

Abstract
Deciphering what governs inflammation and its effects on tissues is vital for understanding many pathologies. The recent discovery that glycogen synthase kinase-3 (GSK3) promotes inflammation reveals a new component of its well-documented actions in several prevalent diseases which involve inflammation, including mood disorders, Alzheimer’s disease, diabetes, and cancer. Involvement in such disparate conditions stems from the widespread influences of GSK3 on many cellular functions, with this review focusing on its regulation of inflammatory processes. GSK3 promotes the production of inflammatory molecules and cell migration, which together make GSK3 a powerful regulator of inflammation, while GSK3 inhibition provides protection from inflammatory conditions in animal models. The involvement of GSK3 and inflammation in these diseases are highlighted. Thus, GSK3 may contribute not only to primary pathologies in these diseases, but also to the associated inflammation, suggesting that GSK3 inhibitors may have multiple effects influencing these conditions.
Keywords: Glycogen synthase kinase-3, Bipolar disorder, Alzheimer’s disease, Diabetes, Cancer, Inflammation, Cell migration, Lithium
Introduction
Few things are more exciting, rewarding, and difficult, than identifying new interactions between previously unrelated fields. Nearly 40 years ago Professor John Blass embarked on a research career aimed at discovering if metabolic dysregulation contributed to the pathophysiology of neurological and psychiatric diseases. Not only did he directly contribute to our current appreciation of these links, but he provided the stimulus for his collaborators and students to follow this direction, as is well-documented in this issue of the journal honoring his achievements. The once revolutionary idea that alterations in metabolic enzymes make a significant contribution to the pathophysiology of neurological and psychiatric diseases is now in the mainstream of neuroscience research. One of the more surprising metabolic enzymes that has been implicated in the pathophysiology of multiple diseases is the formerly inconspicuous enzyme, glycogen synthase kinase-3 (GSK3), a multi-tasking serine/threonine kinase now known to regulate such critical cellular functions as structure, gene expression, mobility, and apoptosis.
Among the diverse functions that are regulated by GSK3, inflammation has recently emerged as one of the most interesting. In this review, we address two major topics that are linked by GSK3. The first is the recent discovery that GSK3 is an important positive regulator of the inflammatory process. The second major topic addressed here is the surprisingly large number of prevalent diseases which share the common feature of chronic inflammation in which very strong connections have been established between GSK3 and pathophysiology and/or therapeutics. These include psychiatric diseases (with a focus on bipolar mood disorder), neurodegenerative diseases [with a focus on Alzheimer’s disease (AD)], diabetes, and cancer. Within these topics, we raise two concepts, that inhibitors of GSK3 may provide an important therapeutic strategy to control inflammation, and that some of the already established associations between disease pathophysiology and GSK3 may be related to, or extended to, the inflammation-promoting effects of GSK3 and thus the anti-inflammatory effects of GSK3 inhibitors.
Regulation of GSK3
To adequately appreciate GSK3, first the initial impression conveyed by its name must be overcome, and second, one must not be overwhelmed by the large number of substrates that it phosphorylates (over 50 known to date) which connects it to a great many cellular processes. GSK3 was named based on its initial identification as a kinase that phosphorylates the glycogen-synthesizing enzyme, glycogen synthase [1, 2]. For many years GSK3 was relegated to this backwater area of glycogen metabolism. Interest began growing when GSK3 was found to be a key member of the insulin and Wnt signaling pathways. This interest was amplified following implications that GSK3 contributed to the neuropathology of AD by regulating phosphorylation of the microtubule-associated protein, tau, that is the predominant component of neurofibrillary tangles in AD. More recently it has become evident that GSK3 is a crucial, and often central, component of many cellular functions, contributing to the regulation of apoptosis, cell cycle, cell polarity and migration, gene expression, and many other functions. This multi-tasking is achieved by the many substrates phosphorylated by GSK3 and the convergence on GSK3 of many regulatory intracellular signaling pathways.
The many functions regulated by GSK3 (through its phosphorylation of numerous substrates) suggests that the activity of GSK3 must be highly regulated. Four key mechanisms have been identified that contribute to regulating the actions of GSK3 in a substrate-specific manner [3]. These include regulation by phosphorylation of GSK3 itself, the subcellular localization of GSK3, the formation of protein complexes containing GSK3, and the phosphorylation state of GSK3 substrates, as depicted in Fig. 1. The most well-defined regulatory mechanism is inhibition of the activity of GSK3 by phosphorylation of a regulatory serine in either of the two isoforms of GSK3, serine-9 in GSK3β or serine-21 in GSK3α [2]. The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway activated in response to insulin and many other growth factors often is a major regulator of GSK3 because Akt phosphorylates GSK3 on these inhibitory serine residues, but several other kinases also can phosphorylate these regulatory serines, such as protein kinase C and protein kinase A [4]. Conversely, the enzymatic activity of GSK3 is enhanced by phosphorylation of tyrosine-216 in GSK3β and tyrosine-279 in GSK3α, but the mechanisms regulating this modification are not well-defined. The actions of GSK3 are also regulated by control of its intracellular localization, as it is dynamically regulated within the nucleus and mitochondria, where it is highly active [5], and in other subcellular regions to selectively control its actions in these compartments. For example, GSK3 is locally inhibited in growth cones [6], whereas conversely, the activity of GSK3 to phosphorylate MAP1B is reported to be spatially restricted to growing axons [7]. Substrate-specific regulation of GSK3 is also achieved by positioning it within protein complexes that direct, or inhibit, its actions toward specific substrates. For example, in the canonical Wnt signaling pathway, GSK3 and the transcriptional co-activator β-catenin are colocalized on the scaffold protein Axin which directs GSK3 to phosphorylate β-catenin, targeting β-catenin for degradation. Activation of Wnt signaling removes GSK3 from accessing β-catenin, causing accumulation of active β-catenin. Finally, the action of GSK3 is usually regulated by the phosphorylation state of its substrate, an indirect mechanism that can regulate how efficiently GSK3 phosphorylates a substrate. This is because most substrates of GSK3 must be “primed,” i.e., prephosphorylated at a residue 4-amino acids C-terminal to the GSK3 phosphorylation site. This necessitates temporal coordination of the activity of the priming kinase along with GSK3 activity for the latter to phosphorylate the primed substrate. These complexities in the mechanisms controlling the actions of GSK3 provide substrate-specific control of its actions, an especially important capacity for an enzyme that can phosphorylate so many substrates that regulate numerous cellular functions.
Fig. 1.
