

Abstract
The bacterial species Vibrio cholerae includes harmless aquatic strains as well as strains capable of causing epidemics and global pandemics of cholera. While investigating the relationship between pathogenic and nonpathogenic strains, we identified a chromosomal pathogenicity island (PAI) that is present in epidemic and pandemic strains but absent from nonpathogenic strains. Initially, two ToxR-regulated genes (aldA and tagA) were studied and were found to be associated with epidemic and pandemic strains but absent in nontoxigenic strains. The region containing aldA and tagA comprises 13 kb of previously unidentified DNA and is part of a PAI that contains a regulator of virulence genes (ToxT) and a gene cluster encoding an essential colonization factor and the cholera toxin phage receptor (toxin-coregulated pilus; TCP). The PAI is 39.5 kb in size, has low %G+C (35%), contains putative integrase and transposase genes, is flanked by att sites, and inserts near a 10Sa RNA gene (ssrA), suggesting it may be of bacteriophage origin. We found this PAI in two clinical non-O1/non-O139 cholera toxin-positive strains, suggesting that it can be transferred within V. cholerae. The sequence within this PAI includes an ORF with homology to a gene associated with the type IV pilus gene cluster of enteropathogenic Escherichia coli, a transposase from Vibrio anguillarum, and several ORFs with no known homology. As the PAI contains the CTXΦ receptor, it may represent the initial genetic factor required for the emergence of epidemic and pandemic cholera. We propose to call this island VPI (V. cholerae pathogenicity island).
Keywords: virulence, colonization, CTXΦ receptor
In the last decade, the life-threatening diarrheal disease cholera has reached a wider distribution than at any other time in the 20th century. Cholera is caused by the bacterium Vibrio cholerae, which can be classified into over 140 serogroups (1). Prior to 1992, it was believed that only V. cholerae of the O1 serogroup were responsible for pandemic cholera and that strains of serogroups other than O1 were avirulent or caused only sporadic illness. However, in 1992 a O139 serogroup strain emerged and caused epidemic disease (2). The factors required for epidemic and pandemic ability are not fully understood, and there have been large outbreaks of cholera caused by toxigenic non-O1/non-O139 strains that have not resulted in significant epidemic or pandemic disease (3, 4).
Epidemic and pandemic strains of V. cholerae secrete cholera toxin (CT), the toxin responsible for the secretory diarrhea that is characteristic of the disease. CT is encoded by the ctxAB genes that are carried on a filamentous bacteriophage designated CTXΦ (5). The bacterial receptor for this phage is the toxin-coregulated pilus (TCP), an essential colonization factor in human and animal models (6, 7). Expression of CT and TCP are coregulated by the ToxR regulatory system consisting of the proteins ToxR, ToxS, and ToxT (8, 9). Recently, it has been shown that the genes tcpP and tcpH within the TCP cluster also regulate virulence factors (10). The gene cluster encoding TCP, an accessory colonization factor (ACF), and the virulence gene regulator ToxT are located near each other (11). Recently Kovach et al. (12) have found that adjacent to the TCP gene cluster is an integrase gene (int) and an att site that marks the right end of a unique locus in pathogenic strains.
Previously, we obtained evidence suggesting that 6th and 7th pandemic strains and U.S. Gulf coast V. cholerae O1 isolates may be derived from nontoxigenic strains and that horizontal transfer occurs in V. cholerae, resulting in the emergence of new pathogenic strains (13, 14). To extend these findings and better understand the genetic population structure of V. cholerae and the factors contributing to epidemic and pandemic ability, we initially studied the prevalence and sequence variation of two genes under control of ToxR (aldA and tagA). aldA encodes a cytoplasmic CoA-independent aldehyde dehydrogenase (EC 1.2.1.3) (15), whereas the adjacent and oppositely transcribed tagA encodes a lipoprotein (16). As no obvious role in virulence was found by using a mouse model (15, 16), these genes were thought to encode metabolic rather than virulence functions, thus being appropriate molecular clocks and were initially studied in that context.
We report that aldA and tagA are part of a 39.5-kb pathogenicity island (PAI) that includes the TCP-ACF cluster and is associated with epidemic and pandemic strains of V. cholerae. We propose to call this locus VPI (for V. cholerae pathogenicity island).
MATERIALS AND METHODS
Bacterial Strains.
A total of 59 wild-type V. cholerae isolates comprising 29 strains of the serogroup O1, 10 of serogroup O139, 20 of non-O1/non-O139 serogroups, and 1 Vibrio mimicus isolate were used in this study. The majority of these isolates have been described (13). V. cholerae N16961 is a CT-positive 7th pandemic O1 El Tor strain isolated in Bangladesh in 1975 (47). Strains were tested for the presence of ctx genes by DNA hybrization to a ctxAB probe. Escherichia coli HB101 and DH5α were used as hosts for maintaining cosmid and plasmid clones, respectively. Cosmid vector pHC79 (17) was used for construction of the N16961 genomic library, whereas pBluescript+ (Stratagene) was used for subcloning cosmid DNA.
Recombinant DNA Techniques.
