

Abstract
Intestinal trefoil factor 3 (TFF3) is a member of the trefoil family of peptides, small molecules constitutively expressed in epithelial tissues, including the gastrointestinal tract. TFF3 has been shown to promote migration of intestinal epithelial cells in vitro and to enhance mucosal healing and epithelial restitution in vivo. In this study, we evaluated the effect of recombinant TFF3 (rTFF3) stimulation on the expression and cellular localization of the epithelial (E)-cadherin–catenin complex, a prime mediator of Ca2+ dependent cell–cell adhesion, and the adenomatous polyposis coli (APC)–catenin complex in HT29, HCT116, and SW480 colorectal carcinoma cell lines. Stimulation by rTFF3 (10−9 M and 10−8 M) for 20–24 hr led to cell detachment and to a reduction in intercellular adhesion in HT29 and HCT116 cells. In both cell lines, E-cadherin expression was down-regulated. The expression of APC, α-catenin and β-catenin also was decreased in HT29 cells, with a translocation of APC into the nucleus. No change in either cell adhesion or in the expression of E-cadherin, the catenins, and APC was detected in SW480 cells. In addition, TFF3 induced DNA fragmentation and morphological changes characteristic of apoptosis in HT29. Tyrphostin, a competitive inhibitor of protein tyrosine kinases, inhibited the effects of TFF3. Our results indicate that by perturbing the complexes between E-cadherin, β-catenin, and associated proteins, TFF3 may modulate epithelial cell adhesion, migration, and survival.
Keywords: trefoil peptides, migration, adhesion, apoptosis, phosphorylation
Interference with cellular attachment and increased migration has been implicated during the neoplastic process (1) and is fundamental during the early reparative response of epithelial restitution in mucosal healing (2). Spatial and temporal changes in cell–cell and cell–matrix interactions occur during migration and several families of adhesion molecules that mediate these interactions have been shown to regulate cell motility (3).
In epithelial cells, cell–cell adhesion is mediated primarily by epithelial (E)-cadherin, a 120-kDa transmembrane glycoprotein localized at the adherens junctions (4). In the presence of Ca2+, E-cadherin’s extracellular domain interacts homotypically with the E-cadherin molecules of neighboring cells to maintain intercellular adhesion, and its cytoplasmic carboxy tail associates with a group of closely related but distinct membrane undercoat proteins, termed the catenins (α, β, and γ) (5). Both β-catenin (92 kDa) and γ-catenin (83 kDa) bind directly to the cytoplasmic domain of E-cadherin, and α-catenin (102 kDa) links the bound β- or γ-catenin to the actin microfilament network of the cellular cytoskeleton (6). Such binding is essential for the adhesive function of E-cadherin and for the establishment of tight physical cell–cell adhesion (7).
A number of studies have confirmed that structural and functional integrity of the components of the E-cadherin–catenin complex are necessary for cell adhesion (8). We have shown that perturbation of E-cadherin–catenin-mediated cell adhesion is associated with cell migration and epithelial restitution in an in vitro model of epithelial injury (9). In vivo, the regenerating epithelium over ulcerated mucosa shows loss of membranous localization and decreased levels of E-cadherin and α-catenin (10). Using chimeric-transgenic mice, Hermiston and Gordon showed that disruption of cadherin mediated cell–cell adhesion in the crypt epithelial cells resulted in altered cell cycle, perturbed mucosal barrier function, and progressive inflammatory changes with features consistent with inflammatory bowel disease (11).
Loss of function or expression of any of the E-cadherin–catenin complex components also has been implicated in leading to loss of epithelial differentiation and normal architecture and the acquisition of a motile and invasive phenotype (8). In vitro studies using human carcinoma cell lines have confirmed this for E-cadherin, α-catenin, and β-catenin (7, 12). In vivo studies of a variety of human malignancies, including oesophageal, gastric, and colonic adenocarcinomas, have shown that reduced E-cadherin–catenin expression is associated with tumor dedifferentiation, infiltrative growth, and lymph node involvement (13, 14).
The cadherins and catenins, however, are more than just intercellular glue. β-catenin seems to participate in signal transduction pathways independently from its role in cell–cell adhesion and thus links the cell surface to downstream cytoplasmic and nuclear events. For instance, β-catenin and its Drosophila homolog, Armadillo, are involved in a Wnt–Wingless signaling pathway critical for developmental patterning at least in Drosophila and Xenopus (15). It also has been discovered that β-catenin can bind not only to E-cadherin but also to other molecules such as the epidermal growth factor receptor (EGFR) (16) and the adenomatous polyposis coli (APC) gene product (17).
The APC gene encodes a 310-kDa cytoplasmic protein (18, 19) and the C-terminal portion of the APC protein is deleted in most of the mutations that are found commonly in inherited familial adenomatous polyposis (FAP) patients and in sporadic colorectal cancers (20, 21). Such mutations appear to be an early event during colorectal tumorigenesis (18, 19, 22, 23). The APC protein competes directly with E-cadherin for binding to β-catenin and can form distinct complexes containing combinations of α-catenin, β-catenin, and γ-catenin that are independent from the cadherin–catenin complexes and through them bind to the cytoskeleton (24, 25). Mutant APC protein may bind more avidly to β-catenin than wild-type APC, and it appears that APC and β-catenin regulate their mutual expressions (26). Phosphorylation of APC by glycogen synthase kinase-3β regulates the interaction of APC with β-catenin and may lead to β-catenin degradation (27). Free β-catenin seems to accumulate in the nucleus (28) and there associate with the DNA binding proteins Tcf-Lef, triggering gene expression leading to proliferation or inhibition of apoptosis (29). It recently has been suggested that APC may act as part of the Wnt–β-catenin signaling cascade in Xenopus (30) and that APC may alter transformation properties, decrease growth rate, or induce apoptosis in colon carcinoma cells (31, 32). It also has been shown that APC can localize in vivo to the end of cell processes and at the tips of microtubule bundles, suggesting a role in the regulation and coordination of directed cell migration (33, 34). Furthermore, Wong et al. (35) have shown that the forced expression of APC protein in transgenic mice leads to a markedly disordered nonadhesive, migratory phenotype. Because molecules such as APC, E-cadherin, and the catenins appear to play an important role in cell adhesion and survival, disruption of their interactions and modulation of their expression and/or function may be a mechanism by which soluble factors promote migration.