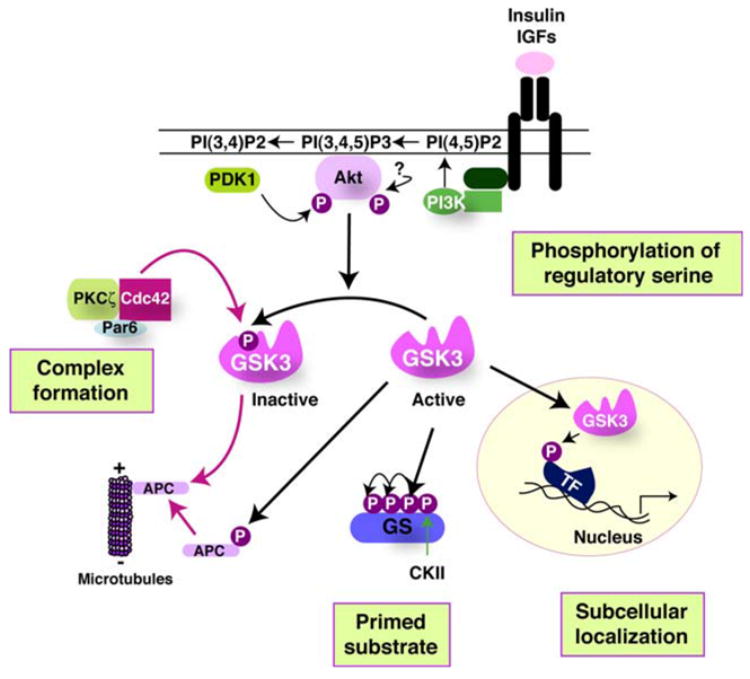
Mechanisms that regulate the actions of GSK3. Four mechanisms act in concert to regulate the phosphorylation of substrates by GSK3. (1) Primed substrate—substrate phosphorylation by GSK3 is limited by the activity of a priming kinase which prepares the substrate for GSK3. This is because GSK3 most often phosphorylates primed substrates that are prephosphorylated four residues C-terminal to the GSK3 phosphorylation site. The example shows the substrate, glycogen synthase (GS), primed by casein kinase II (CKII) followed by sequential phosphorylation by GSK3 at multiple sites spaced four residues apart toward the N-terminal end of GS. (2) Phosphorylation of regulatory serine—a major mechanism for inhibiting the activity of GSK3 is by serine-phosphorylation, so activity is inhibited when serine-9 of GSK3β or serine-21 of GSK3α is phosphorylated. Conversely, the activity of GSK3 is optimal when phosphorylated on tyrosine-216 of GSK3β or tyrosine-279 of GSK3α (not shown). Many kinases are capable of phosphorylating the regulatory serines of GSK3α/β. The example shows this being carried out by Akt (also known as protein kinase B) which itself is activated by phosphorylation on two sites, one mediated by phosphoinositide-dependent kinase-1 (PDK1) and the other by an unidentified kinase. Akt activation follows stimulation-induced activation of PI3K, and its catalysis of the formation of 3′-phosphoinositides, by receptors for insulin, insulin-like growth factors (IGFs), and other receptor sub-types. When the substrate is prephosphorylated and GSK3 is active, with the regulatory serine dephosphorylated, two spatial restrictions also contribute to regulating the actions of GSK3, its subcellular localization and its association with other proteins in regulatory complexes. (3) Subcellular localization—GSK3 is considered to be largely a cytosolic enzyme, but it is also associated with, or internalized in, subcellular compartments such as the nucleus, mitochondria, and growth cones, so dynamic regulation of the subcellular localization of GSK3 can regulate its access to substrates within subcellular compartments. The example depicted shows GSK3 transport into the nucleus where it can phosphorylate a variety of substrates, including several transcription factors (TF). (4) Complex formation—in addition to this gross cellular distribution of GSK3, its distribution in the cell is constrained by its propensity to be associated in protein complexes which provides an important mechanism for regulating its phosphorylation of specific substrates that are colocalized in such complexes. The example shows that a complex of cdc42, Par6, and protein kinase C-ζ (PKCζ) binds GSK3β and catalyzes the phosphorylation of serine-9 to inhibit GSK3β to regulate the phosphorylation of APC and thereby control the association of APC with the plus-end of microtubules. Thus, substrate-specific regulation of phosphorylation by GSK3 is achieved by regulation of the priming kinase activity, phosphorylation of GSK3, the subcellular localization of GSK3, and assembly of GSK3 in protein complexes
Inflammation and migration: regulation by GSK3
A new cellular function regulated by GSK3 was identified by recent findings showing that GSK3 is a vital factor in the inflammation process [8]. Consequently, inhibitors of GSK3 provide strong anti-inflammatory protection. Since inflammation is an important component of many prevalent diseases, including mood disorders, neurodegenerative diseases, diabetes, and cancer (Fig. 2), this close association between GSK3 and inflammation provides a new link between GSK3 and these conditions, an association that should be considered in evaluating the therapeutic potentials of GSK3 inhibitors.
Fig. 2.
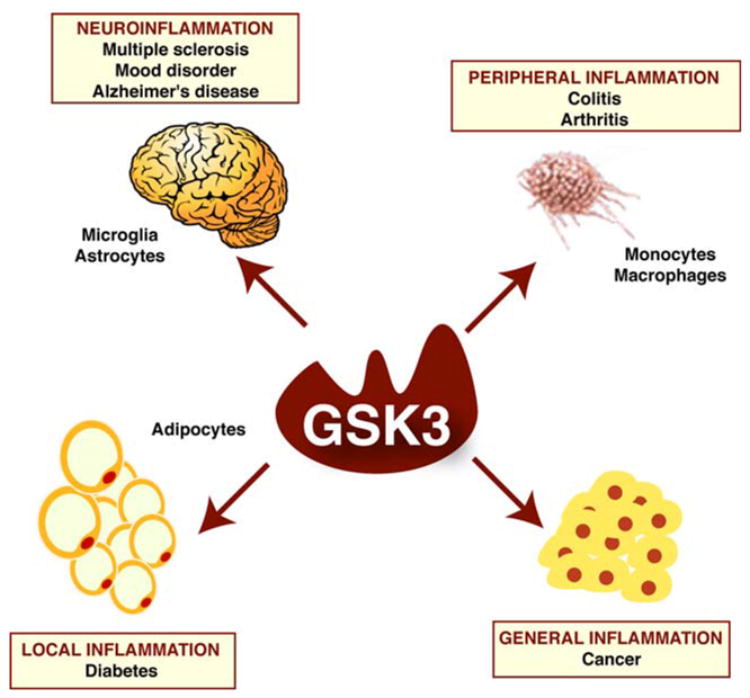
GSK3 may be an important component of multiple inflammatory diseases. GSK3 can influence inflammation throughout the body which may influence many inflammation-associated conditions. Neuroinflammation within the CNS is mediated by microglia and astrocytes, as well as infiltrating cells, which likely contributes to a wide variety of diseases, such as multiple sclerosis, mood disorders, and neurodegenerative disorders, including Alzheimer’s disease (AD). Peripheral inflammation is mediated in part by monocytes and macrophages, and GSK3 promotes inflammatory conditions such as colitis and arthritis. Localized inflammation, such as cytokine production by adipocytes, apparently contributes to the development of diabetic conditions which may be exacerbated by active GSK3. Inflammation also contributes to numerous types of cancer and inhibition of GSK3 exacerbates tumorigenesis
Inflammation is a crucial function of the mammalian immune system involving many types of cells and utilizing many signaling mediators to provide protection from pathogens. Responses of the immune system are classified as adaptive or innate based on the activation timing and antigen. The first line of defense against pathogens is provided by innate immune cells which act as sentinels to detect signs of distress, which includes cells such as macrophages, dendritic cells, neutrophils, and others. When activated, sentinel macrophages and other cells release soluble mediators of inflammation, such as cytokines and chemokines, to promote the infiltration of damaged tissue by other immune cells. Adaptive immunity, on the other hand, is a slower response dependent on T-cells and B-cells reacting with antigen-presenting cells. Thus together, the innate and adaptive immune systems constitute the process of inflammation. Dysregulation of either immune system can contribute to a variety of pathological conditions.
In contrast to other organs, the brain does not display a classical immune response, so for many years it was considered to be immune privileged. This view has changed as much evidence has shown that the brain is capable of mounting its own immune and inflammatory responses. The blood brain barrier serves to separate the brain’s local inflammatory responses from systemic inflammation by limiting the penetration of peripheral inflammatory mediators and only allowing the penetration of leukocytes when appropriate signals are received [9, 10]. This unique protection from systemic inflammation is thought to be necessary because neurons are post-mitotic cells unable to divide and with limited capacity to recover from injury. As a result, autodestructive immune processes could have devastating consequences for the brain even though they may have little effect on other organs.
When this protective barrier is disrupted and/or inflammation invades the brain, there is infiltration of circulating immune cells, such as macrophages and monocytes, and activation of resident glia cells, including astrocytes and microglia. Microglia are generally considered equivalent to macrophages in the periphery and are thought to be the key immune cell of the central nervous system (CNS). A variety of stressors activate glia in the CNS, such as ischemia, trauma, infection, and many neurodegenerative diseases, leading to two major responsive actions of glia, an inflammatory response and migration. Acutely, these glial responses are thought to be beneficial because they allow glia to migrate to afflicted sites where they produce cytokines and growth factors that can contribute to the recovery of affected neurons. Conversely, a long-term inflammatory response mediated by chronically activated glia appears to be detrimental to neuronal function and survival. However, these generalizations remain controversial and variable depending on the cytokines, cell-types, and inflammatory conditions that are involved, as discussed in detail in many excellent reviews [11–16]. There is no doubt, however, that inflammation is linked to many diseases of the nervous system [17–19].
Therefore, it is of interest that GSK3 has been identified as a strong regulator of both inflammation and cell migration. These effects raise the possibilities that GSK3 may contribute to the long-term detrimental effects of unchecked inflammation both in the periphery and in the CNS and that conditions that involve chronic inflammation may be ameliorated by therapeutic application of inhibitors of GSK3.