Genomic, cosmid, and plasmid DNA were prepared by using standard methods (18). A cosmid library was constructed with chromosomal DNA isolated from N16961 partially digested with Sau3AI to give fragments of ≈30 kb. Fragments were ligated into BamHI-digested pHC79, packaged in vitro with the Gigapak lambda packaging kit (Stratagene), and used to infect E. coli HB101. Transformants were selected on Luria-Bertani (LB) agar containing ampicillin (200 μg/ml), and each colony was inoculated into separate wells of a microtiter plate containing LB broth and 5% dimethyl sulfoxide. Microtiter plates were incubated overnight at 37°C and replica plated onto nylon filters (Qiagen, Chatsworth, CA) for subsequent hybridization and identification of cosmid clones containing aldA. pDK8, a cosmid clone of N16961, was selected for further work. Subclones were created by digesting cosmid DNA with HindIII and ligating into HindIII-digested pBluescript+. Ligations were electroporated into DH5α and selected on LB agar containing ampicillin (200 μg/ml).
PCR products from cosmid clone pDK8 and the 7th pandemic strain N16961 were used as probes to determine whether corresponding sequences were present in genomic DNA preparations of various V. cholerae strains. Fragments were labeled with [α-32P]dCTP by using the Ready to Go system (Pharmacia). Southern blot hybridizations were performed by using standard methods (18).
PCR was performed essentially as described by Saiki et al. (19), with primers listed in Table 1. Location of primers is shown schematically in Fig. 1. Extended PCR with the Perkin–Elmer XL PCR kit and extension times of 12 min was used to amplify the tagA–tagD region in pDK8.
Table 1.
Primers used
| Primer | Primer sequence (5′–3′) | Ref. |
|---|---|---|
| KAR3 | AATAGCGAAACTTCGAC | 15 |
| KAR7 | CCTCTAGGTCTATTTTA | 15 |
| KAR8 | GGTGGTAAGATATTCACTC | 16 |
| KAR9 | GTCACAACAGGTACACC | 16 |
| KAR22 | GATAAAGAGATCAAAGCC | 12 |
| KAR23 | ATCTGCTTCCATGTGGG | 12 |
| KAR24 | AAAACCGGTCAAGAGGG | 29 |
| KAR25 | CAAAAGCTACTGTGAATGG | 29 |
| KAR82 | CAAATGCAACGCCGAATGG | 29 |
| KAR85 | CGCCTGCGAACCGACACGC | 12 |
| KAR86 | GCAGCAAGCCTCCACTCCG | 12 |
| KAR87 | CGCACAGCCAAGCGTCCGC | 12 |
| KAR90 | GATGTAATGAGAATGCAACATCCAGTAACGACC | 16 |
| KAR91 | AACTGGTCATAGAAATAAGCGGACATATAGGGC | 46 |
| KAR92 | TAAATTGTTATCATGCATATCCTGCTCATGCGG | 15 |
| KAR94 | TATGATACTGAAAACACCTC | This study |
| KAR95 | GATGCTAACAGCAGAGCATA | This study |
| KAR96 | TGCTACTTACCCAATGGCAC | This study |
| KAR97 | GAGCCAGGCTTATTTGGGCG | This study |
Figure 1.
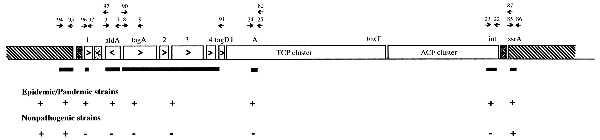
The VPI. Arrows above genes show PCR primers used and are identified by the number of the KAR primer series, whereas arrows within genes show direction of transcription. ▪, Probes used; ▧, common chromosomal flanking DNA; ☒ at each end of VPI denotes att sites; and ░⃞ within the left end of the VPI indicates the defective transposase. The + and − indicate the presence and absence, respectively, of the region in epidemic and pandemic strains and nontoxigenic strains.
Sequencing of double-stranded DNA from cosmid, plasmid, and PCR products was carried out with the Taq Dye-Terminator sequencing kit (Perkin–Elmer) by the Biopolymer laboratory at the University of Maryland by using an automated 373A DNA sequencer (Applied Biosystems). Universal primers corresponding to T7 and T3 promoters were used when needed. Primers for PCR and sequencing were synthesized with an Applied Biosystems DNA synthesizer. Sequence contigs were aligned by using sequencher software version 3.0 (Genecodes, Ann Arbor, MI). Database comparisons were performed with the Basic Local Alignment Search Tool (blast) program (20). Computer analysis was performed by using the Genetics Computer Group (Madison, WI) package (GCG), version 8.0 (21).
Nucleotide Sequence Accession Numbers.
The nucleotide sequences described in this paper have been deposited in the GenBank database (accession no. AF034434).
RESULTS
Association of aldA and tagA with Epidemic and Pandemic V. cholerae.