The intestinal trefoil factor (TFF3) is a member of the trefoil family of peptides, which are small secreted molecules and in which the disulfide bonds between cysteine residues exhibit a common trefoil structure (36). TFF3 is expressed constitutively in the gastrointestinal tract, primarily in the duodenum and colon (36), as well as in other tissues such as brain (37), uterus (38), and breast (39). Trefoil peptides have been shown to be overexpressed within gastrointestinal mucosa affected by chronic ulcerative and inflammatory lesions, suggesting a possible role in mucosal healing (40, 41). Mice lacking TFF3 have impaired mucosal healing and an expanded proliferative compartment in their intestinal epithelium (42). Indeed, recombinant TFF3 as well as TFF2 (human spasmolytic polypeptide) have been shown to stimulate the migration of intestinal epithelial cells, to promote wound healing in vitro (43, 44), and to be irreversibly cross-linked to specific binding sites that are present within gastric, colonic, and jejunal mucosal glands (45, 46).
The exact mechanism of TFF3’s mode of action, however, is not understood. Recently, we have shown that recombinant rat TFF3 induces a rapid tyrosine phosphorylation of β-catenin, the EGFR, and an unknown protein of ≈85 kDa (now thought to be γ-catenin) (47). These changes are associated with decreased calcium-dependent cell–cell-homotypic adhesion and enhanced cell migration on wounded monolayers. In this study, we evaluated the effect of rTFF3 stimulation on the expression and cellular localization of the E-cadherin–catenin and APC–catenin complexes in human-colonic carcinoma cell lines.
MATERIALS AND METHODS
Production and Characterization of a mAb (ALI-12–28) Recognizing the N-Terminal APC Gene product.
The antibody was raised by immunizing BALB/c mice with a N-terminal APC–maltose binding protein (MBP) fusion protein using standard techniques (48). The fusion protein was composed of the first 1.3 kb of APC which includes the first 433 amino acids and the first nine exons of the gene. The hybridoma was selected by differential β-galactosidase/anti-β-galactosidase ELISA (GAG-ELISA) (49) screening using the APC–MBP fusion protein and the MBP alone. The antibody is an IgG1 and is called ALI-12–28.
mAbs Against E-Cadherin, Catenins, and Phosphotyrosine Residues.
A mouse mAb against human E-cadherin (HECD-1) was kindly provided by M. Takeichi (Kyoto University, Kyoto, Japan). Commercially available purified mouse mAbs (1 mg/ml) against human α-, β- and γ-catenins (Transduction Laboratories, Lexington, KY) were used. Mouse anti-phosphotyrosine mAb, 4G10 (1 mg/ml), was purchased from Upstate Biotechnology (Lake Placid, NY).
rTFF3 and Tyrphostin.
Recombinant rat intestinal trefoil factor (rTFF3) was purified as described (47). Immunoblot and laser desorption time of flight mass-spectrometric analyses confirmed the identity of the purified material which was assessed to be ≈99% pure as judged by SDS/PAGE and Coomassie Brilliant blue staining (50). Tyrphostin A25 was obtained from Calbiochem-Novabiochem.
Tissue Culture and Cell Treatment.
Three human colonic carcinoma-derived cell lines, HT29, HCT116, and SW480, which have been confirmed to show mutant (truncated protein product of 110/200 kDa), wild-type (normal protein of 310 kDa) and mutant APC (truncated protein product of ≈170 kDa) respectively, were obtained from the American Type Culture Collection. These cell lines were maintained in 25-cm2 sterile Falcon tissue culture flasks (Beckton Dickinson) in DMEM supplemented with 10% fetal calf serum (FCS) at 37°C in 10% CO2. To investigate the effects of TFF3 stimulation, cells were grown for 24 hr in DMEM/2% FCS and then in either the presence or absence of rTFF3 (10−8 M and 10−9 M) for an additional 12, 20, or 24 hr (unless otherwise indicated). To inhibit tyrosine phosphorylation, tyrphostin (200 mM) was added to the cell culture medium 30 min before cells were stimulated with rTFF3.
Western Blotting.
Cells were lysed in sample buffer (50 mM Tris⋅HCl, pH 6.8/2% SDS/5% β-mercaptoethanol/10% glycerol/1 mM EDTA) containing aprotinin (2 mg/ml), phenylmethylsulfonyl fluoride (100 mg/ml), and trypsin-chymotrypsin inhibitor (10 mg/ml) for 10 min at 100°C. Insoluble proteins were removed by centrifugation. Extracts of total soluble proteins were assayed with the Bio-Rad Protein Assay kit, and equal 30-μg amounts were subjected to SDS/PAGE using 5% or 8% polyacrylamide gels. Gels were transferred to nitrocellulose membranes electrophoretically in a buffer containing 50 mM Tris, 380 mM glycine, 0.01% SDS, and 20% methanol. Blots were stained briefly in Ponceau S (Sigma) to further verify loading equality. Nitrocellulose membranes were blocked for 2 hr in buffer containing Tris-buffered saline (TBS), 0.05% Tween-20 (TBST), 10% powdered milk, and 1% rabbit serum. Primary antibodies were incubated with the blots for 1 hr at room temperature in TBST. Antibodies were used at the following concentrations: ALI-12–28, 4 μg/ml; α-catenin, 4 μg/ml; β- and γ-catenin, 1 μg/ml; and HECD-1, undiluted cultured supernatant. After washing the membranes, rabbit anti-mouse Igs coupled to horseradish peroxidase (Dako) was added at a dilution of 1:2000 for 1 hr. The membranes were washed and antibody reactivity was visualized with the enhanced chemiluminescence (ECL) reagent (Amersham International, Buckinghamshire, UK) against Hyperfilm-MP (Amersham).
