

Abstract
T cell activation through the T cell receptor is necessary to achieve a specific and effective immune response. We report here that stimulation of CD8+ T cells through the T cell receptor complex leads to de novo expression of the CD4 antigen on the cell surface that results in susceptibility of CD8+ T cells to HIV infection. In addition, activation of peripheral blood mononuclear cells from HIV-infected individuals results in the appearance of double-positive CD4+/CD8+ T cells, which become infected by endogenous HIV. HIV DNA sequences could be detected in uncultured and sorted mature CD3+CD8+ T cells from HIV+ individuals. These results suggest a new mechanism by which HIV could attack the immune system and may help to explain the CD8+ T cell defects in AIDS patients.
Keywords: CD8+CD4+ T cells, CD8 infection, cytotoxic T lymphocytes
Despite mounting a strong humoral and cell-mediated immune response against HIV, the host’s immune system is able to only partially control the infection. HIV RNA can be detected in plasma of patients at all stages of disease, with the highest levels observed during initial infection and late stages of disease (1). The generation of viral variants (escape mutants) (2), proviral integration, and infection of key immunocompetent CD4+ T cells and phagocytes may explain how HIV escapes immune surveillance. Furthermore, the effects of HIV proteins (such as gp120 and Tat), as well as elevation of α-interferon further impairs the immune response by suppressing the proliferative response of uninfected T cells (3–10). However, the inability of the immune system to overcome HIV infection is still incompletely understood. For instance, CD8+ cytotoxic T lymphocytes (CTL) are thought to play an important role in the control of viral infection through both HIV-specific cytotoxic activity (11–14) and release of soluble factors capable of suppressing HIV replication (15, 16). In fact, the onset of AIDS is associated with a decline in both the number of CD8+ T cells and specific anti-HIV cytotoxic activity (17–20). Although mature CD8+ T cells are not considered prime targets for HIV infection, several reports have demonstrated that, under certain circumstances, HIV can infect these cells (21–27). However, no satisfactory explanation could be generated as to how HIV may gain entry into CD8+ T cells. In the present work we provide evidence that infection of mature peripheral blood CD8+ T cells by HIV is mediated through the CD4 molecule, whose gene expression is induced following activation through the T cell receptor (TCR) complex. These results will strengthen the notion that CD8+ T cells can become infected by HIV both in vitro and in vivo and provide additional explanations to HIV-associated immunological deterioration.
MATERIALS AND METHODS
Cells, Culture Conditions, and Flow Cytometry.
Peripheral blood mononuclear cells (PBMC) were obtained from healthy donors after centrifugation over Ficoll/Paque gradient (Pharmacia). PBMC were depleted of CD4+ T cells by using anti-CD4 coated magnetic beads (Immunotech, Westbrook, ME). Efficiency of CD4 depletion was monitored by flow cytometry. CD4 depleted cells (107) were stimulated either with 1 μg/ml phytohemagglutinin (PHA)-P (Sigma), 1 μg/ml staphylococcal enterotoxin B (SEB) (Sigma), 200 ng/ml anti-CD3 (Immunotech), or 1 μg/ml toxic shock syndrome toxin 1 (Sigma) in a total volume of 10 ml of RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics (complete medium). Twenty-four hours poststimulation, 20 units/ml of interleukin-2 (Boehringer Mannheim) were added to the cultures and 6 days later cytofluorometric analyses were performed. Cells were simultaneously stained with phycoerythrin (PE)-conjugated anti-CD4 and fluorescein isothiocyanate (FITC)-conjugated anti-CD8 mAb (Becton Dickinson). Control aliquots were stained with FITC- or PE-conjugated mouse IgG1. In all experiments, 10,000 events were accumulated and analyzed. For phenotypic analysis of double positive (DP) cells, CD4-depleted PBMC (0% CD8−CD4+) were stimulated with 1 μg/ml SEB for 7 days and stained with PE-conjugated anti-CD4 and one of the following FITC-labeled antibodies: anti-CD2, -CD3, -CD25, -CD26, -CD27, -CD28, -CD45RO, -CD45 RA, -CD69, -CD71, -CD122, -HLA-DR, -TCR αβ chains (Becton Dickinson), -Vβ 3, -Vβ 6.1, Vβ 13.6, Vβ 14, Vβ 17, and Vβ 21.3 (Immunotech).
CD4 induction was also studied by using the interleukin 2-dependent human T lymphotropic virus, type I-immortalized cell lines 67.I (21), CD8 U-1, and clone D7 derived from the latter. CD4 was induced by stimulating with 1 μg/ml SEB for 4 days and expression studied by flow cytometry following staining with anti-CD4 and anti-CD8 as described above.
Immunoprecipitation and Western Blot Analysis.
PBMC depleted from CD4 were activated with SEB for 6 days. CD8+ T cells (25 × 106), of which 24% were expressing CD4, were lysed and CD4 immunoprecipitated by using rabbit anti-CD4 antibodies and protein A Sepharose. The immunoprecipitate was resolved by SDS/PAGE electrophoresis and proteins transferred to nitrocellulose. Membranes were probed for both CD4 and p56lck. After washing, the membranes were probed with peroxidase-labeled secondary antibodies and signal detected by chemiluminescence (Amersham). SEB (107) activated CD4+ T cells were used as positive controls.
Kinetics of CD4 Protein and mRNA Expression in Activated Mature CD8+ T Cells.
CD4-depleted PBMC from two healthy donors were activated with PHA or SEB. Cells were stained for CD4 and CD8 at various days (from day 0 to day 27) and percentage of CD8+ T cells expressing CD4 calculated.