The prevalence and variation of aldA and tagA in V. cholerae was studied in a large number of strains (pathogenic and nonpathogenic) that were temporally (years 1931–1994) and geographically widespread. PCR analysis with primers KAR3/KAR7 and KAR8/KAR9, and hybridization results with fragments obtained by using these primers, showed that aldA and tagA were associated with CT-positive epidemic and pandemic strains. aldA was found in 24/29 strains of the O1 serogroup including 6th and 7th pandemic isolates, isolates from the U.S. Gulf coast, and strains isolated during two pre-7th pandemic outbreaks (1937 Sulawesi, Indonesia, and 1954 Egypt). Sequence homologous to aldA and tagA was not be detected in 17 nontoxigenic environmental O1 strains, 3 clinical O1 strains including a CT-positive 1961 Indonesian strain with identical ribotype to 7th pandemic strains (13), or in 2 clinical CT-negative strains. aldA was present in 6 serogroup O139 CT-positive strains but not in 4 O139 CT-negative strains, and was identified in 2 non-O1/non-O139 strains. Interestingly, these two latter isolates were both CT-positive, and both strains were associated with explosive outbreaks of diarrhea in Czechoslovakia in 1965 (strain ATCC25872) and the Sudan in 1968 (strain S-21). Neither aldA or tagA were detected in the V. mimicus isolate tested. These results show that aldA and tagA are associated with epidemic and pandemic V. cholerae.
Examination of the aldA gene from 24 isolates showed their sequences were identical. Analysis of the first 1,686 nt of tagA showed near identity among the isolates except that the two non-O1/non-O139 CT-positive strains differed from all other strains at position 786 (nucleotide C → T), which did not alter the amino acid. In addition, at position 1,021 the 6th pandemic strains were unique in that they had a T giving a serine codon whereas 7th pandemic, O139 Bengal, U.S. Gulf coast, the 1937 El Tor strain from Sulawesi, and the two CT-positive non-O1/non-O139 strains had an A giving a threonine codon.
Association of aldA and tagA with the TCP Cluster.
The association of aldA and tagA with epidemic and pandemic strains led us to look in detail at the association between these genes and tcp genes that are associated with pathogenic V. cholerae (22). We first determined whether the tcpA gene, encoding the pilin subunit of the TCP, was present in aldA- and tagA-positive strains. PCR analysis of the tcpA gene with primers KAR24/KAR25 specific for 7th pandemic (El Tor biotype) strains and KAR24/KAR82 specific for 6th pandemic (Classical biotype) strains showed that the presence of tcpA correlated 100% with the presence of aldA and tagA. This finding led us to investigate whether the TCP cluster was located near aldA and tagA. A cosmid library of N16961 was hybridized with probes from aldA, tagA, and tcpA, and eight clones were identified that contained aldA, tagA, and tcpA sequences on the same fragment. PCR analysis of one of these cosmid clones (pDK8) and the wild-type strain N16961 with primers KAR24/KAR25 identified an expected 606-bp fragment, and subsequent hybridization with this fragment confirmed that the TCP cluster was near aldA and tagA. PCR analysis with primers KAR90/KAR91 [located at the 5′ end of tagA and the 5′ end of tagD (adjacent to tcpI)] on the cosmid pDK8, the 7th pandemic strain N16961, and the 6th pandemic strain 395 revealed that tagA was ≈9 kb from the left end of the TCP cluster.
Identification of the VPI and Sequencing of DNA.
Southern hybridizations of chromosomal DNA from a panel of V. cholerae strains with the 9-kb tagA–tagD PCR fragment obtained from N16961 revealed that this region was associated with epidemic and pandemic strains and absent from nontoxigenic environmental strains. Because earlier results showed that aldA and tagA were also only found in epidemic and pandemic V. cholerae, we speculated that their surrounding DNA and the TCP cluster formed part of a locus that had previously been reported to contain a putative integrase gene (int) and an adjacent att site (12). PCR analysis of N16961 with primers KAR22/KAR23 produced an expected 1.2-kb fragment corresponding to the int gene, whereas PCR of the adjacent DNA using KAR85/KAR86 produced a 0.6-kb fragment. Use of the 1.2-kb and 0.6-kb fragments as probes showed that int is associated with epidemic and pandemic strains and absent from nonpathogenic strains, whereas the adjacent 0.6 kb was found in all strains. This confirmed the results of Kovach et al. (12) showing that int is located at the distal end of a unique DNA region that includes the TCP gene cluster.
The DNA downstream of aldA was analyzed to identify the left end of the locus and thus fully define the extent of the unique region in epidemic and pandemic strains. Primers KAR92 (3′ end of aldA) and KAR93 (in the pHC79 cloning vector) yielded a PCR fragment of ≈12 kb from pDK8. Hybridization analysis showed that this fragment contained DNA common to both epidemic and pandemic strains, and that sequences on this fragment were also found in environmental strains. These results showed that the left junction of this unique locus, which appears to be a PAI was downstream of aldA. We propose to call this pathogenicity island VPI.