Immunofluorescence.
Cells were grown on collagen-covered coverslips in DMEM supplemented with 2% fetal calf serum. After washing with TBS, cells were fixed in 100% acetone at room temperature for 10 min. Alternatively, cells were fixed with pH 7.4 paraformaldehyde lysine periodate (2% p-formaldehyde/0.01 M m-sodium periodate/0.075 M lysine/0.037 M sodium phosphate) for 20 min at room temperature. The cells were washed with TBS before incubation in blocking buffer containing TBS, 1% BSA, 5% rabbit serum, and 50 mM NH4Cl for 1h at room temperature. Cells were then incubated with primary antibodies (at the following concentrations: ALI-12–28, 20 μg/ml; β-catenin, 10 μg/ml; and HECD-1, undiluted cultured supernatant) in TBS, 5% rabbit serum, and 0.2% BSA overnight at 4°C. After washing, the binding of primary antibodies was detected with fluorescein-conjugated rabbit anti-mouse Igs (Dako) diluted 1:40 in TBS and incubated for 30–60 min at room temperature. Cells were then washed and slides were mounted in Vectashield (Vector Laboratories) and viewed by three independent observers (J.A.E., M.N., and M.P.) through a fluorescence microscope (Olympus).
Immunoprecipitation.
After stimulation with rTFF3 in either the presence or absence of tyrphostin for 7 min, cells were lysed in vanadate immunoprecipitation buffer (50 mM Tris⋅HCl, pH 7.5/100 mM NaCl/0.5% Nonidet P-40/0.2 mM sodium orthovanadate/50 mM NaF/2 mg/ml aprotinin/2 mg/ml leupeptin) on ice for 30 min and centrifuged at 13,000 rpm for 30 min. After preclearing with protein G-Sepharose beads coated with mouse IgG, the lysates were precipitated with protein G-Sepharose complexed to either anti-β- or -γ-catenin mAb. The protein concentrations of the cell lysates were determined by the Bio-Rad Protein Assay kit. Aliquots of 300 μg of total-cell proteins were incubated with 6 μg of the relevant antibody for 2 hr at 4°C. The beads were collected by centrifugation, washed with detergent buffer, dissolved in the sample buffer by boiling, and the immunoprecipitates were subjected to SDS/PAGE (8% acrylamide). Tyrosine phosphorylation in the immunoprecipitated complex was analyzed by Western blotting using the 4G10 antibody at 1 μg/ml.
Detection of DNA Fragmentation.
Cells were harvested at 24, 36, 42, 48, 60, and 72 hr after stimulation with TFF3 and incubated with lysis buffer (10 mM Tris⋅HCl, pH 8.0/10 mM NaCl/10 mM EDTA/100 mg/ml proteinase K/1% SDS) at 37°C. The DNA was extracted with an equal volume of phenol/chloroform, precipitated in ethanol containing 0.3 M final concentration of sodium acetate (pH 5.2), and centrifuged for 30 min at 4°C. The pellet was resuspended in TE buffer (10 mM Tris⋅HCl/1 mM EDTA, pH 8.0). RNase (100 mg/ml) was added to each sample and incubated for 1 hr at room temperature. The DNA samples were mixed with loading buffer and run on a 1.8% agarose gel. A HindIII digest of Λ-DNA was applied to each gel to provide molecular size markers of 23.5, 9.6, 6.6, 4.3, 2.2, 2.1, and 0.5 kbp. Electrophoresis was carried out in TBE buffer (2 mM EDTA, 89 mM Tris-HCl, 89 mM boric acid), and the DNA was visualized by ethidium bromide and photographed on Polaroid type 667 (3,000 ASA) film.
DNA Labeling with Propidium Iodide (PI).
Cells were grown on sterile glass coverslips in 24-well plates either in the presence or absence of TFF3 for 24 hr. Some coverslips were stained with hematoxylin/eosin to check the morphologic appearance of the cells. The PI staining was carried out as described (51).
RESULTS
Specificity of ALI-12–28.
ALI-12–28 was epitope mapped by differential in vitro expression of the N-terminal region of the APC gene by using the protein truncation test and was found to bind to APC in the region between nucleotides 135 and 422 (exons 2–3). As shown in Fig. 1, ALI-12–28 reacts with normal and mutant-truncated forms of the APC protein in a series of FAP lymphoblastoid cell lines by Western blotting. Mutations that occur at the 5′ end of the APC gene seem to produce an unstable RNA or protein product. As yet, we have been unable to detect small molecular weight truncated products (<65 kDa) by Western blotting techniques. This result may explain why we did not observe a truncated product in all of the FAP cell lines examined (Fig. 1). ALI-12–28 has been used as the initial mutation screen for patients with FAP (data not shown). By calculating the molecular weight of the protein, a rough estimate of the genomic localization of the mutation can be made. The antibody also has specific application in immunoprecipitation and immunocytochemistry. The immunoprecipitating properties of this antibody have been utilized to improve the quality of PTT results covering APC exons 1–14 (A.R. and W.F.B., personal communication).
Figure 1.