The recent identification of GSK3 as a major regulator of peripheral inflammatory responses showed that GSK3 promotes the stimulus-induced production of several cytokines and the subsequent development of disease symptoms in animal models of inflammatory conditions. This role of GSK3 in inflammation was first established by our report that GSK3 activity is necessary for full stimulation of the production of several pro-inflammatory cytokines, such as interleukin-6 (IL-6), IL-1β, and tumor necrosis factor (TNF), following stimulation of several types of Toll-like receptors in monocytes and peripheral blood mononuclear cells [8]. Conversely, GSK3 reduced the production of the anti-inflammatory cytokine IL-10. Reciprocally, inhibitors of GSK3 greatly reduced the production of pro-inflammatory cytokines and increased anti-inflammatory cytokine production. This raised the novel possibility that inhibitors of GSK3 may prove to be beneficial in many conditions involving inflammation. Remarkably, in vivo administration of GSK3 inhibitors provided protection from endotoxin shock sufficiently enough to allow survival of most mice from an otherwise lethal (LD100) dose of lipopolysaccharide [8]. This study showed for the first time the powerful ability of GSK3 inhibitors to shift the balance of the inflammatory response from pro-inflammatory to anti-inflammatory, and revealed the therapeutic potential for these drugs in inflammatory conditions [8].
The reduced Toll-like receptor-induced production of inflammatory cytokines attained by inhibiting GSK3 was shown to be due to inhibition of the transcriptional activity of nuclear factor (NF)-κB [8]. This supported the previous conclusion that GSK3 is necessary for the full transcriptional activity of NF-κB [20]. Interestingly, GSK3 was recently shown to support NF-κB transcriptional activity in a promoter-specific manner, demonstrating that GSK3 selectively supports the expression of a subset of genes activated by NF-κB [21]. For example, NF-κB-mediated expression of IL-6 and monocyte chemoattractant protein-1 required GSK3β for efficient expression, but IκBα and macrophage inflammatory protein-2 did not [21]. This selective action of GSK3β on NF-κB-induced gene expression will likely facilitate the therapeutic anti-inflammatory uses of GSK3 inhibitors since it indicates that inhibition of GSK3 will not interfere with all the actions of NF-κB.
During the last year, knowledge about the crucial involvement of GSK3 in inflammatory responses has been expanded to other conditions. GSK3 inhibitors were reported to reduce the systemic inflammatory response, renal dysfunction, and hepatotoxicity associated with endotoxemia in rats [22]. In other inflammatory conditions, GSK3 inhibitors diminished inflammatory responses in experimentally induced colitis in rats [23] and in arthritis caused by administration of type II collagen in mice [24]. Arthritis and peritonitis in mice were also reduced by in vivo administration of the GSK3 inhibitor lithium [25]. Altogether, it is now evident that GSK3 is an important component of the inflammatory response and that the administration of GSK3 inhibitors can have therapeutic effects in a number of conditions associated with inflammation. Application of this therapeutic strategy may prove beneficial in the treatment of additional chronic diseases that have an associated inflammatory component, potentially including conditions involving inflammation in the CNS, as discussed later in this paper.
Besides producing molecules associated with the inflammatory response, a closely linked important characteristic of inflammatory cells is their capacity to migrate. Migration is a prominent component of immune surveillance, for example, leukocytes in the circulation migrate to sites of infection and tissue damage, as do glia cells in the CNS. Cell migration is not only critical in immune responses, but also during development, for tissue maintenance and repair, as well as during pathological processes such as chronic inflammation, vascular disease, and metastasis invasion. Cell migration has been conceptualized as a cyclic process of cell movement initiated by polarization of a cell [26]. This is followed by polymerization of actin to support protrusions of cells in the direction of migration. This “leading edge” of a migrating cell is stabilized by the formation of adhesions to neighboring cells or to the extracellular matrix which allows the cell to move forward. Concurrently, adhesions at the rear, or “trailing edge,” of the cell disassemble to allow the cell to detach and move forward.
Each step in cell migration is regulated by many intracellular signals, one of which is GSK3. The actions of GSK3 on migration, from the initiation of polarization and the extension of processes to the modulation of adhesions, have been studied in a variety of different cell-types. The most interesting concept that has been established by many of these studies is that GSK3 is regulated very locally within in cells at critical sites in the migratory process. At the leading edge of migrating cells, GSK3 is contained in a protein complex with PKCζ and Par6. Cdc42, a small GTPase which is a key regulator of polarity and directional movement [27], inactivates GSK3 specifically in this complex by stimulating PKCζ-induced inhibitory serine-phosphorylation of GSK3. This inhibition of GSK3 prevents it from phosphorylating adenomatous polyposis coli (APC) protein which allows APC to associate with the plus-ends of microtubules to establish cell polarity and control the direction of cell protrusion [28]. Localized inhibition of GSK3 also occurs in growth cones [6]. PKCζ-induced inhibition of GSK3 has also been shown to abolish random motility of endothelial cells [29]. These studies clearly document that local inhibition of GSK3 promotes migration. Conversely, activation of GSK3 causes growth cone collapse in response to semaphorin stimulation in neurites [6], an effect likely related to phosphorylation by GSK3 of the microtubule-associated proteins CRMP2 [30, 31] and MAP1B [32], indicating that in contrast to local inhibition, local activation of GSK3 blocks the migratory process by controlling microtubule networks. As opposed to local regulation, inhibition of total cellular GSK3 prevented migration of epithelial cells [33] and extension of lamellipodia in keratinocytes [34] and filopodia in neurons [32]. Thus, a selectively inactivated compartment of GSK3 is spatially localized at protrusions and the leading edge of migrating cells to allow movement, but complete inhibition of cellular GSK3 inhibits these events.
Glycogen synthase kinase-3 also plays a role in regulating focal adhesions, or sites of integrin clustering. These structures are important for regulating cell migration since adhesions assemble at the leading edge of migrating cells while disassembling at the trailing edge. Focal adhesion kinase (FAK) plays an important role in regulating protein–protein interactions at sites of cell attachment and also transmits adhesion-regulated signals intracellularly. Thus, FAK phosphorylates adhesion-associated proteins, such as paxillin, to disassemble adhesions and promote migration [35]. GSK3 directly phosphorylates FAK, reducing its activity and consequently impeding cell spreading and migration [36]. Reciprocally, FAK inhibits GSK3 by promoting its serine-phosphorylation which may allow FAK to escape the inhibitory phosphorylation by GSK3 in some situations [37]. In contrast, other evidence indicates that GSK3 is able to promote cell spreading in some situations, for example, by phosphorylating paxillin [38]. GSK3 also binds the adhesion-regulating protein h-prune, such that GSK3 and h-prune cooperatively regulate the disassembly of focal adhesions to promote cell migration [39]. Presumably, these contradictory effects of GSK3 on adhesion are segregated spatially or temporally, or different mechanisms are utilized in a cell-type specific manner to regulate focal adhesions.
Glycogen synthase kinase-3 also can regulate cell adhesion and migration by regulating the degradation of β-catenin in the Wnt pathway. Increasing β-catenin levels by using a selective GSK3 inhibitor or treatment with Wnt-1 inhibited neural crest cell spreading and migration [40]. Activation of the CXCR4 chemokine receptor was also reported to increase β-catenin levels and regulate cell migration [41]. These effects likely depend on the role of β-catenin in adhesion junctions where it links cadherin to α -catenin, because α -catenin associates with the intracellular actin cytoskeleton to make a crucial contribution in the organization of actin at the adherens junction [42]. The integrity of this core protein complex is critical for the formation and maintenance of stable adhesions. Recent evidence, which remains somewhat controversial, indicates that there is cross-talk between the levels of β-catenin in the Wnt pathway with the more stable pools of β-catenin in adhesion complexes [43–46], findings which, with further clarification, may indicate that GSK3 has further influences on adhesion and migration through its regulation of β-catenin.
In conclusion, the roles of GSK3 in cell migration are diverse, and at times conflicting findings have been reported, perhaps in part because these studies used a variety of cell-types expressing different receptors and utilizing a variety of intracellular signaling mechanisms. Despite these differences, it is now evident that GSK3 plays many important roles in cell migration, from initiation to termination. These studies also indicate that spatial and temporal regulation are critical in specifying the actions of GSK3. Thus, GSK3 has strong regulatory effects on both inflammation and migration, actions that are likely to be important in inflammation-associated diseases and which may indicate that targeting GSK3 pharmacologically could provide a therapeutic mechanism to control inflammation and migration.