For CD4 mRNA studies, PBMC from three healthy donors were depleted of CD4+ T cells by using magnetic beads coated with anti-CD4 antibodies (Immunotech). The CD4 depleted cells were stained for both CD8 and CD4 and further purified by cell sorting. Sorted cells were lysed in Trizol and RNA extracted. CD4 depleted cells were also stimulated with 1 μg/ml SEB for 6 days before phenotyping for CD8 and CD4 coexpression and RNA extraction. CD4+ T cells were used as positive controls. The RNA was extracted and analyzed for CD4 and glyceraldehyde-3-phosphate dehydrogenase (G3PDH) expression by reverse transcriptase (RT)–PCR. The following primer pairs were used: CD4 5′-GGAGTCCCTTTTAGGCACTTGC-3′ and 5′-AAGACAGTGCATGTC-CAGGTG-3′; G3PDH 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCT-GTTGCTGTA-3′. After 30 cycles of amplification, 15% of the products were resolved by electrophoresis, transferred to nylon membranes and hybridized with specific 32P-labeled oligonucleotide probes. The CD4 probe sequence is 5′-GCCACTCAGGGAAAGAAAGT-3′ and the G3PDH probe sequence is 5′-CACGGAAGGCCATGCCAGTGAGCTTC-CCGT-3′. After hybridization, the membranes were washed and exposed to x-ray films.
Infection of Activated CD8+ T Cells by HIV-1.
CD4-depleted PBMC were stimulated with PHA for 7 days. Phenotypic analysis indicates that 96.5% of the cells were CD8+ T cells, of which 24% coexpressed the CD4 molecule. CD8−CD4+ (1.6%) and CD4−CD8− (1.9%) represented the remaining cell populations. Cells (107) were centrifuged, washed with PBS and resuspended in 0.1 ml PBS-1% fetal bovine serum with or without 5 μg/ml OKT4A or 5 μg/ml anti-CD28 and incubated for 1 hr at 4°C. After an additional wash, cell pellets were treated with HIV-IIIB (1 ng p24 equivalent) for 1 hr at 4°C then washed trice with PBS and resuspended in complete medium supplemented with 10 units/ml interleukin 2. Corresponding cultures were also conditioned with 2 μg/ml OKT4A or 2 μg/ml anti-CD28. Cell-free supernatants were collected at various days and assayed for p24 antigen.
Intracellular expression of HIV-1 antigen in CD8+ T cells was detected by partial cytoplasmic membrane solubilization (0.1% Triton X-100 in PBS) of paraformaldehyde-fixed infected cells by using FITC-labeled anti-HIV p24 (Coulter) and PE-labeled anti-CD8 (Becton Dickinson). Controls consisted of infected cells treated as above and incubated with matching isotypes FITC- and PE-labeled mAb.
For electron microscopy studies, PHA-activated CD4-depleted PBMC were treated with HIV-1 for 6 days. Cells (99% CD8+) were surface labeled with anti-CD8, washed twice with PBS followed by incubation with gold-labeled (10-nm particle diameter) goat anti-mouse antibodies. Cells were fixed in 2.5% glutaraldehyde and processed for electron microscopy.
HIV DNA PCR.
PBMC from HIV-infected individuals with CD4 counts ranging from 50 to 300 were isolated and parts thereof were immediately frozen to −70°C. The remainder were surface stained with PE-labeled anti-CD3 and FITC-labeled CD8 antibodies. Mature T cells (i.e., CD3+CD8+) were sorted out from the remaining cells and stored frozen until used. Precautions to avoid contamination included separate chambers for DNA isolation and reactions setup. Crude DNA (20 μl) was added to 80 μl of PCR mixture containing 100 pmol of each oligo (SK38 and SK39), 200 μM dNTP, and 2.5 units of TAQ gold enzyme (Perkin–Elmer). A negative control consisting of water along with the PCR mix were included in each run. An initial denaturation step of 10 min at 95°C was performed, followed by 45 cycles of 1 min 94°C denaturation, 1 min at 55°C for annealing, and 1 min at 72°C for elongation. Thirty microliters of the PCR mixture was then mixed with a 32P-labeled (105 cpm) internal probe (SK19). The mixtures were heated to 95°C for 5 min and incubated at 55°C for 15 min. Ten microliters was then electrophoresed through a 10% nondenaturing Tris⋅borate EDTA gel and exposed to x-ray films. All samples were also amplified by using a primer pair for the G3PDH gene to show the presence of DNA and the absence of inhibitors.
RESULTS
Activation Through the TCR Induces Cell Surface CD4 Expression in Mature CD8+ T Cells.
While studying T cells, we observed that cellular activation induces cell surface expression of CD4 molecules at the cell surface of CD8+ T cells. PBMC depleted of CD4+ T cells were stimulated with mitogens, including the plant lectin PHA, the bacterial superantigen SEB, and the anti-CD3 mAb OKT3. As shown in Fig. 1A, resting CD8 T cells do not express CD4 antigen (Upper Left). However, 7 days after activation with such mitogens, expression of CD4 could be detected, by using two-color flowcytometric analysis, at the cell surface of CD8+ T cells. The percentage of CD8+ T cells expressing the CD4 molecule varied from 26–59%. Similar results were obtained with another bacterial superantigen, toxic shock syndrome toxin 1 (data not shown). Although variations were observed in the intensity of CD4 expression between different PBMC donors, CD4 expression on the surface of CD8+ T cells upon cell activation was consistently observed. CD4 induction by mitogens was also observed in unfractionated PBMC (not shown). We next determined whether the CD4 receptor expressed in CD8+ T cells was functionally associated with its signaling enzyme, the p56lck tyrosine kinase. The CD4 antigen was immunoprecipitated from SEB-activated CD4-depleted PBMC (<0.5% CD3+CD4+) and probed by Western blot for CD4 and p56lck. As shown in Fig. 1B, CD4 could be immunoprecipitated from CD8+CD4+ DP cells and these CD4 molecules were physically associated with p56lck, suggesting that CD8+ T cells express a functional CD4 capable of signaling through this kinase. SEB-activated CD4+ T cells were used as positive control.