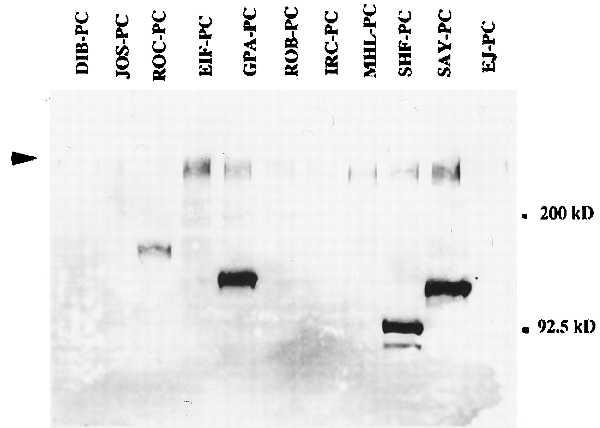
Western blot (7.5% PAGE) using ALI-12–28 showing specificity of the antibody for both wild-type and mutant-truncated forms of the APC product in a series of FAP lymphoblastoid cell lines derived from FAP patients. These cell lines were established by introducing Epstein–Barr virus into isolated patient B cells. Arrowhead indicates the wild-type product of ≈310 kDa.
TFF3 Modulates Cellular Adhesion of Colonic Carcinoma Cell Lines.
Stimulation by rTFF3 (10−9 M and 10−8 M) led to decreased cell substratum and cell–cell adhesion in HT29 (Fig. 2) and HCT116 (data not shown) with an increase in the number of floating cells. There was no change in the morphology nor the adhesion of SW480 cells. A time course experiment revealed that this cellular detachment and reduction in adhesion was seen when rTFF3 had been administered continuously for at least 20 hr. Interestingly, stimulation of HT29 for only 1 hr at 10−8 M or 10−9 M showed similar but delayed (3–10 days) cell detachment.
Figure 2.

Phase-contrast photograph of HT29 cells unstimulated (A) and stimulated (B) with rTFF3 (10−8 M) for 24 hr. Stimulation by rTFF3 led to the detachment of cells, reduction in cell adhesion, and an increase in the number of floating cells.
TFF3 Affects the Expression and Cellular Localization of E-Cadherin, APC, and Catenins.
HT29, HCT116, and SW480 cells were examined for APC, E-cadherin, and catenin expression by Western blotting. In rTTF3 stimulated HT29 cells, E-cadherin and α-catenin expression virtually disappeared whereas APC and β-catenin decreased markedly. However, γ-catenin expression was preserved largely (Fig. 3). In HCT116 cells, only E-cadherin expression was down-regulated, and in SW480 cells, none of the proteins we examined were affected by the administration of rTFF3 (data not shown). Down-regulation of E-cadherin expression in HT29 was the earliest event, detectable after 12 hr, whereas the effect on APC and catenin expression was seen after stimulation for 20–24 hr.
Figure 3.
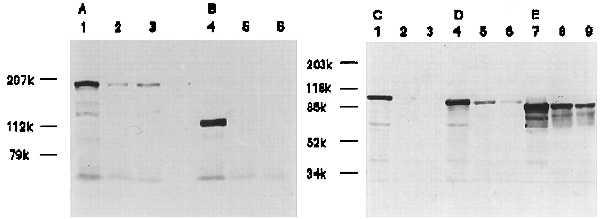
Western blot analysis of APC, E-cadherin, and α-, β-, and γ-catenins. Twenty-four hours after HT29 cells were stimulated with 10−9 M (lanes 2, 5, and 8) or 10−8 M (lanes 3, 6, and 9) or without rTFF3 (lanes 1, 4, and 7), total cell lysates were subjected to Western blotting with ALI-12–28 (A), HECD-1 (B), anti-α- (C), β- (D), or γ-catenin (E). The expression of APC, E-cadherin, α-catenin, and β-catenin significantly decreased in HT29 cells at both concentrations of rTFF3. The expression of γ-catenin was preserved largely.
The cellular localization of E-cadherin, APC, and catenins was examined by indirect immunofluorescence in HT29 cells in the presence or absence of rTFF3. In unstimulated conditions, APC showed intense and diffuse cytoplasmic localization (Fig. 4A). β-catenin expression was also cytoplasmic with a clear submembranous localization at intercellular junctions, as shown (47). Following rTFF3 stimulation for 20–24 hr, APC immunostaining was heterogeneous, reduced, and showed a granular pattern. This was accompanied by translocation of the protein from the cytoplasm to the nucleus with nucleolar accentuation (Fig. 4B). The change in APC cellular localization appeared to commence gradually after 12 hr. β-catenin expression virtually was undetectable by immunofluorescence (data not shown) in HT29 cells stimulated with rTFF3 for 20–24h.
Figure 4.
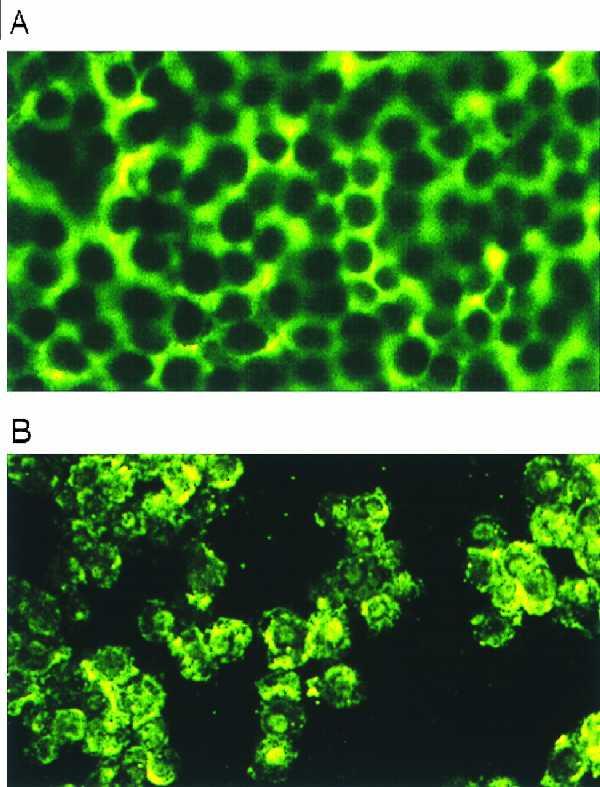
Immunofluorescence staining of APC in HT29 cells unstimulated (A) and stimulated (B) with rTFF3 (10−8 M) for 24 hr. After treatment with rTFF3, APC showed heterogeneous and granular staining with nuclear/nucleolar localization.
rTFF3 Induces Apoptosis in HT29 Cells.