Psychiatric diseases: GSK3 and mood disorders
Lithium has been used therapeutically for 50 years as a mood stabilizer in patients with bipolar mood disorder, previously called manic-depression, but its therapeutic mechanism of action was unknown. However, in 1996 lithium was discovered to be a selective inhibitor of GSK3 and evidence is growing that this may be the basis for lithium’s mood stabilizing effects in bipolar disorder [47]. Thus, it is interesting to note that humans were unknowingly treated with a GSK3 inhibitor, lithium, for many years.
Lithium binds to GSK3 to directly inhibit its activity, acting as a competitive inhibitor with respect to magnesium [48]. However, at a therapeutic concentration of lithium, which is approximately 1 mM, GSK3 is only moderately inhibited because lithium’s Ki for inhibition of GSK3 is approximately 2 mM. However, in addition to this direct inhibitory action of lithium on GSK3, treatment with lithium of mice, cultured cells, or human blood cells increases the inhibitory serine-phosphorylation of GSK3 [49, 50; reviewed in Ref. 51]. This serine-phosphorylation of GSK3 following its initial direct inhibition by lithium appears to be mediated by a feedback loop involving regulation of protein phosphatase-1 [52] or by increased Akt activity following lithium treatment [53]. This phosphorylation-mediated amplification mechanism allows the low (approximately 1 mM) concentrations of lithium that are achieved in patients undergoing lithium therapy to reduce the activity of GSK3 to a greater extent than would be possible based on only the direct inhibitory action of lithium. Thus, the current concept is that lithium-induced serine-phosphorylation of GSK3 amplifies the direct inhibitory effect of lithium on GSK3 to allow therapeutic levels of lithium to moderately inhibit GSK3. However, since complete inhibition of GSK3 would have widespread consequences on many cellular functions [3], even with both mechanisms of inhibition acting together a therapeutic level of lithium does not completely inhibit GSK3 but only reduces its maximal activity.
Although much has been learned about lithium’s inhibitory effect on GSK3 and the cellular actions of GSK3 during the last 10 years, many questions remain about the relationship between lithium’s inhibition of GSK3 and the mood stabilizing action of lithium in the treatment of bipolar disorder. First, it is not known which action of GSK3 that is inhibited by lithium is critical for lithium’s therapeutic mood stabilization. Second, until other GSK3 inhibitors are tested as therapeutic agents, it cannot be concluded unequivocally that GSK3 is the therapeutic target of lithium. Third, it is unknown what aberrations linked to GSK3 occur in bipolar disorder and how these are manifested as the disease. These are all difficult questions to address experimentally, so most evidence is based on correlative data. For example, lithium inhibits GSK3 both in mouse brain [49] and in human blood cells [50], which has led to the hypothesis that lithium also inhibits GSK3 in human brain. Since impaired serotonergic activity has long been linked to depression, the finding that enhanced serotonergic activity or anti-depressants acting on serotonergic systems cause inhibition of GSK3 in mouse brain in vivo lends further support to the possibility that impaired control of GSK3 is a component of mood disorders [54]. That GSK3 is important therapeutically is also supported by recent behavioral studies in rodents based on the hypothesis that hyperactive GSK3 may partially contribute to mood disorders. Using the forced swim test, which is used to assess depressive behavior and the effects of therapeutic agents, administration of a peptide inhibitor of GSK3 rapidly induced anti-depressant-like behavioral effects [55]. Furthermore, large reductions in immobilization time in the forced swim test were induced not only by inhibition of GSK3 by lithium administration, but also by the lowered GSK3β levels expressed in heterozygote GSK3β+/− mice [56]. These reports demonstrate convincingly that GSK3 has a critical role in this widely used paradigm to assess depressive activity. But the key actions of GSK3 that are attenuated by therapeutically administered lithium remain unknown. Many actions of GSK3 lend themselves to intriguing hypotheses about how inhibition of GSK3 may be therapeutic. GSK3 regulates many transcription factors, and by doing so can regulate the expression of numerous genes, so it is tempting to speculate that altered gene expression resulting from lithium’s inhibition of GSK3 may contribute to lithium’s therapeutic effects. But equally possible are regulation by lithium of GSK3’s actions on cell survival, synaptic activity, cellular plasticity, and others. Thus, we are beginning to understand the functions of GSK3 and the ramifications of inhibiting GSK3 with lithium, but much remains to be learned about how these are linked to the therapeutic use of lithium in mood disorders.
The discovery that inhibition of GSK3 reduces inflammation, taken in conjunction with evidence of inflammation associated with mood disorders, raises the new possibility that part of the therapeutic effect of lithium-induced inhibition of GSK3 may result from reduction of inflammation, as reported to occur in mice treated with a clinically relevant dose of lithium [8]. There is a large literature linking inflammation with mood disorders. For example, many studies have reported increased levels of inflammatory cytokines in patients with major depression or bipolar disorder [e.g., Refs. 57–59]. Furthermore, administration or increased production of cytokines in humans or animals can induce biochemical and behavioral changes having many similarities to those characteristic of mood disorders [60–62]. Although it is not known if the inflammatory condition contributes to the causes or is an effect of these diseases, many investigators suggest that controlling inflammation may contribute to therapy based on both the (a) depressive effects that cytokines can produce, as well as (b) on much evidence that classical anti-depressants have anti-inflammatory effects [59, 63]. Thus, the new finding that lithium and other GSK3 inhibitors have anti-inflammatory properties raises the possibility that this contributes to therapeutic effects. Furthermore, sodium valproate has recently become very widely used as a mood stabilizer to treat bipolar disorder [64]. Although, like lithium, the therapeutic mechanism of action of valproate is not known, it is of considerable interest that recent reports indicate that valproate also shares with lithium the ability to reduce inflammation and ameliorate inflammation-associated conditions in experimental animals [65, 66]. Thus, the two most widely used mood stabilizers may have in common the capacity to reduce inflammation. However, other anti-inflammatory drugs do not share lithium’s mood stabilizing action, so it is clear that reducing inflammation alone is not enough to achieve mood stabilization. Nonetheless, reducing inflammation by inhibiting GSK3 may contribute some part of lithium’s actions in controlling mood fluctuations and its long-term prophylactic action in preventing recurrences of episodes.
Neurodegenerative diseases: GSK3 and AD
A remarkable number of connections between GSK3 and the neuropathological hallmarks of AD have been identified, as reviewed previously [3, 4]. After a brief review of these links between GSK3 and AD, we discuss a more insidious interaction that has previously not been recognized, the promotion by GSK3 of inflammation, a potentially serious influence of GSK3 on the progression of AD.
Two pathological hallmarks define AD, plaques composed primarily of the amyloid β-peptide (Aβ), and neurofibrillary tangles consisting predominantly of hyperphosphorylated tau, a microtubule-binding protein. Although familial AD is much less common than sporadic AD, mutations causing familial AD have provided tremendous insight into the pathological process. Known mutations include those in amyloid precursor protein, the protein from which Aβ is derived via cleavage by proteases, and in presenilin-1, an enzyme involved in proteolysis of amyloid precursor protein. Current concepts of the pathological process suggest that abnormal accumulation of toxic oligomers of Aβ lead to both plaque formation and to tau hyperphosphorylation, which evolves into tangles. Furthermore, toxic oligomers of both Aβ and tau lead to synaptic dysfunction and cell death. GSK3 has been found to promote most of the key steps in these pathological process in AD.
Abnormal accumulation of Aβ is a critical early stage in AD neuropathology, and several studies have shown that Aβ production is promoted by GSK3 and reduced by GSK3 inhibitors [67–70]. Nevertheless, the mechanism whereby GSK3 promotes Aβ production remains to be clarified, perhaps being associated with the reported phosphorylation of the amyloid precursor protein by GSK3 [71] or the binding of GSK3 to presenilin-1 [72], because GSK3 phosphorylates and regulates the processing of presenilin-1 [73]. Furthermore, expression of an AD-related mutated presenilin-1 activates GSK3 [72, 74, 75]. In addition to Aβ, other cleavage products of the amyloid precursor protein also bind and/or activate GSK3, indicating multiple points of contact between GSK3 and metabolism of the amyloid precursor protein [76–78]. Not only is GSK3 intimately involved in many aspects of amyloid precursor protein metabolism and Aβ production, but GSK3 also contributes to Aβ-induced neuronal toxicity. Aβ is well-known to cause neuronal death when applied to cultured cells or to brain in vivo, and inhibition of GSK3 by a variety of methods significantly attenuates the neurotoxicity caused by Aβ [79–85]. This appears not to be a “bystander” effect of GSK3 because treatment with Aβ activates GSK3 in a number of conditions, indicating a direct link between Aβ-induced GSK3 activation and neurotoxicity [79, 86, 87]. These many interactions of GSK3 with Aβ suggest that GSK3 inhibitors may be therapeutic, and this has been demonstrated in transgenic mouse models of AD in which administration of GSK3 inhibitors reduced the Aβ burden [68, 70, 88]. Thus, GSK3 regulates many aspects of amyloid precursor protein metabolism and promotes the production and toxicity of Aβ.