To characterize the VPI, pDK8 was digested with HindIII, and the resulting fragments were ligated into pBluescript+. DNA sequence for several clones was initially determined by using flanking vector sequences as universal primer binding sites followed by a walking strategy with forward and reverse primers designed from the ends of each sequencing run. Primers downstream of aldA were designed and used in PCRs with V. cholerae strains to identify the left end of the VPI. Primers KAR96/KAR97 generated a single fragment of the expected 0.8-kb size that was specific to epidemic and pandemic strains, whereas the adjacent fragment produced by primers KAR94/KAR95 gave a single fragment of 1.4 kb that was common to all strains tested. Hybridization with these fragments confirmed that we had identified the region containing the left end of the VPI and that there was ≈13 kb between the left junction and tagD.
Sequence Analysis of Novel DNA.
The VPI is shown in Fig. 1. The DNA sequence of the 13 kb between the left junction and tagD and the several kilobases flanking the left end of the VPI was compared with the GenBank database by using the blast algorithm (20). Within the VPI near the left junction, an ORF (ORF1) was identified with significant homology (58% identity over 109 aa) to the bfpM gene of enteropathogenic E. coli (EPEC) (GenBank accession no. U27184) (23). In EPEC this gene is located on a plasmid and is associated with a cluster of genes encoding the type IV bundle forming pilus (BFP) that is thought to be involved in the initial attachment of EPEC strains to eukaryotic host cells (23, 24). After four independent sequencing runs at this region (twice in each direction) we found that ORF1 (325 codons encoding a potential protein of 38.2 kDa) is considerably larger than the published EPEC bfpM gene (109 codons) (GenBank accession no. L07028) (23). As the relevant E. coli sequence downstream of bfpM is not in the database, we are unable to speculate whether these investigators have found only part of the E. coli gene. ORF1 also showed significant homology to several transposases found in other bacterial species such as the tnpA gene of Arthrobacter nicotinovorans (31% identity over 324 aa) (GenBank accession no. X97015), suggesting that ORF1 is a transposase. It is quite possible that the bfpM in EPEC functions as a transposase and is involved in the transfer of the BFP gene cluster. Adjacent to ORF1 and downstream of aldA is a region showing homology to a transposase (ISV-3L) found in Vibrio anguillarum (88% identity) (GenBank accession no. L40498) (25) and the transposase of Tn903 found in E. coli (48% identity) (GenBank accession no. I77546) (26). This transposase appears to be nonfunctional as there is one stop codon and a deletion of 30 aa in the central region compared with the transposase found in V. anguillarum.
Analysis of the DNA at the 3′ end of tagA revealed a discrepancy between the published sequence generated from the 6th pandemic strain 395 (16) and that of the 7th pandemic strain N16961 used in this study. Multiple sequencing runs of both DNA strands from the tagA gene in pDK8 (a cosmid clone of the 7th pandemic strain N16961) and confirmation of the tagA sequence from pDK29 (a cosmid clone of the 6th pandemic strain 395) showed that between nucleotides 1685 and 1692 (numbers as in the published sequence) there were seven T residues in contrast to the six reported for the published 395 tagA gene. This additional nucleotide alters the reading frame and dramatically increases the size of the putative encoded protein to a large gene of 1,002 aa (predicted molecular weight of 114.6 kDa) instead of 568 aa (64 kDa) previously reported for the predicted tagA protein from 395. The DNA between tagA and tagD contains three ORFs (including another very large ORF) with no obvious homology to any sequence in the database. These included ORF2, ORF3, and ORF4, which encode potential proteins of 379 aa (43.9 kDa), 1111 aa (126.4 kDa), and 285–332 aa (32.8–37.6 kDa, depending upon which start codon is used), respectively. ORF4 contains a motif associated with metalloproteases (27). The tagA gene, ORF2, ORF3, ORF4, and tagD are transcribed in the same direction with no obvious transcription terminator, suggesting they are an operon. tagD is adjacent to tcpI, the first gene of the TCP cluster (28). We found no compelling database homology in the 2 kb adjacent to the left junction of the VPI that was common to all strains.
Analysis of Left and Right Junction Sequences.
We examined the insertion site of the VPI in several epidemic and pandemic strains and found it was identical in all VPI-positive strains (Fig. 2). The insertion site of the left junction of the VPI was first investigated by using primers KAR94/KAR97. These PCR primers generated an expected 3-kb fragment in all VPI-positive strains tested (6th pandemic, 7th pandemic, U.S. Gulf, O139, the two non-O1/non-O139 strains, and a 1937 Sulawesi strain). The insertion site of the right junction was then studied by using primers KAR87 (outside the right junction in the ssrA gene) and KAR23 (within int). These primers generated an expected 1.6-kb fragment in all of the above strains. No fragments were produced in any of the eight nontoxigenic strains tested. These results show that the VPI is inserted into the identical chromosomal site in all VPI-positive strains tested. To determine whether the actual sequence at the insertion site was identical in all strains, we sequenced the left and right junctions contained in the PCR products of KAR94/KAR97 and KAR23/KAR87 obtained from the VPI-positive strains. Sequencing confirmed that the sequence at the left and right junction is identical among all VPI-positive strains (Fig. 3A).
Figure 2.