Figure 1.
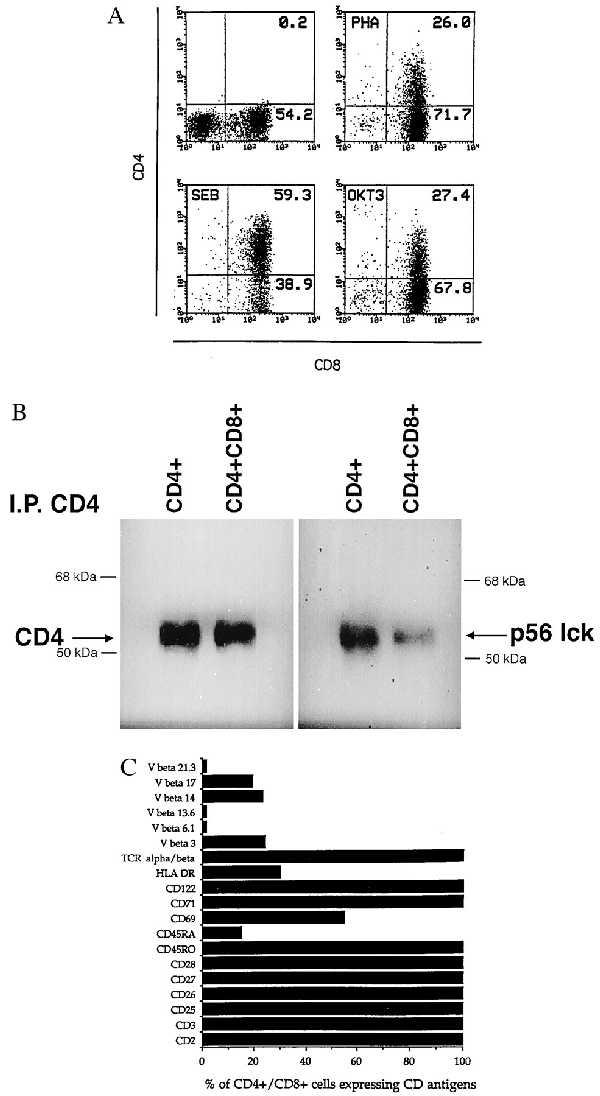
(A) CD4 antigen induction in activated mature CD8+ T cells. Resting, or 6-day-old PHA-P (1 μg/ml), SEB (1 μg/ml), or anti-CD3 (200 ng/ml) activated cells were stained for both CD4 and CD8 antigen expression. Control aliquots were stained with FITC- or PE-conjugated irrelevant mouse IgG1. (B) Association of p56lck to CD4 in activated mature CD8+ T cells. PBMC depleted CD4+ T cells were activated with SEB for 6 days. CD8+ T cells (25 × 106), of which 24% were expressing CD4, were lysed and CD4 immunoprecipitated. Following SDS/PAGE electrophoresis, proteins were transferred to nitrocellulose and probed for both CD4 and p56lck. SEB (107) activated CD4+ T cells were used as positive controls. (C) Phenotypic analysis of CD8+ T cells coexpressing CD4. Seven-day-old SEB activated CD4-depleted PBMC (0% CD8−CD4+) were simultaneously stained with PE-conjugated anti-CD4 and various FITC-conjugated mAb. The percentage of cells expressing a specific marker was determined after acquisition and analysis of 10,000 events.
Because only a portion of SEB-activated CD8+ T cells acquire the CD4 antigen after activation we proceeded to phenotype these DP cells (Fig. 1C). These DP cells express common activation markers (CD25, CD26, CD27, CD28, CD69, CD71, CD122, and HLA-DR) as well as T cell markers (CD2, CD3, and TCR αβ chains). Interestingly, the majority of the DP CD8+CD4+ cells were also coexpressing TCR Vβ chains that are reactive with SEB (Vβ 3, Vβ 14, and Vβ 17) while low percentages of CD4+/CD8+ coexpression were observed with SEB unreactive Vβ s (6.1, 13.6, and 21.3), suggesting that direct activation by SEB is required for CD4 induction. Consistent with this observation, conditioned media from PHA- or SEB-stimulated CD8+ T cells were not found to induce CD4 on resting CD8+ T cells (data not shown), suggesting that a soluble factor released by activated cells is not responsible for CD4 induction. In addition to primary cells, similar results were obtained with SEB-activated human T lymphotropic virus type I-immortalized CD8+ T cell lines expressing SEB-reactive Vβ TCR chains (data not shown). These cell lines express major histocompatibility complex class II antigens thereby bypassing the need for professional antigen presenting cells to obtain proper activation stimuli. Although these cell lines express minimal levels of CD4 under basal conditions, upon activation with SEB, the vast majority of CD8+ T cells were found to express the CD4 antigen. A selective expansion of the few DP initially present is unlikely as these cultures were kept for short periods of time (3–4 days) after stimulation.
Kinetics of CD4 Expression in CD8+ T Cells.