Abnormal hyperphosphorylation of tau and its aggregation into oligomers and eventually neurofibrillary tangles is the second pathological hallmark of AD, and many reports have found GSK3 to be involved in this pathological process. Dephosphorylated tau binds to microtubules to contribute to the stability of microtubules and thus to neuronal structure [89]. Tau was identified as a substrate of GSK3β in vitro [90, 91] and in intact cells [92, 93] and GSK3 is now widely acknowledged to be a bona fide tau kinase. Recently it was found specifically that the phosphorylation of Thr231 in tau by GSK3β is a critical site in impairing the ability of tau to bind and stabilize microtubules [94]. GSK3β can phosphorylate many sites on tau, some of which are the same sites that are abnormally hyperphosphorylated in AD brain [89, 95]. By impairing the binding of tau to microtubules this action of GSK3β can impact neuronal structure and plasticity, may cause abnormal localization and/or accumulation of tau, and may contribute to tau aggregation and eventual tangle formation. GSK3 can also regulate the splicing of tau [96], hyperphosphorylated tau itself can be neurotoxic [97], and inhibition of GSK3 protects cells from aggregated tau-induced neurotoxicity [98]. Furthermore, tau phosphorylation is increased in transgenic mice overexpressing GSK3β [99, 100]. Thus, GSK3 may contribute to the abnormal hyperphosphorylation of tau, the major component of neurofibrillary tangles in AD brain, which promotes tau aggregation and neurotoxicity.
It is well-established that inflammation occurs in AD, raising a new connection between GSK3, which promotes inflammation, and pathological mechanisms in AD. As previously reviewed [101–103], the levels of many cytokines and chemokines are upregulated in AD, such as IL-1β, IL-6, and TNF, among others. Even transgenic mouse models of AD display inflammation and increased cytokine production [104, 105]. Activation of astrocytes and microglia and the ensuing production of inflammatory molecules has been reported to be stimulated by degenerating neurons, neurofibrillary tangles, Aβ and other cleavage products of amyloid precursor protein, and activated astrocytes and microglia are found surrounding amyloid plaques in AD brain [106–108]. Evidence indicates that astrocytes beneficially contribute to clearance of accumulated Aβ [109, 110], but also contribute to inflammatory-induced neuronal damage such as by producing neurotoxic nitric oxide [111] and cytokines. Activated microglia also release inflammatory cytokines and nitric oxide, as well as reactive oxygen species, and activate the complement system [112]. Thus, on one hand, these inflammatory molecules produced by glia cells are thought to contribute to the pathological progression of AD, while on the other hand, by contributing to the clearance of Aβ and secreting growth factors, both activated astrocytes and microglia are not thought to be entirely detrimental in AD [113, 114]. In fact, therapeutic intervention by administration of Aβ antibodies appears to increase the clearance of Aβ by activated microglia [115, 116]. However, the general consensus remains that inflammation associated with activated astrocytes and microglia contributes to the pathological progression of AD [101]. Therefore, blocking components of the inflammatory system in the brain is a potential therapeutic avenue that is being explored, particularly with the use of non-steroidal anti-inflammatory drugs (NSAIDs). Although epidemiological evidence and studies in animal models provide a strong case for NSAID use in AD, clinical trials have provided mixed, and controversial, results [117]. It has been suggested that the variable results obtained in clinical trials are in part because long-term administration of NSAIDs is necessary to achieve beneficial effects, such as delayed progression and reduced symptomology, including a slower rate of cognitive decline [103, 118]. While the effectiveness and mechanisms of NSAIDs remain to be more clearly defined, it is notable that reduced inflammation in the brain also might be achieved by inhibitors of GSK3. Thus, not only might GSK3 inhibitors reduce Aβ production and tau phosphorylation, they also may contribute therapeutic anti-inflammatory actions in AD brain.
Diabetes and GSK3
Impaired regulation of glucose production and utilization is associated with insulin-resistance and diabetes, including both type 1 insulin-dependent and type 2 insulin-independent diabetes, which are increasingly prevalent conditions [119–121]. Insulin receptors in most peripheral tissues, as well as in the brain, stimulate a signaling cascade leading to activation of Akt which phosphorylates, to inhibit, GSK3 [122]. By this signaling mechanism insulin stimulates increases in the phosphorylation levels of both Akt and GSK3, which increases and decreases, respectively, their activities [123]. Since GSK3 contributes to the regulation of glucose homeostasis, during the last few years increasing attention has been focused on diabetes-associated changes in GSK3 and the possibility that GSK3 may be a useful therapeutic target in diabetes to facilitate control of glucose levels [124].
Despite its being a potential therapeutic target, there have been only a few studies of the in vivo phosphorylation state of GSK3 in peripheral tissues in insulin-resistant animal models, and these have produced variable results. In mice with high fat diet-induced diabetes, the activity of GSK3β was increased in epididymal fat, slightly decreased in liver, and not changed in skeletal muscle, demonstrating that regulation of GSK3 in diabetic conditions can differ dramatically among tissues [125]. Others also found no changes in diabetic animals in skeletal muscle GSK3β activity [126, 127]. However, GSK3 activity was elevated approximately twofold in skeletal muscle samples from human patients with type 2 diabetes [128]. There was also decreased insulin-induced serine-phosphorylation of GSK3 in the myocardium of diabetic rats [129]. Thus, while still few, these studies indicate that the effects of insulin-resistance on GSK3 vary considerably among different tissues with some evidence of dephosphorylated, hyperactive GSK3 associated with insulin deficiency.
In contrast to studies of the effects of diabetic conditions on peripheral tissues, little is understood about diabetes-associated changes in the brain even though cognition is often impaired in diabetic subjects [130]. Recently, two related mechanisms have been found to contribute to the regulation of GSK3 in the brain: insulin and glucose levels. First, it has been demonstrated that insulin activates the same signaling pathway leading to phosphorylation of Akt and GSK3 in brain as in peripheral tissues. Thus, injection of insulin increased GSK3 phosphorylation in mouse brain [131] and brain GSK3 phosphorylation was reduced by blockade of insulin signaling by knockout of the insulin receptor [132] and in mice with diet-induced insulin-resistance [133]. It is important to note that glucose levels were not changed in these latter two studies, because glucose levels were recently found to also regulate GSK3 phosphorylation. Thus, it was initially surprising to find that insulin depletion, which is associated with hyperglycemia, greatly increased the phosphorylation of GSK3 in mouse brain [134]. These results revealed that after insulin depletion changes of GSK3 in the brain are relatively large and opposite to changes in peripheral tissues. The insulin-depletion-induced increases in the phosphorylation of GSK3 in mouse brain were found to result from the hyperglycemia associated with insulin depletion. Thus, strong roles were found for physiological and pathological changes in the circulating concentrations of both insulin and glucose in regulating GSK3 in mouse brain. Hyperglycemia in the presence of normal or deficient insulin levels caused large increases in the phosphorylation level of GSK3 in the brain, as did acute insulin administration, whereas it was reduced by hypoglycemia. Since both fasting and acute glucose administration altered the phosphorylation level of GSK3, it appears that GSK3 phosphorylation is perpetually fluctuating in response to food intake or deprivation. The regulation of brain GSK3 differentiates the response of this enzyme in the brain from changes in peripheral tissues in insulin-resistant conditions, as GSK3 in epididymal fat was dephosphorylated while the phosphorylation level of GSK3 in the brain was dramatically increased in diabetic mice. These results show that glucose as well as insulin contributes to the in vivo regulation of the phosphorylation of GSK3 in mouse brain, with hyperglycemia ensuing from insulin depletion causing large increases in the phosphorylation level of GSK3 in the brain. Similar effects on brain GSK3 phosphorylation were observed in the IRS-2-deficient mouse model of diabetes which had increased brain phospho-Ser9-GSK3β [135] and these mice are hyperglycemic [136]. These findings indicate that insulin has a significant influence on the phosphorylation level of GSK3 in the brain, which decrease with deficient insulin signaling if glucose levels remain normal. However, if hyperglycemia is associated with insulin deficiency, the elevated glucose levels cause large increases in the phosphorylation level of brain GSK3. Thus, both insulin and glucose contribute to regulating the phosphorylation level of GSK3 in the brain.