To explore whether the cell detachment induced by rTTF3 triggered apoptotic changes, we analyzed cellular morphology using PI and the appearance of DNA fragmentation with agarose gel analysis. The PI-stained HT29 cell nuclei showed a much higher number of apoptotic nuclei with marked chromatin condensation and nuclear fragmentation compared with unstimulated cells (data not shown). The agarose gel analysis showed that DNA fragmentation was present in the DNA samples extracted from HT29 (floater and attached cell populations) when the cells had been stimulated with rTFF3 for 36 hr (Fig. 5).
Figure 5.
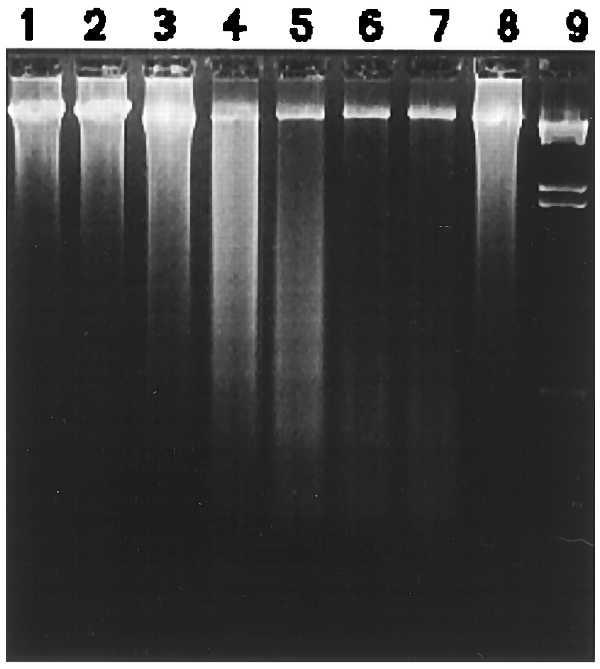
Time-dependent effects of rTFF3 on DNA fragmentation. Agarose gel electrophoresis of DNA extracted from HT29 cells 24 hr (lane 2), 36 hr (lane 3), 42 hr (lane 4), 48 hr (lane 5), 60 hr (lane 6), and 72 hr (lane 7) after cells were stimulated with 10−8 M rTFF3. Control experiment was carried out under the same conditions for 24 hr (lane 1). Tyrphostin was added into the cell culture medium (200 mM) 30 min before cells were stimulated with rTFF3 and thereafter coincubated with cells for 48 hr (lane 8). Lane 9 is a HindIII digest of λ DNA providing a molecular size marker. HT29 cells showed DNA fragmentations <2-kbp in size after 36 hr and <500 bp in size after 48 hr. Administration of tyrphostin largely inhibited these changes. After 60 hr (lanes 6 and 7), no DNA laddering was detected probably due to cells undergoing necrosis.
Inhibition of rTFF3-Induced Changes by Tyrphostin 25.
To examine whether tyrosine phosphorylation is a possible mechanism by which rTFF3 mediates its effects on cell adhesion and apoptosis, HT29 cells were stimulated with rTFF3 in the presence or absence of tyrphostin, a competitive inhibitor of substrate binding on protein tyrosine kinases (52). Expression and cellular localization of E-cadherin, APC, α-catenin, and β-catenin was preserved in HT29 cells treated with rTFF3 and tyrphostin (Fig. 6). Tyrphostin inhibited cell detachment and reversed DNA fragmentation induced by rTFF3 (Fig. 5, lane 8).
Figure 6.
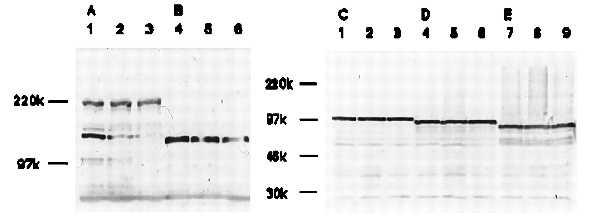
Western blot analysis of APC, E-cadherin, and α-, β-, and γ-catenins. Twenty-four hours after HT29 cells were stimulated with 10−9 M (lanes 2, 5, and 8) or 10−8 M (lanes 3, 6, and 9) rTFF3 in the presence of 200 nM tyrphostin or without rTFF3 (lanes 1, 4, and 7), total cell lysates were subjected to Western blotting with ALI-12–28 (A), HECD-1 (B), anti-α- (C), β- (D), or γ-catenin (E). Tyrphostin inhibited the down-regulation of APC, E-cadherin, α-catenin, and β-catenin induced by rTFF3 as shown in Fig. 3.
To assess whether tyrphostin inhibited tyrosine phosphorylation of β- and γ-catenin, which we have shown (47) to be induced by rTFF3 (10−6 M for 7 min), we examined the change of phosphorylated tyrosine residues by immunoprecipitation. In HT29 cells stimulated either with 10−8 or 10−9 M rTFF3, tyrphostin did not appear to have a significant effect on the levels of tyrosine phosphorylation of β-catenin or γ-catenin (data not shown).