Next, we determined the kinetics of CD4 induction on CD8+ T cells following mitogen activation. As shown in Fig. 2A, no DP cells were observed until day 3 poststimulation. The maximum number of DP cells, varying between 10 and 30% in this experiment, was observed on day 6. DP cells (range 7–17%) were observed for up to 27 days poststimulation, at which time cultures were terminated. We also studied CD4 mRNA in both resting CD8+CD4− and SEB activated CD8+ T cells obtained from three normal individuals. To ensure that no residual CD4+ T cells remained, we extensively purified single positive CD8+ T cells by both magnetic bead selection and cell sorting. The resulting cells (0% CD4+) were lysed and RNA extracted. In parallel, CD4-depleted PBMC were activated with SEB for 6 days, phenotyped by flow cytometry for CD4 and CD8 coexpression followed by RNA extraction. CD4+ T cells were used as positive control. Flow cytometry profiles of cells before and after SEB activation from one donor, representative of three different donors, are shown in Fig. 2B. We then analyzed, by RT-PCR, the expression of the CD4 mRNA along that of the housekeeping G3PDH gene in these three cell populations. As shown in Fig. 2B, CD4 mRNA can be detected in mature CD4+ T cells while no expression of CD4 mRNA could be detected in freshly isolated CD8+CD4− cells. In contrast, SEB-activated CD8+ T cells contained an easily detectable level of CD4 mRNA. The G3PDH mRNA amplification was used to assess the relative amount of RNA used for amplification. A minus RT reaction tube was included for each sample to assess for possible contamination by genomic DNA. In such samples, no amplicons were detected. The inability to detect CD4 mRNA in resting CD8+CD4− T cells, by using the sensitive PCR technique, confirms previous work of others (22, 28), and supports the notion of a transcriptionally controlled CD4 gene.
Figure 2.
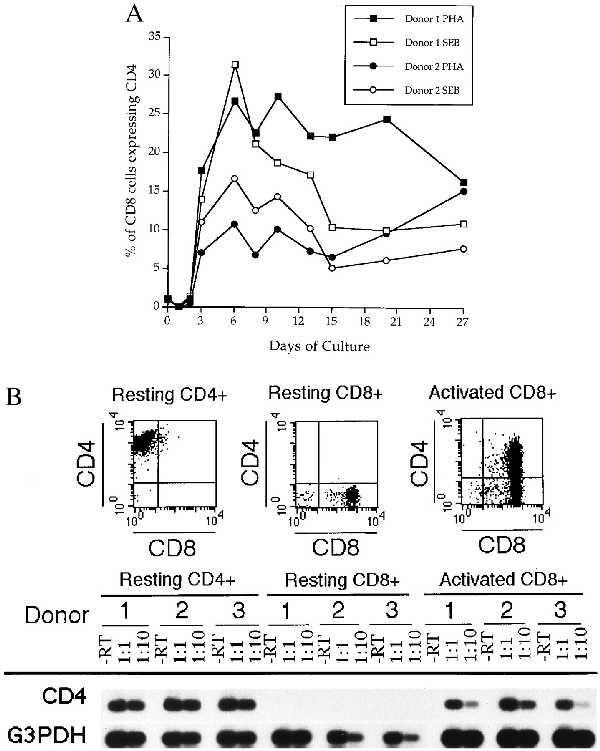
(A) Kinetics of de novo surface CD4 expression of activated CD8+ T cells. CD4-depleted PBMC were obtained from two healthy donors and activated with PHA or SEB. Cells were stained for CD4 and CD8 at various days (from day 0 to day 27) as described in Materials and Methods. Percentages of CD8+ T cells expressing the CD4 antigen were determined after analysis of 10,000 events. (B) CD4 mRNA levels before and after CD8+ T cell activation. Resting CD4+, resting CD8+ and SEB activated CD8+ T cells from three healthy donors were analyzed for CD4 protein and CD4 mRNA expression. (Top) The CD4/CD8 flow cytometry profiles of one of the donors, which is representative of the two other donors. RNA from each category (resting CD4+, resting CD8+, and activated CD8+) was extracted and subjected to RT-PCR for both CD4 mRNA and G3PDH mRNA expression. For each sample, a minus RT control was included to monitor for possible genomic contamination.
CD4-Mediated Infection of Activated Mature CD8+ T Cells.
We next determined the susceptibility of activated CD8+ T cells to HIV infection. Seven-day-old PHA-activated CD8+ cultures (99% CD8+) were treated with HIV-IIIB in the presence or the absence of an anti-CD4 or anti-CD28 mAb. As shown in Fig. 3A, HIV was capable of productively infecting activated CD8+ T cells, as determined by p24 antigen production, and this infection could be abrogated by treatment with anti-CD4 mAb but not with anti-CD28 mAb, suggesting that infection proceeded through a CD4 receptor-mediated pathway. Furthermore, just as the case with CD4+ T cells, HIV infection of DP cells was associated with reduction of CD4 expression. Twelve days postinfection both uninfected and HIV-infected DP cells were stained for CD8 and CD4 antigens. As previously reported for CD4+ T cells (29, 30), HIV-IIIB was able to decrease CD4 expression on CD8+ T cells (Fig. 3B). Whether this finding reflects selective depletion of CD4-expressing cells or receptor down-regulation remains to be established. In addition, infection of CD8+ T cells by HIV was conclusively established by p24 antigen staining and CD8-ImmunoGold-labeled electron microscopic analyses. Approximately 20% of CD8+ T cells were positive for intracellular HIV p24 (Fig. 3C), indicative of a productive type of infection. These results were also confirmed by detection of HIV budding particles from ImmunoGold-labeled CD8+ T cells. Budding and more mature HIV particles could be found associated in 10% of CD8+ gold-labeled cells in agreement with the number of DP cells from this particular experiment (not shown).
Figure 3.