Although as noted above there is limited data indicating that GSK3 in peripheral tissues is abnormally activated in diabetes, this is not a requirement for GSK3 inhibitors to contribute to the control of glucose levels, an outcome reported in several recent studies. Administration of a peptide inhibitor of GSK3 increased glucose tolerance in diabetic mice [137]. Also, rosiglitazone, a peroxisome proliferator-activated receptor agonist that also inhibits GSK3, reduced circulating blood glucose levels in Zucker diabetic fatty rats and in patients with type 2 diabetes [138, 139]. In several models of diabetes, two GSK3 inhibitors lowered plasma glucose levels in a dose-dependent manner by as much as 50% [140]. Additionally, the GSK3 inhibitor CHIR98023 lowered glucose concentrations in Zucker diabetic fatty rats during a glucose tolerance test [127, 141]. These results demonstrate that inhibition of GSK3 improves glucose regulation in diabetic animals. Consistent with this, overexpression of GSK3β in skeletal muscle was sufficient to cause glucose intolerance [142]. Taken together it appears that GSK3 may be hyperactivated in some peripheral tissues during diabetes, and that inhibition of GSK3 may be able to contribute to controlling glucose levels in diabetic subjects.
Therapeutic intervention with GSK3 inhibitors in diabetes has been based on the regulatory role of GSK3 in glucose utilization and production, however it is also interesting to note an additional link, that inflammation is often associated with diabetes and that GSK3 is a pro-inflammatory enzyme. The levels of cytokines such as TNF and IL-6 often are elevated in insulin-resistant conditions and much evidence has shown that these cytokines can impair insulin signaling in cells and animals [143, 144]. For example, TNF levels in the circulation are elevated in obese subjects and blocking the action of TNF can promote insulin signaling [145, 146]. Additionally, adipose tissue is the source of a large portion of IL-6 in the blood and elevated IL-6 levels predict the development of type 2 diabetes [147], an interesting link to the finding that GSK3 promotes IL-6 production [8]. IL-6 is well-documented to cause insulin-resistance in liver and fat [148, 149]. TNF and IL-6 represent only two examples of numerous inflammatory molecules for which elevations have been linked to the development of type 2 diabetes, and several anti-inflammatory drugs have been reported to decrease the risk of developing type 2 diabetes [150]. Overall, many studies have shown that in obesity and type 2 diabetes the plasma levels of inflammatory molecules are increased and these molecules can impede the actions of insulin, and similar studies suggest that elevated cytokine levels promote the development of type 2 diabetes [151]. Taken along with the finding that GSK3 inhibitors reduce the production of inflammatory molecules, therapeutically GSK3 inhibitors may prove to be useful for both contributing to the control of blood glucose levels and reducing inflammation associated with, or preceding, insulin-resistance.
Cancer and GSK3
Glycogen synthase kinase-3 has arisen as a potential mediator contributing to neoplastic transformation in part because it belongs to both the canonical Wnt/β-catenin and the PI3K/Akt signaling systems, two major pathways often dysregulated in cancer. In the Wnt signaling pathway, the transcriptional co-activator β-catenin promotes growth and survival. The Wnt signaling protein complex composed of the tumor suppressor APC, the scaffold protein Axin, β-catenin, and GSK3 promotes the phosphorylation of β-catenin by GSK3 to target it for degradation by the proteasome [152]. Activation of Wnt signaling inhibits GSK3 selectively in this complex, causing accumulation of β-catenin and the translocation of β-catenin to the nucleus where it interacts with the T-cell factor/lymphoid enhancer factor transcription factors to activate gene expression, including oncogenes such as cyclin D1, Myc, and c-jun [153]. Thus, activation of the Wnt signaling pathway has emerged as an oncogenic pathway which, for example, is associated with colorectal cancer [154]. Therefore, GSK3 acts as a potential tumor suppressor protein in the Wnt signaling pathway, since its inhibition when this pathway is activated promotes the stabilization, as well as expression, of oncogenic proteins. In the PI3K/Akt signaling pathway, which is activated by growth factors such as insulin or insulin-like growth factors (IGFs), GSK3 is a substrate of Akt which phosphorylates GSK3 on its regulatory serine residues (Ser21 in GSK3α and Ser9 in GSK3β) causing inhibition of the activity of GSK3 [4]. Upon activation of growth factor receptors, PI3K and subsequently Akt are activated leading to the inhibition of GSK3. The PI3K/Akt signaling pathway is frequently activated in cancers [155], suggesting again that GSK3 functions as a putative tumor suppressor. Thus, in these two major signaling pathways cancer cell viability is associated with inhibition of GSK3.
Considering the possibility that GSK3 inhibition may contribute to tumorigenesis, it is particularly interesting that no inactivating mutations in GSK3 have been found yet in human cancers [156]. This is in marked contrast to the relatively frequent mutations that have been found in human cancers in the genes encoding β-catenin or APC in the Wnt pathway or in components of the PI3K/Akt pathway [153, 155]. This suggests that GSK3 is fundamentally required for cell survival, which is consistent with a study showing that a knockout of the GSK3β gene in mice is lethal at developmental day E14 due to massive hepatocyte apoptosis because of hypersensitivity to TNF [20]. Alternatively, it may be that mutations in GSK3 have not been found in association with cancers because each of the isoforms of GSK3 are encoded by separate genes and spontaneous mutations in both genes would appear with a rare frequency. Moreover, since each isoform can compensate for the other in most known cellular functions, a spontaneous mutation in the gene of one isoform may be unlikely to be sufficient for tumorigenesis.
Therefore, the involvement of GSK3 in tumorigenesis may be linked to the dysregulation of its activity rather than to inactivating mutations, a hypothesis that has been tested in many carcinogenic models in mice. Thus, mice developing either DEN-induced or spontaneous hepatocellular carcinoma show an early inhibitory phosphorylation of Ser9-GSK3β which correlates with the appearance of neoplastic nodules [157]. In chemically induced two-stage mouse skin carcinogenesis and in tumors generated by subcutaneous injection of Akt-transformed keratinocytes, GSK3β activity is downregulated by Tyr216 dephosphorylation and increased Ser9 phosphorylation [158]. Similarly, constitutive inhibition of GSK3β has been reported in human hepatocellular carcinoma [157, 159] and in human hepatoma cell lines [160]. Not only its activity but also the expression level of GSK3 has been reported to be dysregulated in tumors, as downregulation of GSK3 expression occurs in squamous cell carcinoma of the tongue [161]. Furthermore, silencing of GSK3β expression is frequently associated with advanced prostate cancer [162]. In contrast, in colo-rectal cancer cell lines and colorectal tumors, the levels of GSK3β expression and its active form are higher in tumor cells than in their normal counterparts, but these cancers are often associated with mutations in β-catenin or APC which abrogate the normal function of GSK3 in the Wnt signaling pathway [163, 164]. Therefore, while there appears to be some selectivity based on the tumor cell-type, the consensus concept indicates that GSK3 is inhibited during the process of carcinogenesis. Consistent with this conclusion that GSK3 is inhibited in cancer, evidence indicates that inhibition of GSK3 might promote tumorigenesis processes in vivo. Thus, transgenic mice overexpressing a kinase-inactive GSK3β under the control of the mouse mammary tumor virus-long terminal repeat develop more mammary tumors than control mice [165]. In addition, the localization of GSK3 in the cell has emerged as an important process contributing to tumorigenesis. Thus, redistribution of GSK3 in the cell based on the loss of cytoplasmic GSK3 activity has been identified as another mechanism by which, for example, β-catenin can be dysregulated and contribute to human cancers [166]. However, the inhibition of GSK3 does not seem to be sufficient to transform cells since patients with mood disorders treated with the GSK3 inhibitor lithium for many years do not have an increased risk of developing cancer compared with the general population [167], although this result should be interpreted cautiously because it still needs to be repeated in a larger population. Consistent with this, genetically predisposed APC mutant mice treated with lithium for 60 days do not develop a significantly increased number of tumors [168]. Altogether these data suggest that the role of GSK3 in neoplastic transformation remains to be more completely clarified since by participating in oncogenic signaling pathways GSK3 should function as a tumor suppressor but its inhibition alone is not sufficient to increase the incidence of cancer.
