC), IVS6–2A->T (AG to TG), Asn321(1-bp ins) (AAT to AAAT), and Val473Asp (GTT to GAT). Family members from four of these cases participated in segregation analyses (families 9148, 3867, 7775, and 9190; Fig. 2B). In these families, affected individuals were homozygotes or compound heterozygotes and unaffected siblings and parents were either wild-type homozygotes or heterozygotes. Among these four families, family 3867 is distinctive in that one of the mutations, Glu102Lys, present in the affected patient is a new mutation that arose on the maternal allele. The families of three of these cases (families 9404, 0748, and 9145) were unavailable or unwilling to participate in segregation analysis. However, the index case in each family was homozygous for a sequence change that would be predicted to have a major impact on the encoded protein. One of the mutations affected the initiating methionine codon (Met1Thr, family 9404), another affected a base in the canonical intron donor site of intron 6 (IVS6–2A->T, family 0748), and one was a missense change that converted an uncharged residue to a charged one (Val473Asp, family 9145). Both parents of the index case in family 0748 had retinitis pigmentosa by history.
DISCUSSION
The data presented here provide compelling evidence that defects in RPE65 cause retinal degeneration, with pathogenic mutations discovered in 3 of 162 index patients (≈2%) with recessive or isolate RP and 7 of 45 index patients (16%) with LCA. Especially convincing are the data from family 7828 (Fig. 2A) indicating that individuals with RP resulting from three separate parental pairs carry the same mutations. This family is from the Dominican Republic, and it is likely that the Leu341Ser allele is fortuitously present in the two unrelated individuals (I-1 and I-8) in the first generation of the pedigree who do not report any consanguinity with their spouses who presumably carry either the same allele (individual I-7) or the mutation Glu404(4-bp ins) (individual I-2). Although family 7828 was originally categorized as showing a recessive inheritance pattern, it was only through analysis of RPE65 that the possibility of different genetic loci causing RP in the three branches was ruled out. Another piece of unusual and convincing evidence for the pathogenicity of RPE65 mutants comes from family 3867 (Fig. 2B), in which a new gene defect arises concomitantly with LCA in the only affected offspring of an unaffected couple.
The clinical criteria distinguishing RP from LCA deserve special attention in the light of these findings. RP is the diagnosis given to patients with photoreceptor degeneration who have good central vision within the first decade of life, and the diagnosis of LCA is given to patients who are born blind or lose vision within a few months after birth. Both diagnostic entities feature attenuated retinal vessels and a variable amount of retinal pigmentation in older patients and a reduced or nondetectable electroretinogram at all ages. Both exhibit nonallelic heterogeneity (10, 11). LCA is almost always recessively inherited, whereas families with RP can show any of the commonly recognized Mendelian inheritance patterns or maternal (mitochondrial) or digenic inheritance.
There is no universally accepted diagnostic term for those patients with retinal degeneration who lose useful (i.e., ambulatory) vision during the first few years of life, with ophthalmologists considering such cases as either LCA or severe RP. Clinicians have commented that “there is in principle no essential difference between Leber’s congenital amaurosis and early pigmentary retinopathy [i.e., retinitis pigmentosa]” (12). The lack of features discriminating between the two entities, together with the observed variation and apparent overlap in their severity, suggests that some cases of LCA represent one extreme of a spectrum of diseases, categorized for the most part as RP, featuring panretinal photoreceptor degeneration. The data presented in this paper and in two recent reports from other groups (6, 7) support this view. Some of the patients with photoreceptor degeneration due to mutations in the RPE65 gene had the clinical diagnosis of RP, others that of LCA. Family 0748 (Fig. 2B) is particularly illustrative in this regard because a child with LCA is the offspring of two parents with RP. Although the two parents did not participate in this study, it is reasonable to speculate that they both have RP due to compound heterozygosity including the mutation found in the child.
We found overlap even among the specific mutations found in patients with RP and LCA (Fig. 3), with the missense change Arg91Trp found heterozygously in one patient with RP (together with the Val452Gly mutation in family E070, Fig. 2A) and heterozygously in one patient with LCA (together with the Glu102Lys mutation in family 3867); the mutation also was found homozygously in another patient with LCA (family 7775). It is still possible that the types of mutations found in the patients may explain some of the variation in the severity of disease due to RPE65 gene defects. In particular, all three index patients with RP had at least one missense mutation that did not alter the charge of the affected amino acid residue. Missense mutations in this category would be the ones most likely to produce mutant versions of the encoded protein with some residual function (i.e., hypomorphic alleles). All but one of the seven index patients with LCA were homozygotes or compound heterozygotes with nonsense, frameshift, or splice-site mutations that changed many amino acid residues or with missense mutations that altered the charge of an amino acid residue. All of these types of mutations are more likely to produce alleles that would express no protein or one with little or no residual function (i.e., null alleles). The single case of LCA due to mutations reported by Marlhens et al. (6) fits within this scheme because the index patient was a compound heterozygote for two presumed null mutations in RPE65, a nonsense (Arg234End) and a frameshift [ASN356(1-bp del)]. However, some of the mutations in cases of RP reported by Gu et al. (7) are difficult to evaluate because they do not perfectly cosegregate with disease, they affect intron sequence apart from canonical splice donor or acceptor sites, or they are found in index cases who are heterozygotes and in whom no mutation of the other allele was discovered.