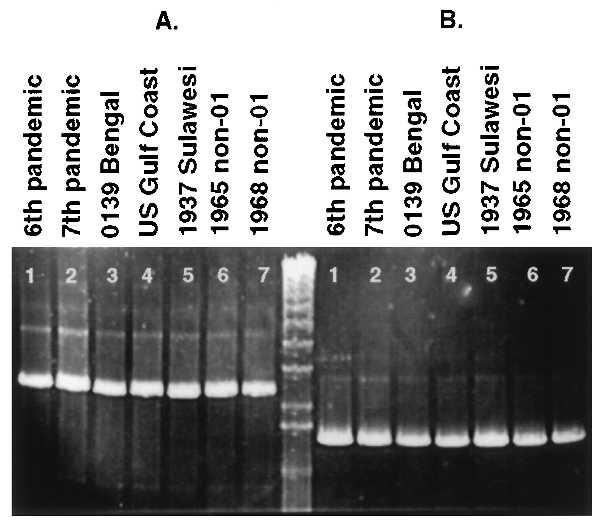
PCR analysis of the left and right junctions of the VPI. (A) Similar sized fragments containing the left junction for all strains with primers KAR94/KAR97. (B) Similar sized fragments containing the right junction for all strains with KAR23/KAR87. Lanes: 1, 6th pandemic strain 395; 2, 7th pandemic strain N16961; 3, O139 Bengal strain AI1837; 4, U.S. Gulf coast strain E506; 5, 1937 Sulawesi strain 66–2; 6, non-O1/non-O139 CT-positive strain ATCC25872 from Czechoslovakia; 7, non-O1/non-O139 CT-positive strain S-21 from Sudan; center lane, 1-kb marker.
Figure 3.

Sequence of the left and right junctions of the VPI and its insertion site into the V. cholerae chromosome. (A) The left and right junctions of VPI. (B) att site at the 3′ end of the ssrA gene in three VPI-negative strains. Bold text indicates att sites. Note that the absence of the adenine in the att site at the left junction of VPI-positive strains (A) is also found in the VPI-negative CT-positive strain E9120 (B). Differences in sequence flanking the att site in the VPI-negative strain DK63 are underlined (B).
We then compared the putative insertion site of VPI-positive and VPI-negative strains. By using primers KAR94/KAR87 that flank the left and right junctions, none of the VPI-positive strains yielded a PCR product. This result was expected because these strains contain 40 kb between the primers which is beyond the capabilities of PCR under the conditions employed. Strains lacking VPI would be expected to yield a 1.7-kb product when amplified with these primers but of eight VPI-negative strains analyzed, only three yielded the expected 1.7-kb fragment. However, all eight strains appear to have similar flanking regions because the ssrA gene (to the right of VPI) could be amplified by using primers KAR85/KAR86 and the region to the left of the VPI could be amplified by using KAR94/KAR95. This result suggests that there is sequence variation in the region corresponding to the 3′ end of primer KAR87 or there is another DNA fragment of considerable size inserted at this site in these strains. To investigate these possibilities, PCR was conducted with primers KAR94 and KAR86, both of which successfully amplified fragments from all eight strains when combined with other primers (see above). The KAR94/KAR86 primer pair amplified fragments only from those three strains which produced products with KAR94/KAR87 (data not shown). Southern blot analysis with the PCR product of KAR94/KAR87 from strain E92120 as a probe revealed distinct differences among the banding patterns of the strains (data not shown). These results suggest that another fragment may be inserted at this site and raise the possibility that the region of the V. cholerae chromosome containing VPI may be a “hot spot” for the insertion of other DNA elements.
The three VPI-negative strains that yielded the expected 1.7-kb PCR product with primers KAR94/KAR87 were further analyzed for the sequence around the att site. The sequence of the amplified products showed that all three strains possessed a single att site at the 3′ end of the ssrA gene. Alignment of these sequences with those of the left and right junctions of VPI-positive strains showed slight differences within and adjacent to their att site (Fig. 3B). The att sites of the left and right junctions of VPI-positive strains share 13 of 20 nt; a single nucleotide is also deleted from the left junction compared with the right junction (Fig. 3A). Two of the three VPI-negative strains contain att sequences identical to those found at the right junction of VPI-positive strains (Fig. 3B). The third VPI-negative strain contains an att site in which an adenine is deleted, similar to the deletion seen in the left junction of VPI-positive strains. Interestingly, this VPI-negative strain (E9120) is CT-positive, whereas the other two VPI-negative strains are CT-negative. Strain E9120 was isolated in Indonesia at the start of the 7th pandemic (1961) and has a ribotype identical to that of typical 7th pandemic strains. These results suggest that E9120 is an isolate of the 7th pandemic that has lost the VPI or is an isolate related to the ancestor of the 7th pandemic strain that has not yet acquired VPI. Because VPI contains the TCP cluster that is required for acquisition of CTXΦ, it is more likely that this strain once contained VPI and then lost it.
These results indicate that the insertion site and sequence at the left and right ends of the VPI is identical in all VPI-positive strains, although some sequence variation is found in the sequence adjacent to the att site in VPI-negative strains. The excision of VPI appears to be specific and does not disrupt adjacent DNA, suggesting that insertion of VPI involves a partial duplication of the att site with loss of the adenine and excision of VPI restores the single att site except for the adenine.