DISCUSSION
In this study, we evaluated the effects of rTFF3 on the expression and function of the E-cadherin–catenin and APC–catenin complexes in human colorectal carcinoma cell lines. Such a study requires careful consideration of the mutational background on which these observations are made. Our results demonstrate that in HT29 cells, which harbor an APC mutation but have a normal E-cadherin–catenin complex (47), rTFF3 leads to down-regulated expression of E-cadherin (after 12 hr), decreased cell–cell and cell–substratum adhesion, down-regulated expression of APC and α- and β-catenin, translocation of APC from the cytoplasm to the nucleus (after ≈20 hr), and the induction of apoptotic changes (after 36 hr). It is interesting to note that loss of adhesion was triggered irreversibly within 1 hr after rTFF3 administration and is thus an early event, though it did not materialize morphologically until at least 20 hr after stimulation. Therefore, the APC mutation present in HT29 cells does not appear to affect their ability to respond to TFF3 and to translocate the APC protein into the nucleus and nucleoli. While this study was in progress, Neufeld and White (53) demonstrated that full length APC also can localize in the nucleus of unstimulated colon carcinoma-derived cell lines.
With regard to the other cell lines examined, HCT116, which has wild-type E-cadherin, responded to TFF3 with a down-regulation in E-cadherin and loss of cell–cell adhesion. HCT116, however, is known to harbor a mutation in β-catenin (54), which may explain its limited responsiveness to TFF3 as compared with HT29. In addition, SW480, which has low levels of E-cadherin and α-catenin, has been shown to be unresponsive not only to TFF3 but also to TFF2 (44). Interestingly, we recently have shown that transfection of E-cadherin into LS174T colon carcinoma cells (which are E-cadherin negative yet have wild-type APC and β-catenin) restores their lost sensitivity to TFF2 (M.P., unpublished data). It is thus likely that the action of trefoil peptides, such as TFF2 and TFF3, requires the presence of an intact E-cadherin–catenin complex as seen in HT29 cells (47).
TFF3 has been shown to be overexpressed in mammary (39), gastric (M.N., personal communication), and colonic carcinomas (55) where loss of the E-cadherin–catenin complex commonly is seen (13, 14). Therefore, resistance to TFF3 may confer a selective advantage to carcinoma cells that already possess a disrupted E-cadherin–catenin complex by allowing them to escape from the apoptotic pathway.
Although TFF3 appears to be overexpressed in neoplastic tissue and appears to play an important role during the early stages of epithelial restitution when viable epithelial cells rapidly migrate from the ulcer margins over the denuded area (8), its function in the normal gastrointestinal tract largely is unknown. In normal colonic mucosa, TFF3 has been shown to localize to the superficial columnar epithelial cells (55) where APC (56) and the classical E-cadherin–catenin complex (14) also are expressed at high levels. Because our results suggest that rTFF3 reduces E-cadherin-mediated cell–cell adhesion and induces apoptosis (probably as a secondary response), it is conceivable that it may play a role during normal intestinal epithelial cell kinetics, which involves migration-associated differentiation, programmed cell death, and cell shedding at the top of the crypt (57, 58). This hypothesis is consistent with the observed expansion of the proliferative compartment seen in the TFF3 knock-out mice (42) and with the increased epithelial cell apoptosis seen in the dominant-negative N-cadherin mutant transgene mice (59).
Other studies support the notion that cell–cell junctional proteins can be common targets for motogenic factors when initiating cell migration. For example, hepatocyte growth factor/scatter factor and EGF have been shown to induce tyrosine phosphorylation of β-catenin, γ-catenin, and p120 in HT29 cells. This induction was associated with cellular redistribution of E-cadherin, resulting in increased motility (60). Similarly, as we have described, rTFF3 induces tyrosine phosphorylation of catenins as well as EGFR leading to loss of E-cadherin-mediated adhesion and promotion of cell migration (47). In this study, tyrphostin successfully inhibited most of the effects induced by rTFF3 and re-established normal cell interactions confirming that tyrosine phosphorylation is an important mechanism by which rTFF3-mediated changes are regulated.
Components of the E-cadherin–catenin and the APC–catenin complex have been shown to associate with molecules which either express tyrosine kinase activity or act as substrates for various receptor tyrosine kinases, such as EGFR and p120 (16, 61). Furthermore, in cells transfected with the v-src oncogene, increased tyrosine phosphorylation of β-catenin and E-cadherin was observed resulting in functional changes such as decreased adhesion, increased migration, and increased invasiveness, although the overall expression of neither the catenins nor the cadherins changed (62). Because in this study we detected no significant inhibition of catenin tyrosine phosphorylation by tyrphostin, it is likely that tyrosine phosphorylation of other molecules, such as EGFR or the putative TFF3 receptor, plays an important role in mediating the effects of TFF3. It is interesting to note that EGF has been shown to potentiate the effects of rTFF3 (63). The synergism between TFF3 and EGF suggests potential complementary roles and linked receptor signaling pathways, possibly through β-catenin. Elucidation of the respective pathways and the mechanisms and the molecules involved may lead to a greater understanding of normal intestinal epithelial cell kinetics, mucosal repair, and colorectal tumorigenesis.
Acknowledgments
We are grateful to Amanda Littler for the APC–MBP fusion protein, to Ragai Mitry for advice on the apoptotic assays, to Helen Cox for advice on the tyrphostin experiments, and to William Gullick and Roy Bicknell for the critical reading of the manuscript. We also thank Richard Poulsom for his assistance in the preparation of the photographs. We are grateful to the Medical Research Council, Wellcome Trust, and Crohn’s in Childhood Research Association for their financial support. J.A.E. was supported by a Wellcome Trust Vacation Scholarship, M.N. was supported by a Rotary Foundation Scholarship, and A.J. was supported by a Wellcome Trust Clinical Training Fellowship.