Figure 3.
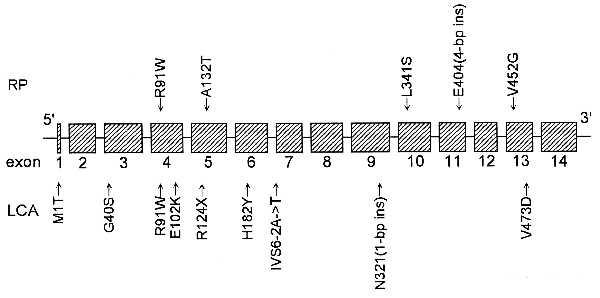
Schematic model of the exons of the human RPE65 locus with the locations of the mutations found in patients with RP (top) and LCA (bottom).
Future work is needed to determine whether and to what extent the severity of disease caused by RPE65 gene defects correlates with the residual function of the mutant alleles. In this respect, it will be of paramount importance to determine the role of the encoded protein in the retinal pigment epithelium, a role that is now made especially intriguing in view of the severe retinal degeneration caused by defects in it. Unfortunately, the locations of the mutations found by us in the RPE65 gene so far (Fig. 3) do not seem to indicate a functional domain that is a common target.
Acknowledgments
This work was supported by grants from the National Institutes of Health (EY00169 and EY08683) and from the Foundation Fighting Blindness and by gifts to the Taylor Smith Laboratory and the Ocular Molecular Genetics Institute. T.P.D. is a Research to Prevent Blindness Senior Scientific Investigator.
ABBREVIATIONS
- RP
retinitis pigmentosa
- LCA
Leber congenital amaurosis
Footnotes
References
- 1.Dryja T P. Proc Natl Acad Sci USA. 1997;94:12117–12121. doi: 10.1073/pnas.94.22.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamel C P, Jenkins N A, Gilbert D J, Copeland N G, Redmond T M. Genomics. 1994;20:509–512. doi: 10.1006/geno.1994.1212. [DOI] [PubMed] [Google Scholar]
- 3.Hamel C P, Tsilou E, Harris E, Pfeffer B A, Hooks J J, Detrick B, Redmond T M. J Neurosci Res. 1993;34:414–425. doi: 10.1002/jnr.490340406. [DOI] [PubMed] [Google Scholar]
- 4.Hamel C P, Tsilou E, Pfeffer B A, Hooks J J, Detrick B, Redmond T M. J Biol Chem. 1993;268:15751–15757. [PubMed] [Google Scholar]
- 5.Nicoletti A, Wong D J, Kawase K, Gibson L H, Yang-Feng T L, Richards J E, Thompson D A. Hum Mol Genet. 1995;4:641–649. doi: 10.1093/hmg/4.4.641. [DOI] [PubMed] [Google Scholar]
- 6.Marlhens F, Bareil C, Griffoin J M, Zrenner E, Amalric P, Eliaou C, Liu S Y, Harris E, Redmond T M, Arnaud B, et al. Nat Genet. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 7.Gu S M, Thompson D A, Srikumari C R S, Lorenz B, Finckh U, Nicoletti A, Murthy K R, Rathmann M, Kumaramanickavel G, Denton M J, et al. Nat Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 8.Berson E L, Rosen J B, Simonoff E A. Am J Ophthalmol. 1979;87:460–468. doi: 10.1016/0002-9394(79)90231-9. [DOI] [PubMed] [Google Scholar]
- 9.Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Proc Natl Acad Sci USA. 1989;86:2766–2770. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrault I, Rozet J M, Calvas P, Gerber S, Camuzat A, Dollfus H, Châtelin S, Souied E, Ghazi I, Leowski C, et al. Nat Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 11.Dryja T P, Li T. Hum Mol Genet. 1995;4:1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- 12.Franceschetti A, Francois J, Babel J. Chorioretinal Heredodegenerations. Springfield, IL: Charles C. Thomas; 1963. [Google Scholar]