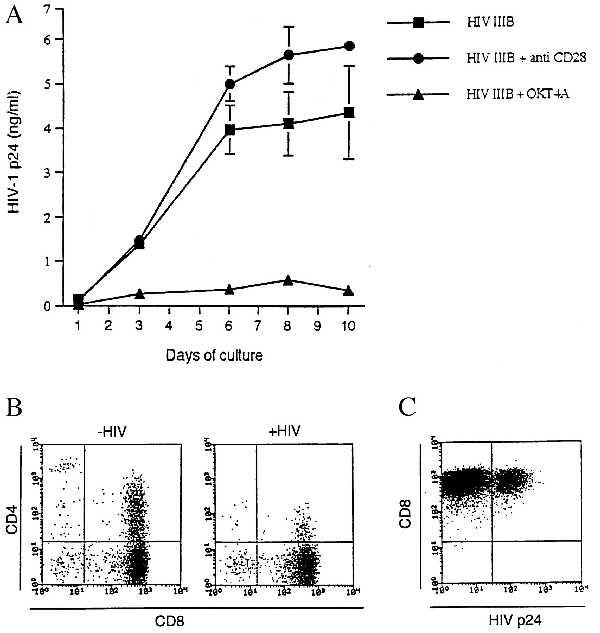
(A) Infection of activated mature CD8+ T cells by HIV-1. Seven-day-old PHA-activated CD4-depleted PBMC were infected with HIV-1 IIIB in the absence or the presence of OKT4A or anti-CD28. At various times after infection, cell-free supernatants were collected and stored until HIV-1 p24 antigen determination. Results (mean ± SD) were calculated from triplicate cultures and are representative of two separate experiments. (B) Disappearance of CD4+CD8+ T cells following HIV-1 infection. Twelve days post-HIV-1 infection of activated CD8+ T cells, cells were stained for both CD8 and CD4. Cells from uninfected cultures were simultaneously analyzed for CD4 and CD8 markers. (C) Intracellular detection of HIV p24 in CD8+ T cells. Six-day-old PHA-activated CD4-depleted PBMC were treated with HIV-1 for 5 days and analyzed for cell surface CD8 and intracellular HIV-1 p24 antigen expression.
Next, we determined whether in vitro activation of PBMC from HIV-infected patients with PHA or SEB would induce DP cells and subsequently whether these DP cells could be infected by endogenous HIV. PBMC from HIV-infected patients with circulating CD4 counts ranging from 5–1,200 per μl were isolated and stimulated with mitogens. Between days 12–19 of culture, when HIV-1 p24 could be detected in the culture supernatant, cells were doubly stained by using PE-conjugated anti-CD4 and FITC-conjugated anti-CD8 mAb. As shown in Fig. 4A, 79% of the CD8+ T cells from one HIV-infected individual coexpressed CD4. The other six donors had a percentage of DP cells ranging from 18–90%. Next, we determined whether the DP cells could be infected by endogenous HIV. Duplicate cultures were also stained with PE-conjugated anti-CD8 and FITC-conjugated anti-HIV gp120 mAb. In most patients studied (5/7) we could demonstrate the presence of HIV-infected CD8+ T cells. The two patients from which we could not show evidence of infection of CD8+ T cells may be explained by the fact that we could not rescue HIV from these two patients as determined by p24 antigen production. An example of HIV-infected CD8+ T cells from one patient is shown in Fig. 4B. A low proportion of the CD8+ T cells (1–3%) were found to express HIV gp120 at their surface. Similar results were obtained when cells were stained for intracellular HIV p24 (data not shown) confirming that detection of gp120 on the cell surface was not due to virus bound to the cell surface. These DP cells could also be infected with exogenously added HIV-IIIB (Fig. 4C). Five days following infection with HIV-IIIB (multiplicity of infection, 0.1) 7–12% of CD8+ T cells were expressing HIV gp120.
Figure 4.
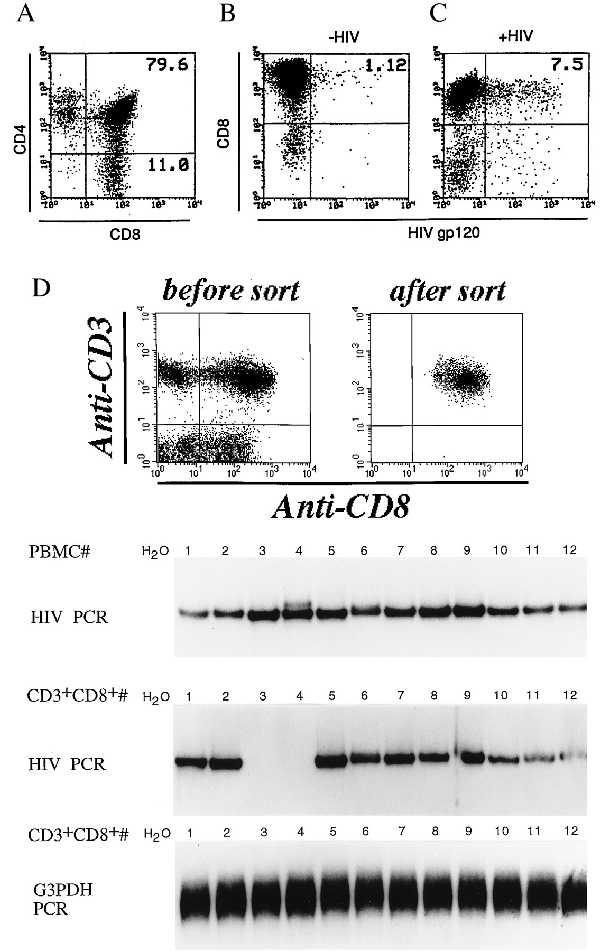
(A) Analysis of DP cells in activated PBMC cultures from HIV-infected individuals. PBMC from HIV-infected patients were isolated and stimulated with PHA. On days 12–19, cells were stained and analyzed for CD4 and CD8 coexpression. (B) Infection of CD8+ T cells by endogenous HIV. On days 12–19 poststimulation cells were stained for both CD8 and HIV gp120 expression and analyzed by flow cytometry. Results from one donor, representative of five, is presented. (C) Infection of CD8+ T cells from HIV-infected individuals with HIV-1 IIIB. PBMC from HIV-infected individuals were stimulated for 12–19 days with PHA and then treated with HIV-1 IIIB (multiplicity of infection, 0.1). After five days, cells were stained for surface expression of both CD8 and HIV-1 gp120 and analyzed by flow cytometry. (D) HIV DNA detection in PBMC and sorted CD3+CD8+ T cells from HIV-infected individuals. DNA from PBMC or CD3+CD8+ sorted T cell population from 12 HIV-infected individuals was isolated and subjected to PCR for the presence of HIV DNA sequences. G3PDH gene was amplified as positive control and water as negative control. (Top) The dot plot profile of one donor, representative of 12, before and after sorting of CD3+CD8+ T cells.