ABBREVIATIONS
- TFF3
intestinal trefoil factor 3
- rTFF3
recombinant TFF3
- E
epithelial
- EGF
epidermal growth factor
- EGFR
EGF receptor
- APC
adenomatous polyposis coli
- MBP
maltose binding protein
- TBST
Tris buffered saline Tween 20
- FAP
familial adenomatous polyposis
- PI
propidium iodide
References
- 1.Pignatelli M, Vessey C J. Hum Pathol. 1994;25:849–856. doi: 10.1016/0046-8177(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 2.Pignatelli M. Yale J Biol Med. 1996;69:131–135. [PMC free article] [PubMed] [Google Scholar]
- 3.Huttenlocher A, Sandborg R R, Horwitz A F. Curr Opin Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- 4.Takeichi M. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 5.Gumbiner B M, Mc Crea P D. J Cell Sci Suppl. 1993;17:155–158. doi: 10.1242/jcs.1993.supplement_17.22. [DOI] [PubMed] [Google Scholar]
- 6.Nieset J E, Redfield A R, Jin F, Knudsen K A, Johnson K R, Wheelock M J. J Cell Sci. 1997;110:1013–1022. doi: 10.1242/jcs.110.8.1013. [DOI] [PubMed] [Google Scholar]
- 7.Shimoyama Y, Nagafuchi A, Fujita S, Gotoh M, Takeichi M, Tsukita S, Hirohashi S. Cancer Res. 1992;52:5770–5774. [PubMed] [Google Scholar]
- 8.Pignatelli M, Karayiannakis A J, Noda M, Efstathiou J, Kmiot W A. In: The Gut as a Model in Cell and Molecular Biology. Halter F, Winton D, Wright N A, editors. Boston: Kluwer; 1997. pp. 194–203. [Google Scholar]
- 9.Hanby A M, Chinery R, Poulsom R, Playford R J, Pignatelli M. Am J Pathol. 1996;148:723–729. [PMC free article] [PubMed] [Google Scholar]
- 10.Karayiannakis, A. J., Syrigos, K. N., Efstathiou, J., Valizadeh, A., Noda, M., Playford, R. J., Kmiot, W. A. & Pignatelli, M. (1998) J. Pathol., in press. [DOI] [PubMed]
- 11.Hermiston M L, Gordon J I. Science. 1995;270:1203–1207. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 12.Pignatelli M, Liu D, Nasim M M, Stamp G W H, Hirano S, Takeichi M. Br J Cancer. 1992;66:629–634. doi: 10.1038/bjc.1992.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jawhari A, Jordan S, Poole S, Browne P, Pignatelli M, Farthing M J G. Gastroenterology. 1997;112:46–54. doi: 10.1016/s0016-5085(97)70218-x. [DOI] [PubMed] [Google Scholar]
- 14.Gagliardi G, Kandemir O, Liu D, Guida M, Benvestito S, Ruers T G M, Benjamin I S, Northover J M A, Stamp G W H, Talbot I C, Pignatelli M. Virchows Archiv. 1995;426:149–154. doi: 10.1007/BF00192636. [DOI] [PubMed] [Google Scholar]
- 15.Gumbiner B M. Curr Opin Cell Biol. 1995;7:634–640. doi: 10.1016/0955-0674(95)80104-9. [DOI] [PubMed] [Google Scholar]
- 16.Hoschuetzky H, Aberle H, Kemler R. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su L K, Vogelstein B, Kinzler K W. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 18.Kinzler K W, Nilbert M C, Su L K, Vogelstein B, Bryan T M, Levy D B, Smith K J, Preisinger A C, Hedge P, McKechnie D, et al. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 19.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 20.Bodmer W F, Bishop T, Karran P. Nat Genet. 1994;6:217–219. doi: 10.1038/ng0394-217. [DOI] [PubMed] [Google Scholar]
- 21.Bodmer W F, Bailey C J, Bodmer J, Bussey H J, Ellis A, Gorman P, Lucibello F C, Murday V A, Rider S H, Scambler P, et al. Nature (London) 1987;328:614–616. doi: 10.1038/328614a0. [DOI] [PubMed] [Google Scholar]
- 22.Powell S M, Zilz N, Beazer B J, Bryan T M, Hamilton S R, Thibodeau S N, Vogelstein B, Kinzler K W. Nature (London) 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 23.Solomon E, Voss R, Hall V, Bodmer W F, Jass J R, Jeffreys A J, Lucibello F C, Patel I, Rider S H. Nature (London) 1987;328:616–619. doi: 10.1038/328616a0. [DOI] [PubMed] [Google Scholar]
- 24.Hulsken J, Birchmeier W, Behrens J. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- 26.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 28.Valizadeh A, Karayiannakis A J, El-Hariry I, Kmiot W A, Pignatelli M. Am J Pathol. 1997;150:902–907. [PMC free article] [PubMed] [Google Scholar]
- 29.Korinek V, Barker N, Morin P J, van-Wicken D, de Weger R, Kinzler K, Vogelstein B, Clevers H. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 30.Vleminckx K, Wong E, Guger K, Rubinfeld B, Polakis P, Gumbiner B M. J Cell Biol. 1997;136:411–420. doi: 10.1083/jcb.136.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groden J, Joslyn G, Samowitz W, Jones D, Bhattachryya N, Spirio L, Thliveris A, Robertson M, Egan S, Meuth M, White R. Cancer Res. 1995;55:1531–1539. [PubMed] [Google Scholar]