With the identification of att sites flanking this unique DNA locus and the newly obtained sequence, we calculated (by using the known TCP/ACF cluster sequence from strain 395; GenBank accession nos. X64098 and U39068) that VPI in the 7th pandemic El Tor O1 strain N16961 is 39.5 kb in size. In addition, we determined that the VPI has strikingly low %G+C content (35%) compared with the average genomic %G+C of V. cholerae (47–49%).
DISCUSSION
We report the identification, sequence, and analysis of V. cholerae pathogenicity island (VPI). We show that the VPI is clearly associated with epidemic and pandemic strains of V. cholerae. VPI contains the previously described V. cholerae virulence determinants ToxT, TCP, and ACF. In addition, we show that aldA and tagA, as well as several ORFs with homologies to sequences not previously known to occur in V. cholerae, are also located on the VPI.
Our initial finding that aldA sequences are identical among 6th pandemic, 7th pandemic, U.S. Gulf coast isolates, and serogroup O139 Bengal strains was in striking contrast to our results with the asd gene (14), which differed between 6th and 7th pandemic strains. The asd gene is found in all V. cholerae strains unlike the aldA gene, which is confined to epidemic and pandemic strains. The aldA sequence is identical among strains, whereas other genes on the VPI such as tcpA (29) and tagA (this study) vary, suggesting aldA may be a useful genetic marker for identifying potentially epidemic and pandemic strains.
We show that the VPI is 39.5 kb in size and includes 13 kb of previously unidentified DNA. We hypothesize that all the genes on the VPI are likely to be important in disease, either having a direct role in cholera pathogenesis or an indirect role in the transfer and mobility of the VPI, thereby creating the potential for the emergence of new epidemic and pandemic strains. The VPI contains genes such as tcpA that encodes an important colonization factor and the receptor for the CTXΦ; toxT, tcpP, and tcpH that encode regulators of virulence genes; genes that may be required for the transfer and integration of the VPI (ORF1 and int); and DNA of uncharacterized but potentially important function. The finding of two very large potential proteins encoded on the VPI (1,002 and 1,111 aa in size) is interesting as ORFs of this size have not been reported previously for V. cholerae and are not common in general. The VPI is flanked by att sites that presumably function as specific attachment sites between this element and the host bacterial chromosome. It appears that possession of the VPI has allowed specific strains of V. cholerae (which is normally an aquatic or estuarine water bacterium) to become adapted to the human intestinal environment and successfully colonize it. In addition, the VPI allows these strains to become toxigenic following infection by CTXΦ. The ability to colonize and secrete CT results in copious numbers of V. cholerae cells being excreted during diarrheal disease and would thereby allow for the organism’s continual survival in nature and its selective advantage over nonpathogenic strains. The identification of potential integrase and transposase genes at each end of the VPI suggests that these genes may have had a role in the transfer and integration of the VPI into epidemic and pandemic strains.
Recently, it has been found that genes involved in virulence, the presence of which distinguish pathogenic from nonpathogenic strains of a species, are often clustered into PAIs. These PAIs often insert near tRNA genes on the bacterial chromosome (30–32). PAIs have been found in bacterial pathogens including enteropathogenic E. coli (EPEC) (33), enterohemorrhagic E. coli O157:H7 (EHEC) (33), uropathogenic E. coli (31, 34, 35), Yersinia pestis (36), Salmonella typhimurium (37, 38), and Dichelobacter nodosus (39, 40). The V. cholerae PAI is 39.5 kb in size and is associated with epidemic and pandemic strains of the species. The VPI is similar to other PAIs in that (i) it contains clusters of known virulence genes (tcp and acf), including a regulator of virulence genes (toxT); (ii) it has both a putative integrase and transposase gene and is flanked by att sites; (iii) it has low %G+C relative to the overall genomic content; and (iv) it is inserted at a site adjacent to a tRNA-like gene (ssrA). Recently it has been shown that 10Sa RNA acts to provide a “termination” function for incomplete mRNAs without stop codons and is a widely present function in Gram-positive and Gram-negative bacteria (41, 42). Interestingly, ssrA is also the site of insertion of the PAI from D. nodosus (40). It has been suggested that PAIs could be transferred by transducing phages (43), and given the findings in this study, it is quite possible that the VPI is of phage origin. One may imagine that phages select these types of sites because they are widely present and probably strongly conserved in sequence. As the VPI contains no obvious excision genes it is possible that the VPI was originally a functional phage that was acquired by V. cholerae and then modified making it defective and unable to excise. This would provide a selective advantage to these strains over nonpathogenic strains in that they would permanently have the ability to colonize the human intestine, become toxigenic through the subsequent acquisition of CTXΦ, and thus maintain their numbers in nature.