Because CD4 expression can be induced by T cell activation, one should expect to find a portion of the CD8+ T cells to be infected by HIV in vivo. To test this hypothesis, we isolated PBMC from 12 HIV+ individuals, selectively isolated their CD3+CD8+ mature T cells by sorting and analyzed these samples for HIV DNA sequences (Fig. 4D). For detection, we used the PCR technique with conditions that allowed detection of as low as one to five integrated copies of HIV (data not shown). An example from one sample of PBMC before and after sorting for CD3+CD8+ is shown in Fig. 4D. Typically, <1% contaminating CD3+CD8− were present in the sorted samples. After PCR and hybridization, a positive signal was detected in all of the unfractionated PBMC samples. In the sorted CD3+CD8+ T cells, HIV DNA was detected in 10 out of 12 samples. PCR was also performed by using a primer pair specific for the G3PDH gene to show the presence of amplifiable DNA.
DISCUSSION
The results presented here show that a significant proportion of activated CD8+ T cells are induced to express the CD4 molecule at their surface. This is the first conclusive demonstration that CD4 can be expressed by CD8+ T cells under physiological conditions, i.e., through direct triggering of the TCR. Previous observations had shown CD4 induction by treating cells with 5-azacytidine (31), by infecting cells with human herpesvirus 6 (23) or by stimulation with the plant lectin Con A (32). DP cells were tested for the presence of human herpesvirus 6, which may have accounted for CD4 induction, and no evidence of infection could be demonstrated (data not shown).
Although CD4 induction on CD8+ T cells has yet to be studied in classical T cell activation, i.e., by TCR interaction with peptides presented by major histocompatibility complex class 1 molecules, similar results are expected as SEB, toxic shock syndrome toxin 1 and anti-CD3 mAb can mimic activation of T cells through TCR engagement. It is noteworthy that a low percentage (1–3%) of DP cells can be observed in the peripheral blood of healthy individuals (33). Although these cells are commonly referred to as immature DP cells, our results suggest that, more than likely, these cells may represent activated peripheral blood CD8+ T cells.
To better understand CD4 gene expression in mature CD8+ T cells, we have generated a series of plasmid constructs in which the expression of the luciferase reporter gene is driven by genetic elements controlling CD4 gene expression. Such elements include the CD4 promoter (34), the CD4 enhancer (35), and the CD4 silencer (36–38). All of these CD4 constructs, when transfected in CD4+ T cells such as Jurkat, showed considerable activity (not shown). These constructs were therefore tested for inducibility by SEB following transfection in the 67.I CD8+ T cell line. Although basal activity was detected, no significant increase in reporter gene activity could be detected following SEB activation (data not shown). As a positive control, the cells were transfected with the pGL3 control plasmid in which luciferase gene expression is driven by the simian virus 40 promoter. Thus, the mechanisms leading to CD4 gene expression in mature CD8+ T cells remains unclear. It is possible that our reporter constructs, although active in CD4+ T cells, may not adequately mimic the genetic organization of the CD4 gene. The CD4 gene is complex with an enhancer element located 6.5 kb upstream of the first exon (35) and a silencer element located within the first intron (36–38). Characterization of the human CD4 silencer indicates that this element is active in vivo and much less effective in suppressing heterologous promoter in vitro, suggesting that the simple presence of this regulatory sequence may not be sufficient to observe the silencing effect (38). In all likelihood, CD8+ T cell activation turns on the CD4 gene through silencing of the silencer. Further studies are needed to characterize the factors interacting with this regulatory element.
Although it is not yet known whether this phenomenon has any role in the physiology of cytotoxic T cells, the relevance of this observation to HIV pathogenesis is of obvious importance. In HIV-infected individuals, reactive cytotoxic T cells recruited to kill HIV-infected cells would, as a consequence of specific antigenic activation, express the CD4 antigen and become targets for HIV infection. Moreover, CD8+ T cells reactive against any pathogens, once activated, may become susceptible to HIV infection. A recent report indicates that CD8+ T cells from macaques can be infected in vivo by simian immunodeficiency virus (39). A proportion of these CD8+ T cells were also found to coexpress the CD4 antigen that, in most likelihood, is the mean by which simian immunodeficiency virus gained entry into these cells. In the past, it has been difficult to show the presence of HIV in CD8+ human T cells. The reasons for this may be accounted for by the less sensitive detection methods used or by the fact that positive results were thought to arise from contaminating CD4+ T cells in the samples. However, more recent studies indicate that HIV can be found in uncultured human CD8+ T cells (40, 41). Our results are supportive of such findings.