Therefore, while the direct involvement of GSK3 in tumorigenesis remains to be clarified, much evidence suggests that oncogenic processes target GSK3 to inhibit its activity in order to promote proliferative or invasive signals or to inhibit apoptotic signals. GSK3 phosphorylates many oncogenic factors such as the transcription factors c-Myc, β-catenin, Snail, and BCL3 to target them for degradation through the proteasome pathway [153, 169, 170]. Therefore, inhibiting GSK3 can benefit transformed cells by maintaining the stability of these proliferative signals. For example, in colon cancer oncogenic K-Ras stimulates Wnt signaling through inhibition of GSK3 to promote tumorigenesis [171]. Additionally, since GSK3 also phosphorylates the translation factor eIF2B to inhibit protein synthesis [172], inhibition of GSK3 is favorable for cancerous proliferation.
Besides its role in tumor formation, recently evidence has appeared indicating that inhibition of GSK3 plays a fundamental role in metastasis. The metastatic cascade is generally believed to start with the escape of a tumor cell from the primary tumor into the bloodstream or lymphatic system where it migrates to establish a colony at a distant organ. Cells acquire an invasive capacity through changes in adhesiveness by proteolysis of the extracellular matrix and increased motility [173]. The increased motility and invasiveness of cancer cells in the first phase of metastasis resemble the epithelial-mesenchymal transition during embryonic development [174]. The transcription factor Snail has a fundamental role in the epithelial-mesenchymal transition by suppressing expression of the adhesion protein E-cadherin, and the level of Snail correlates with breast cancer metastases [170]. In tumor cells, since GSK3 promotes degradation of Snail, inhibition of GSK3β causes upregulation of Snail and downregulation of E-cadherin. Therefore, the regulation of Snail by GSK3β, as well as the other migration mechanisms regulated by GSK3 discussed above, reveals that GSK3 inhibition in tumors would promote cancer cell invasion and metastases [170].
An intrinsic feature of cancer cells is their resistance to apoptosis, so in addition to proliferation and migration, GSK3 may also be important in cancerous tissues because of the widespread regulatory role of GSK3 in apoptosis [175]. For example, in neuroblastoma SH-SY5Y cells [176] or in hepatoma cell lines [177, 178], inhibition of GSK3 impedes the apoptotic signaling pathway and thereby induces resistance to chemotherapeutic agents, such as camptothecin, etoposide, and doxorubicin. This is in part because GSK3 promotes the actions of major tumor suppressor proteins such as p53 [177, 179]. The short-lived p53 protein was linked to GSK3 by the finding that following treatment with chemotherapeutic agents, GSK3β forms a complex with nuclear p53 that both increases GSK3 activity and promotes p53-induced gene expression and apoptosis [176, 177, 179]. Considering the major role of p53 in tumorigenesis, this evidence indicates that GSK3 can play a role in neoplastic transformation by influencing tumor suppressors such as p53.
To summarize, many putative oncogenic signaling pathways converge on GSK3 to inhibit its activity. Additionally, the apoptotic role of GSK3 may also be targeted during tumor progression [157]. These findings indicate that inhibition of GSK3 can contribute to tumor development by promoting proliferation and inhibiting apoptosis of tumor cells. However, presently it appears that inhibition of GSK3 alone is not sufficient to cause to cellular transformation because even with GSK3 inhibition at very early stages in tumor formation [157], all the studies on the role of GSK3 inhibition in tumorigenesis indicate that a prior induction of the tumor is required. This is in agreement with the observations that patients treated with lithium do not spontaneously develop more tumors than the general population and that lithium by itself is not tumorigenic. It could also be speculated that inhibition of GSK3 is likely the result of simultaneous activation of many oncogenic pathways, so examination of one pathway at a time cannot provide a complete picture of the complex biology of the tumor.
Inflammation is closely linked to cancer, as the association between immune cells and cancer has been known for over a century [180]. As early as 1863, Virshow noted that cancers tend to occur at sites of chronic inflammation [180]. On the other hand, acute inflammation may contribute to the regression of cancer [181]. Thus, whereas acute activation of adaptive immune cells in response to tumors should promote eradication of malignant cells, chronic activation of innate immune cells at premalignant tissues promotes tumor development [182]. Therefore, by reducing cytokine production mediated by monocytes, inhibited GSK3 could prevent the deleterious effects of acute inflammation on tumor progression. On the other hand, attenuation of innate inflammation following GSK3 inhibition could explain the absence of an effect of lithium alone on tumor formation, since in the absence of infiltration of immune cells, components of tumor development, such as increased cell survival, tissue remodeling, angiogenesis, and suppression of anti-tumor adaptive immune responses, are perturbed [182, 183]. Of particular note, GSK3 inhibition leads to a decrease of Fas receptor expression [177]. The Fas ligand/Fas receptor system is a mechanism used by lymphocytes to eliminate non-self cells since lymphocytes secrete Fas ligand which once recognized by its receptor on the receptive cell triggers a signaling pathway leading to apoptosis. This suggests that by decreasing Fas receptor expression, inhibition of GSK3 could render tumor cells more resistant to eradication by the adaptive immune system and thus promote the persistence of tumor cells. However, although cancer, inflammation, and GSK3 are complexly intertwined, the role of GSK3 in inflammation during tumorigenesis remains to be clarified.
Conclusion
Chronic inflammation is a common feature of many pathological conditions. Therefore, deciphering the mechanisms that govern inflammation in tissues and diseases should provide new strategies for therapeutic intervention. The recent finding that GSK3 is a powerful regulator of inflammation has increased the focus on this Ser/Thr kinase that was already characterized as having a tentacular capacity to influence numerous aspects of cell function, often acting as a centralized integrator of many intracellular signals. Abnormal function of GSK3 was already implicated in a large number of prevalent diseases, such as mood disorders, AD, diabetes, and cancer. These conditions are associated with a high rate of mortality and inadequate therapeutic options. Taking into account that these pathologies are linked to abnormal functioning of GSK3 and are associated with inflammation, the new revelations showing that GSK3 promotes inflammation raises the possibility that inhibitors of GSK3 may provide therapeutic anti-inflammatory effects in a broad range of conditions.
Acknowledgments
Research in the authors’ laboratory was supported by grants from the National Institutes of Health.
Footnotes
Special issue dedicated to John P. Blass.