Deletions of short DNA regions containing virulence factors have been reported for a few pathogens such as Haemophilus influenzae and Streptococcus pyogenes (44). However, prior to this study the only other reports of deletions of whole chromosomal PAIs from bacteria have been for Y. pestis (36) and uropathogenic E. coli (31). The absence of the VPI from a CT-positive O1 strain isolated in 1961 in Indonesia with a ribotype identical to 7th pandemic strains represents the first finding of a CT-positive pandemic strain that has apparently lost the VPI. Sequence analysis of the excision site in the VPI-negative CT-positive strain suggests that excision is specific and does not alter DNA adjacent to the att site; however, it apparently retains the sequence of the VPI left att site following excision. This situation differs from that found after excision of the PAI in uropathogenic E. coli where excision results in deletion of part of the adjacent tRNA gene (31).
It is noteworthy that the VPI is found in two non-O1/non-O139 CT-positive strains. One of these strains caused an explosive outbreak of diarrheal disease in Czechoslovakia in 1965 (3), whereas the other caused an outbreak in the Sudan in 1968 (45). The O139 Bengal strain, which has recently emerged and caused large epidemics of cholera, is at present the best known example showing that a strain of a non-O1 serogroup can cause epidemic disease. Our finding of epidemiologically unassociated VPI- and CT-positive non-O1/non-O139 strains that were isolated from different continents in different years highlights the importance of horizontal transfer of gene clusters in the emergence and creation of pathogenic organisms. It is quite possible that previous and future epidemics of cholera may have been and could be caused by non-O1/non-O139 strains if they acquire VPI and CTXΦ.
The VPI clearly is a requirement for epidemic and pandemic cholera, the emergence of which appears to be a multistep process. Prior to a strain becoming toxigenic via infection with the CTXΦ, the TCP receptor encoded on the VPI is required. Thus the VPI may be the initial and essential genetic factor required for epidemic and pandemic disease. The results in this study identify the complete VPI and show the existence of potential virulence genes that were previously unknown in V. cholerae. As the VPI is one of the requirements for epidemic and pandemic ability and appears to be able to undergo horizontal transfer, our study provides important insights into the emergence and pathogenesis of epidemic and pandemic cholera.
Acknowledgments
We are grateful to Lisa Sadzewicz of the Biopolymer Core Facility, University of Maryland, for excellent technical assistance in the sequencing component of this study. This work was supported by grants from the Public Health Service (AI35729) and the Department of Veterans Affairs (to J.A.J.), the National Institutes of Health (AI19716 to J.B.K.), and the Australian Research Council (A1913857 to P.R.R.).
ABBREVIATIONS
- CT
cholera toxin
- TCP
toxin-coregulated pilus
- ACF
accessory colonization factor
- PAI
pathogenicity island
- VPI
V. cholerae pathogenicity island
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF034434).
References
- 1.Shimada T, Arakawa E, Itoh K, Okitsu T, Matsushima A, Asai Y, Yamai S, Nakazoto T, Nair G B, Albert M J, Takeda Y. Curr Microbiol. 1994;28:175–178. [Google Scholar]
- 2.Albert M J, Siddique A K, Islam M S, Faruque A S G, Ansaruzzaman M, Faruque S M, Sack R B. Lancet. 1993;341:704. doi: 10.1016/0140-6736(93)90481-u. [DOI] [PubMed] [Google Scholar]
- 3.Aldova E, Laznickova K, Stepankova E, Lietava J. J Infect Dis. 1968;118:25–31. doi: 10.1093/infdis/118.1.25. [DOI] [PubMed] [Google Scholar]
- 4.Morris Jr J M. In: Vibrio cholerae and Cholera: Molecular to Global Perspectives. Wachsmuth I K, Blake P A, Ølsvik Ø, editors. Washington, DC: Am. Soc. for Microbiol.; 1994. pp. 103–115. [Google Scholar]
- 5.Waldor M K, Mekalanos J J. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 6.Taylor R K, Miller V L, Furlong D B, Mekalanos J J. Proc Natl Acad Sci USA. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrington D A, Hall R H, Losonsky G A, Mekalanos J J, Taylor R K, Levine M M. J Exp Med. 1988;168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiRita V J, Parsot C, Jander G, Mekalanos J J. Proc Natl Acad Sci USA. 1991;88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiRita V J. Mol Microbiol. 1992;6:451–458. doi: 10.1111/j.1365-2958.1992.tb01489.x. [DOI] [PubMed] [Google Scholar]
- 10.Carroll P A, Tashima K T, Rogers M B, DiRita V J, Calderwood S B. Mol Microbiol. 1997;25:1099–1111. doi: 10.1046/j.1365-2958.1997.5371901.x. [DOI] [PubMed] [Google Scholar]