To gain entry into cells, HIV-1 must bind and interact with at least two distinct molecules: CD4 and a seven transmembrane G-protein coupled chemokine receptor. Most nonsyncytium-inducing primary HIV-1 isolates and monocytotropic strains of HIV-1 interact with the CCR5 chemokine receptor (42–44) while syncytium-inducing or T cell tropic HIV-1 strains bind to CXCR4 (45). CCR5 is expressed on both CD4+ T lymphocytes and monocytes/macrophages (46) while CXCR4 is predominantly expressed on T cells (47). Resting and activated primary CD8+ T lymphocytes do also express CXCR4 and CCR5 (46, 47), suggesting that both syncytium-inducing and nonsyncytium-inducing strains of HIV-1 can infect these cells, once the CD4 gene has been turned on. Our data on infection of activated CD8+ T cells by HIV-1 IIIB (Fig. 3) and Ba-L (not shown) correlates with CXCR4 and CCR5 coreceptor expression.
HIV infection is not dormant at any time and many infected cells are continuously producing virus leading to a perpetual immune solicitation of HIV reactive cells spanning several years (1, 48). During this time, HIV-specific CD8+ T cells could be eliminated, both directly by HIV infection and indirectly by immune exhaustion, leaving a gap in the repertoire of HIV-reactive cells leading to uncontrolled viral expression as seen in the late stage of the disease. Infection of CD8+ T cells by HIV is likely to happen during all stages of disease with perhaps the greatest rate occurring during the initial phase of infection when an intact repertoire of reactive cells is present. However, because of the high number of CD4+ T cells present, and the low percentage of susceptible CD8+ T cells, the infection of CD8+ T cells may go undetected with current technology. Interestingly, the disappearance of HIV CTL during the early phase of infection was recently reported (49). These results suggest that CTL depletion does occur after HIV infection and massive antigenic stimulation is undoubtedly responsible for specific CTL clone exhaustion. However, the potential of HIV to infect and kill activated CD8+ T cells can only accentuate the turnover rate of reactive clones and further contribute to CTL exhaustion.
In summary, several hypotheses such as anergy, apoptosis and extensive activation/differentiation have been made to explain the depletion of HIV-specific CTL observed in AIDS. The results presented here suggest that HIV may impair the effector arm of the immune system through an additional mechanism, by direct infection and thus selective depletion of HIV reactive CTL.
Acknowledgments
Throughout these studies, L.F. was supported by fellowships from the Medical Research Council of Canada and the National Health Research and Development Program of Canada.
ABBREVIATIONS
- CTL
cytotoxic T lymphocytes
- DP
double positive
- FITC
fluorescein isothiocyanate
- G3PDH
glyceraldehyde-3-phosphate dehydrogenase
- PE
phycoerythrin
- PBMC
peripheral blood mononuclear cells
- PHA
phytohemagglutinin
- RT
reverse transcriptase
- SEB
staphylococcal enterotoxin B
- TCR
T cell receptor
References
- 1.Piatak M, Jr, Saag M S, Yang L C, Clark S J, Kappes J C, Luk K C, Hahn B H, Shaw G M, Lifson J D. Science. 1993;259:1749–1754. doi: 10.1126/science.8096089. [DOI] [PubMed] [Google Scholar]
- 2.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel R M. Nature (London) 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 3.Chanh T C, Kennedy R C, Kanda P. Cell Immunol. 1988;111:77–786. doi: 10.1016/0008-8749(88)90052-4. [DOI] [PubMed] [Google Scholar]
- 4.Diamond D C, Sleckman B P, Gregory T, Lasky L A, Greenstein J L, Burakoff S J. J Immunol. 1988;141:3715–3717. [PubMed] [Google Scholar]
- 5.Lachgar A, Bizzini B. Biomed Pharmacother. 1994;48:73–77. doi: 10.1016/0753-3322(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 6.Lachgar A, Bernard J, Bizzini B, Astgen A, LeCoq H, Fouchard M, Chams V, Feldman M, Burny A, Zagury J F. Biomed Pharmacother. 1996;50:13–18. doi: 10.1016/0753-3322(96)85092-x. [DOI] [PubMed] [Google Scholar]
- 7.Schols D, DeClercq E. J Virol. 1996;70:4953–4960. doi: 10.1128/jvi.70.8.4953-4960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viscidi R P, Mayur K, Lederman H M, Frankel A D. Science. 1989;246:1606–1608. doi: 10.1126/science.2556795. [DOI] [PubMed] [Google Scholar]
- 9.Weinhold K J, Lyerly H K, Stanley S D, Austin A A, Matthews T J, Bolognesi D P. J Immunol. 1989;142:3091–3097. [PubMed] [Google Scholar]
- 10.Zagury J F, Lachgar A, Achour A, Chams-Harvey V, Cho Y Y, LeCoq H, Bizzini B, Feldman M, Burny A, Zagury D. Biomed Pharmacother. 1994;48:11–16. doi: 10.1016/0753-3322(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 11.Walker B D, Chakrabarti S, Moss B, Paradis T J, Flynn T, Durno A G, Blumberg R S, Kaplan J C, Hirsch M S, Schooley R T. Nature (London) 1987;328:345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- 12.Plata F, Autran B, Martins L P, Wain-Hobson S, Raphael M, Mayaud C, Denis M, Guillon J M, Debre P. Nature (London) 1987;328:348–351. doi: 10.1038/328348a0. [DOI] [PubMed] [Google Scholar]
- 13.Hoffenbach A, Langlade-Demoyen P, Dadaglio G, Vilmer E, Michel F, Mayaud C, Autran B, Plata F. J Immunol. 1989;142:452–462. [PubMed] [Google Scholar]