- 32.Morin P J, Vogelstein B, Kinzler K W. Proc Natl Acad Sci USA. 1996;93:7950–7954. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathke I S, Adams C L, Polakis P, Sellin J H, Nelson W J. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollack A L, Barth A I M, Altschuler Y, Nelson W J, Mostov K E. J Cell Biol. 1997;137:1651–1662. doi: 10.1083/jcb.137.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong M H, Hermiston M L, Syder A J, Gordon J I. Proc Natl Acad Sci USA. 1996;93:9588–9593. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suemori S, Lynch-Devaney K, Podolsky D K. Proc Natl Acad Sci USA. 1991;88:11017–11021. doi: 10.1073/pnas.88.24.11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Probst J C, Skutella T, Muller-Schmid A, Jirikowski G F, Hoffman W. Mol Brain Res. 1995;33:269–276. doi: 10.1016/0169-328x(95)00137-h. [DOI] [PubMed] [Google Scholar]
- 38.Hauser F, Poulsom R, Chinery R, Rogers L A, Hanby A M, Wright N A, Hoffmann W. Proc Natl Acad Sci USA. 1993;90:6961–6965. doi: 10.1073/pnas.90.15.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poulsom R, Hanby A M, Lalani E-N, Hauser F, Hoffmann W, Stamp G W H. J Pathol. 1997;183:30–38. doi: 10.1002/(SICI)1096-9896(199709)183:1<30::AID-PATH1085>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Wright N A, Poulsom R, Stamp G, Noorden S V, Sarraf C, Elia G, Ahnen D, Jeffery R, Longcroft J, Pike C, et al. Gastroenterology. 1993;104:12–20. doi: 10.1016/0016-5085(93)90830-6. [DOI] [PubMed] [Google Scholar]
- 41.Alison M R, Chinery R, Poulsom R, Ashwood P, Longcroft J M, Wright N A. J Pathol. 1995;175:405–414. doi: 10.1002/path.1711750408. [DOI] [PubMed] [Google Scholar]
- 42.Mashimo H, Wu D C, Podolsky D K, Fishman M C. Science. 1996;274:262–265. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 43.Dignass A, Lynch-Devaney K, Kindon H, Thim L, Podolsky D K. J Clin Invest. 1994;94:376–383. doi: 10.1172/JCI117332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Playford R J, Marchbank T, Chinery R, Evison R, Pignatelli M, Boulton R A, Thim L, Hanby A M. Gastroenterology. 1995;108:108–116. doi: 10.1016/0016-5085(95)90014-4. [DOI] [PubMed] [Google Scholar]
- 45.Chinery R, Cox H M. Peptides (Tarrytown, NY) 1995;16:749–755. doi: 10.1016/0196-9781(95)00045-l. [DOI] [PubMed] [Google Scholar]
- 46.Tan X D, Hsueh W, Chang H, Wei K R, Gonzalezcrussi F. Biochem Biophys Res Commun. 1997;237:673–677. doi: 10.1006/bbrc.1997.7144. [DOI] [PubMed] [Google Scholar]
- 47.Liu D, El-Hariry I, Karayiannakis A J, Wilding J, Chinery R, Kmiot W, McCrea P D, Gullick W J, Pignatelli M. Lab Invest. 1997;77:557–563. [PubMed] [Google Scholar]
- 48.Galfré G, Howe S C, Milstein C, Butcher G W, Howard J C. Nature (London) 1977;266:550–552. doi: 10.1038/266550a0. [DOI] [PubMed] [Google Scholar]
- 49.Durbin H, Bodmer W F. J Immunol Methods. 1987;97:19–27. doi: 10.1016/0022-1759(87)90100-1. [DOI] [PubMed] [Google Scholar]
- 50.Chinery R, Poulsom R, Elia G, Hanby A M, Wright N A. Eur J Biochem. 1993;212:557–563. doi: 10.1111/j.1432-1033.1993.tb17693.x. [DOI] [PubMed] [Google Scholar]
- 51.Mitry R, Sarraf C E, Wu C-G, Pignatelli M, Habib N A. Lab Invest. 1997;77:369–378. [PubMed] [Google Scholar]
- 52.Levitzki A. FASEB J. 1992;6:3275–3282. doi: 10.1096/fasebj.6.14.1426765. [DOI] [PubMed] [Google Scholar]
- 53.Neufeld K L, White R L. Proc Natl Acad Sci USA. 1997;94:3034–3039. doi: 10.1073/pnas.94.7.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ilyas M, Tomlinson I P M, Rowan A, Pignatelli M, Bodmer W F. Proc Natl Acad Sci USA. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taupin D, Ooi K, Yeomans N, Giraud A. Lab Invest. 1996;75:25–32. [PubMed] [Google Scholar]
- 56.Smith K J, Johnson K A, Bryan T M, Hill D E, Markowitz S, Willson J K, Paraskeva C, Peterson G M, Hamilton S R, Vogelstein B, et al. Proc Natl Acad Sci USA. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Potten C S, Allen T D. J Ultrastruct Res. 1977;60:272–277. doi: 10.1016/s0022-5320(77)80071-3. [DOI] [PubMed] [Google Scholar]
- 58.Wright N A, Irwin M. Cell Tissue Kinet. 1982;15:595–609. doi: 10.1111/j.1365-2184.1982.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 59.Hermiston M L, Gordon J I. J Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Oku N, Miyazawa K, Kitamura N, Takeichi M, Ito F. Cell Adhes Commun. 1994;1:295–305. doi: 10.3109/15419069409097261. [DOI] [PubMed] [Google Scholar]
- 61.Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Miyazawa K, Kitamura N, Johnson K R, Wheelock M J, Matsuyoshi N, Takeichi M, et al. J Cell Biol. 1995;128:949–957. doi: 10.1083/jcb.128.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Behrens J, Vakaet L, Friis R, Winterhagel E, Roy F V, Mareel M M, Birchmeier W. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chinery R, Playford R J. Clin Sci (Lond) 1995;88:401–403. doi: 10.1042/cs0880401. [DOI] [PubMed] [Google Scholar]