- 11.Brown R C, Taylor R K. Mol Microbiol. 1995;16:425–439. doi: 10.1111/j.1365-2958.1995.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 12.Kovach M E, Shaffer M D, Peterson K M. Microbiology. 1996;142:2165–2174. doi: 10.1099/13500872-142-8-2165. [DOI] [PubMed] [Google Scholar]
- 13.Karaolis D K R, Lan R, Reeves P R. J Bacteriol. 1994;176:6199–6206. doi: 10.1128/jb.176.20.6199-6206.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karaolis D K R, Lan R, Reeves P R. J Bacteriol. 1995;177:3191–3198. doi: 10.1128/jb.177.11.3191-3198.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsot C, Mekalanos J J. J Bacteriol. 1991;173:2842–2851. doi: 10.1128/jb.173.9.2842-2851.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harkey C W, Everiss K D, Peterson K M. Gene. 1995;153:81–84. doi: 10.1016/0378-1119(94)00811-6. [DOI] [PubMed] [Google Scholar]
- 17.Hohn B, Collins J. Gene. 1980;11:291–298. doi: 10.1016/0378-1119(80)90069-4. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 19.Saiki R K, Gelfand D H, Stofell S, Scharf S J, Higuchi R, Horn G T, Mullis K B, Erlich H A. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 20.Altschul A F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor R K, Shaw C, Peterson K M, Spears P, Mekalanos J J. Vaccine. 1988;6:151–154. doi: 10.1016/s0264-410x(88)80019-7. [DOI] [PubMed] [Google Scholar]
- 23.Sohel I, Puente J L, Ramer S W, Bieber D, Wu C-Y, Schoolnik G K. J Bacteriol. 1996;178:2613–2628. doi: 10.1128/jb.178.9.2613-2628.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giron J A, Ho A S Y, Schoolnik G K. Science. 1991;254:710–713. doi: 10.1126/science.1683004. [DOI] [PubMed] [Google Scholar]
- 25.Tolmasky M E, Crosa J H. Plasmid. 1995;33:180–190. doi: 10.1006/plas.1995.1019. [DOI] [PubMed] [Google Scholar]
- 26.Grindley N D, Joyce C M. Proc Natl Acad Sci USA. 1980;77:7176–7180. doi: 10.1073/pnas.77.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jongeneel C V, Bouvier J, Bairoch A. FEBS Lett. 1989;242:211–214. doi: 10.1016/0014-5793(89)80471-5. [DOI] [PubMed] [Google Scholar]
- 28.Harkey C W, Everiss K D, Peterson K M. Infect Immun. 1994;62:2669–2678. doi: 10.1128/iai.62.7.2669-2678.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhine J A, Taylor R K. Mol Microbiol. 1994;13:1013–1020. doi: 10.1111/j.1365-2958.1994.tb00492.x. [DOI] [PubMed] [Google Scholar]
- 30.Hacker J, Bender L, Ott M, Wingender J, Lund B, Marre R, Goebel W. Microb Pathog. 1990;8:213–225. doi: 10.1016/0882-4010(90)90048-u. [DOI] [PubMed] [Google Scholar]
- 31.Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Hacker J. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Mol Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- 33.McDaniel T K, Jarvis K G, Donnenberg M S, Kaper J B. Proc Natl Acad Sci USA. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swenson D L, Bukanov N O, Berg D E, Welch R A. Infect Immun. 1996;64:3736–3743. doi: 10.1128/iai.64.9.3736-3743.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kao J-S, Stucker D M, Warren J W, Mobley H L T. Infect Immun. 1997;65:2812–2820. doi: 10.1128/iai.65.7.2812-2820.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fetherston J D, Schuetze P, Perry R D. Mol Microbiol. 1992;6:2693–2704. doi: 10.1111/j.1365-2958.1992.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 37.Mills D M, Bajaj V, Lee C A. Mol Microbiol. 1995;15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 38.Shea J E, Hensel M, Gleeson C, Holden D W. Proc Natl Acad Sci USA. 1996;93:2593–2597. doi: 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katz M E, Strugnell R A, Rood J I. Infect Immun. 1992;60:4586–4592. doi: 10.1128/iai.60.11.4586-4592.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheetham B F, Tattersall D B, Bloomfield G A, Rood J I, Kaetz M E. Gene. 1995;162:53–58. doi: 10.1016/0378-1119(95)00315-w. [DOI] [PubMed] [Google Scholar]
- 41.Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. Proc Natl Acad Sci USA. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keiler K C, Waller P R H, Sauer R T. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 43.Cheetham B F, Kaetz M E. Mol Microbiol. 1995;18:201–208. doi: 10.1111/j.1365-2958.1995.mmi_18020201.x. [DOI] [PubMed] [Google Scholar]
- 44.Ott M. Zentralbl Bakteriol. 1993;278:457–468. doi: 10.1016/s0934-8840(11)80817-0. [DOI] [PubMed] [Google Scholar]
- 45.Zinnaka Y, Carpenter C C J., Jr Hopkins Med J. 1972;131:403–411. [PubMed] [Google Scholar]
- 46.Hughes K J, Everiss K D, Harkey C W, Peterson K M. Gene. 1994;148:97–100. doi: 10.1016/0378-1119(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 47.Levine M M, Black R E, Clements M L, Nalin D R, Cisneros L, Finkelstein R A. In: Acute Enteric Infections in Children: New Prospects for Treatment and Prevention. Holme T, Holmgren J, Merson M H, Möllby R, editors. Amsterdam: Elsevier-North-Holland; 1981. [Google Scholar]