- 14.Koenig S, Earl P, Powell D, Pantaleo G, Merli S, Moss B, Fauci A S. Proc Natl Acad Sci USA. 1988;85:8638–8642. doi: 10.1073/pnas.85.22.8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker C M, Moody D J, Stites D P, Levy J A. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 16.Cocchi F, DeVico A L, Garzino-Demo A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 17.Giorgi J V, Detels R. Clin Immunol Immunopathol. 1989;52:10–18. doi: 10.1016/0090-1229(89)90188-8. [DOI] [PubMed] [Google Scholar]
- 18.Pantaleo G, De Maria A, Koenig S, Butini L, Moss B, Baseler M, Lane H C, Fauci A S. Proc Natl Acad Sci USA. 1990;87:4818–4822. doi: 10.1073/pnas.87.12.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson R E, Shiboski S C, Royce R, Jewell N P, Lang W, Winkelstein W., Jr AIDS. 1991;5:213–215. doi: 10.1097/00002030-199102000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Carmichael A, Jin X, Sissons P, Borysiewicz L. J Exp Med. 1993;177:249–256. doi: 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeRossi A, Franchini G, Aldovini A, Del Mistro A, Chieco-Bianchi L, Gallo R C, Wong-Staal F. Proc Natl Acad Sci USA. 1986;83:4297–4301. doi: 10.1073/pnas.83.12.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lusso P, Lori F, Gallo R C. J Virol. 1990;64:6341–6344. doi: 10.1128/jvi.64.12.6341-6344.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lusso P, DeMaria A, Malnati M, Lori F, DeRocco S E, Baseler M, Gallo R C. Nature (London) 1991;349:533–535. doi: 10.1038/349533a0. [DOI] [PubMed] [Google Scholar]
- 24.DeMaria A, Pantaleo G, Schnittman S M, Greenhouse J J, Baseler M, Orenstein J M, Fauci A S. J Immunol. 1991;146:2220–2226. [PubMed] [Google Scholar]
- 25.DeMaria A, Colombini S, Schnittman S M, Moretta L. Eur J Immunol. 1994;24:531–536. doi: 10.1002/eji.1830240306. [DOI] [PubMed] [Google Scholar]
- 26.Mercure L, Brenner B J, Phaneuf D, Tsoukas C, Wainberg M A. Antimicrob Agents Chemother. 1994;38:986–990. doi: 10.1128/aac.38.5.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsubota H, Ringler D J, Kannagi M, King N W, Solomon K R, MacKey J J, Walsh D G, Letvin N L. J Immunol. 1989;143:858–863. [PubMed] [Google Scholar]
- 28.Maddon P J, Littman D R, Godfrey M, Maddon D E, Chess L, Axel R. Cell. 1985;42:93–104. doi: 10.1016/s0092-8674(85)80105-7. [DOI] [PubMed] [Google Scholar]
- 29.Agy M B, Wambach M, Foy K, Katze M G. Virology. 1990;177:251–258. doi: 10.1016/0042-6822(90)90478-a. [DOI] [PubMed] [Google Scholar]
- 30.Kawamura I, Koga Y, Oh-Hori N, Onodera K, Kimura G, Nomoto K. J Virol. 1989;63:3748–3754. doi: 10.1128/jvi.63.9.3748-3754.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson B, Kahn L, Lovett E J, Hudson J. J Immunol. 1986;137:35–39. [PubMed] [Google Scholar]
- 32.Blue M L, Daley J F, Levine H, Craig K A, Schlossman S F. J Immunol. 1986;137:1202–1207. [PubMed] [Google Scholar]
- 33.Blue M L, Daley J F, Levine H, Schlossman S F. J Immunol. 1985;134:2281–2286. [PubMed] [Google Scholar]
- 34.Salmon P, Giovane A, Wasylyk B, Klatzmann D. Proc Natl Acad Sci USA. 1993;90:7739–7743. doi: 10.1073/pnas.90.16.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blum M D, Wong G T, Higgins K M, Sunshine M J, Lacy E. J Exp Med. 1993;177:1343–1358. doi: 10.1084/jem.177.5.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawada S, Scarborough J D, Killeen N, Littman D R. Cell. 1994;77:917–929. doi: 10.1016/0092-8674(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 37.Siu G, Wurster A L, Duncan D D, Soliman T M, Hedrick S M. EMBO J. 1994;13:3570–3579. doi: 10.1002/j.1460-2075.1994.tb06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donda A, Schulz M, Burki K, DeLibero G, Uematsu Y. Eur J Immunol. 1996;26:493–500. doi: 10.1002/eji.1830260232. [DOI] [PubMed] [Google Scholar]
- 39.Dean G A, Reubel G H, Pedersen N C. J Virol. 1996;70:5646–5650. doi: 10.1128/jvi.70.8.5646-5650.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livingstone W J, Moore M, Innes D, Bell J E, Simmonds P. Lancet. 1996;348:649–654. doi: 10.1016/s0140-6736(96)02091-0. [DOI] [PubMed] [Google Scholar]
- 41.Semenzato G, Agostini C, Ometto L, Zambello R, Trentin L, Chieco-Bianchi L, DeRossi A. Blood. 1995;85:2308–2314. [PubMed] [Google Scholar]
- 42.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 43.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRosa G, Newman W, et al. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 44.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, DiMarzio P, Marmon S, Sutton R E, Hill C M, Davis C B, et al. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 45.Feng Y, Broder C C, Kennedy P E, Berger E A. Science. 1996;272:872–877. [Google Scholar]
- 46.Wu L, Paxton W A, Kassam N, Ruffing N, Rottman J B, Sullivan N, Choe H, Sodroski J, Newman W, Koup R A, et al. J Exp Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bleul C C, Wu L, Hoxie J A, Springer T A, Mackay C R. Proc Natl Acad Sci USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. Nature (London) 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 49.Pantaleo G, Soudeyns H, Demarest J F, Vaccarezza M, Graziosi C, Paolucci S, Daucher M, Cohen O J, Denis F, Biddison W E, et al. Proc Natl Acad Sci USA. 1997;94:9848–9853. doi: 10.1073/pnas.94.18.9848. [DOI] [PMC free article] [PubMed] [Google Scholar]