References
- 1.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- 2.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3 (GSK3) Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Grimes CA, Jope RS. The multi-faceted roles of glycogen synthase kinase-3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 5.Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- 6.Eickholt BJ, Walsh FS, Doherty P. An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J Cell Biol. 2002;157:211–217. doi: 10.1083/jcb.200201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3β phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993–1005. doi: 10.1242/jcs.01697. [DOI] [PubMed] [Google Scholar]
- 8.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lampson LA. Molecular bases of the immune response to neural antigens. Trends Neurosci. 1987;10:211–216. [Google Scholar]
- 10.Fuchs HE, Bullard DE. Immunology of transplantation in the central nervous system. Appl Neurophysiol. 1988;51:278–296. doi: 10.1159/000099973. [DOI] [PubMed] [Google Scholar]
- 11.Matyszak MK. Inflammation in the CNS: balance between immunological privilege and immune responses. Prog Neurobiol. 1998;56:19–35. doi: 10.1016/s0301-0082(98)00014-8. [DOI] [PubMed] [Google Scholar]
- 12.Correale J, Villa A. The neuroprotective role of inflammation in nervous system injuries. J Neurol. 2004;251:1304–1316. doi: 10.1007/s00415-004-0649-z. [DOI] [PubMed] [Google Scholar]
- 13.Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 14.Hauwel M, Furon E, Canova C, Griffiths M, Neal J, Gasque P. Innate (inherent) control of brain infection, brain inflammation and brain repair: the role of microglia, astrocytes, “protective” glial stem cells and stromal ependymal cells. Brain Res Rev. 2005;48:220–233. doi: 10.1016/j.brainresrev.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Rev. 2005;48:166–177. doi: 10.1016/j.brainresrev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Imitola J, Chitnis T, Khoury SJ. Insights into the molecular pathogenesis of progression in multiple sclerosis: potential implications for future therapies. Arch Neurol. 2006;63:25–33. doi: 10.1001/archneur.63.1.25. [DOI] [PubMed] [Google Scholar]
- 19.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoeflich KP, Luo J, Rubie EA, Tsao M-S, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 21.Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Mol Cell Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, Louhelainen M, Foster SJ, Yaqoob MM, Thiemermann C. GSK-3β inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- 23.Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3β. Br J Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuzzocrea S, Mazzon E, Di Paola R, Muia C, Crisafulli C, Dugo L, Collin M, Britti D, Caputi AP, Thiemermann C. Glycogen synthase kinase-3β inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin Immunol. 2006;120:57–67. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Vicente-Manzanares M, Webb DJ, Horwitz AR. Cell migration at a glance. J Cell Sci. 2005;118:4917–4919. doi: 10.1242/jcs.02662. [DOI] [PubMed] [Google Scholar]
- 27.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCκ. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 28.Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 29.Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci. 2005;118:623–632. doi: 10.1242/jcs.01661. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3β phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 32.Owen R, Gordon-Weeks PR. Inhibition of glycogen synthase kinase 3β in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/s1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 33.Farooqui R, Zhu S, Fenteany G. Glycogen synthase kinase-3 acts upstream of ADP-ribosylation factor 6 and Rac1 to regulate epithelial cell migration. Exp Cell Res. 2006;312:1514–1525. doi: 10.1016/j.yexcr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Koivisto L, Hakkinen L, Matsumoto K, McCulloch CA, Yamada KM, Larjava H. Glycogen synthase kinase-3 regulates cytoskeleton and translocation of Rac1 in long cellular extensions of human keratinocytes. Exp Cell Res. 2004;293:68–80. doi: 10.1016/j.yexcr.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 35.Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Bianchi M, De Lucchini S, Marin O, Turner DL, Hanks SK, Villa-Moruzzi E. Regulation of FAK Ser-722 phosphorylation and kinase activity by GSK3 and PP1 during cell spreading and migration. Biochem J. 2005;391:359–370. doi: 10.1042/BJ20050282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang D, Cheung AT, Parsons JT, Bryer-Ash M. Focal adhesion kinase (FAK) regulates insulin-stimulated glycogen synthesis in hepatocytes. J Biol Chem. 2002;277:18151–18160. doi: 10.1074/jbc.M104252200. [DOI] [PubMed] [Google Scholar]
- 38.Cai X, Li M, Vrana J, Schaller MD. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol Cell Biol. 2006;26:2857–2868. doi: 10.1128/MCB.26.7.2857-2868.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, Kikuchi A. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol Cell Biol. 2006;26:898–911. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Melker AA, Desban N, Duband JL. Cellular localization and signaling activity of β-catenin in migrating neural crest cells. Dev Dyn. 2004;230:708–726. doi: 10.1002/dvdy.20091. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y, Cai J, Xue H, Mattson MP, Rao MS. SDF1α/CXCR4 signaling stimulates β-catenin transcriptional activity in rat neural progenitors. Neurosci Lett. 2006;398:291–295. doi: 10.1016/j.neulet.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 42.Gates J, Peifer M. Can 1000 reviews be wrong? Actin, α-catenin, and adherens junctions. Cell. 2005;123:769–772. doi: 10.1016/j.cell.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Bek S, Kemler R. Protein kinase CKII regulates the interaction of β-catenin with α-catenin and its protein stability. J Cell Sci. 2002;115:4743–4753. doi: 10.1242/jcs.00154. [DOI] [PubMed] [Google Scholar]
- 44.Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004;18:2225–2230. doi: 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottardi CJ, Gumbiner BM. Distinct molecular forms of β-catenin are targeted to adhesive or transcriptional complexes. J Cell Biol. 2004;167:339–349. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris TJC, Peifer M. Decisions, decisions: β-catenin chooses between adhesion and transcription. Trends Cell Biol. 2005;15:234–237. doi: 10.1016/j.tcb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- 49.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS, Davis LL, Jope RS. Lithium regulates glycogen synthase kinase-3β in human peripheral blood mononuclear cells—implications in the treatment of bipolar disorder. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.02.027. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jope RS. Lithium and GSK3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 52.Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium: evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
- 53.Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuro-psychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 56.O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35:2–11. doi: 10.1080/07853890310004075. [DOI] [PubMed] [Google Scholar]
- 58.O’Brien SM, Scott LV, Dinan TG. Cytokines: abnormalities in major depression and implications for pharmacological treatment. Hum Psychopharmacol. 2004;19:397–403. doi: 10.1002/hup.609. [DOI] [PubMed] [Google Scholar]
- 59.Schiepers OJG, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:201–217. doi: 10.1016/j.pnpbp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Licinio J, Wong ML. The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry. 1999;4:317–327. doi: 10.1038/sj.mp.4000586. [DOI] [PubMed] [Google Scholar]
- 61.Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience. 2005;135:659–678. doi: 10.1016/j.neuroscience.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 62.Simen BB, Duman CH, Simen AA, Duman RS. TNFα signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 63.Reynolds JL, Ignatowski TA, Sud R, Spengler RN. An antidepressant mechanism of desipramine is to decrease tumor necrosis factor-α production culminating in increases in noradrenergic neurotransmission. Neuroscience. 2005;133:519–531. doi: 10.1016/j.neuroscience.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 64.Li X, Ketter TA, Frye MA. Synaptic, intracellular, and neuroprotective mechanisms of anticonvulsants: are they relevant for the treatment and course of bipolar disorders? J Affect Disord. 2002;69:1–14. doi: 10.1016/s0165-0327(00)00361-x. [DOI] [PubMed] [Google Scholar]
- 65.Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, Yang S, Hong JS. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Mol Brain Res. 2005;134:162–169. doi: 10.1016/j.molbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 66.Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F, Mascagni P, Fantuzzi G, Dinarello CA, Siegmund B. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 67.Sun X, Sato S, Murayama O, Murayama M, Park JM, Yamaguchi H, Takashima A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci Lett. 2002;321:61–64. doi: 10.1016/s0304-3940(01)02583-6. [DOI] [PubMed] [Google Scholar]
- 68.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 69.Li B, Ryder J, Su Y, Zhou Y, Liu F, Ni B. FRAT1 peptide decreases Aβ production in swAPP(751) cells. FEBS Lett. 2003;553:347–350. doi: 10.1016/s0014-5793(03)01042-1. [DOI] [PubMed] [Google Scholar]
- 70.Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, Brune K, Paul S, Zhou Y, Liu F, Ni B. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-β precursor protein processing. Biochemistry. 2004;43:6899–6908. doi: 10.1021/bi035627j. [DOI] [PubMed] [Google Scholar]
- 71.Aplin AE, Jacobsen JS, Anderton BH, Gallo JM. Effect of increased glycogen synthase kinase-3 activity upon the maturation of the amyloid precursor protein in trans-fected cells. Neuroreport. 1997;8:639–643. doi: 10.1097/00001756-199702100-00012. [DOI] [PubMed] [Google Scholar]
- 72.Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kirschenbaum F, Hsu SC, Cordell B, McCarthy JV. Glycogen synthase kinase-3β regulates presenilin 1 C-terminal fragment levels. J Biol Chem. 2001;276:30701–30707. doi: 10.1074/jbc.M102849200. [DOI] [PubMed] [Google Scholar]
- 74.Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- 76.Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh YH. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FAS-EB J. 2003;17:1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 77.Akiyama H, Shin RW, Uchida C, Kitamoto T, Uchida T. Pin1 promotes production of Alzheimer’s amyloid β from β-cleaved amyloid precursor protein. Biochem Biophys Res Commun. 2005;336:521–529. doi: 10.1016/j.bbrc.2005.08.130. [DOI] [PubMed] [Google Scholar]
- 78.Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol. 2005;171:327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid β-protein-induced neurotoxicity. Proc Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takashima A, Yamaguchi H, Noguchi K, Michel G, Ishiguro K, Sato K, Hoshino T, Hoshi M, Imahori K. Amyloid β peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3β in rat hippocampal neurons. Neurosci Lett. 1995;198:83–86. doi: 10.1016/0304-3940(95)11964-x. [DOI] [PubMed] [Google Scholar]
- 81.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- 82.Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. β-amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur J Pharmacol. 2000;392:117–123. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- 83.De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- 84.Ghribi O, Herman MM, Savory J. Lithium inhibits Aβ-induced stress in endoplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage and tau phosphorylation. J Neurosci Res. 2003;71:853–862. doi: 10.1002/jnr.10511. [DOI] [PubMed] [Google Scholar]
- 85.Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-β-peptide neurotoxicity: role in Alzheimer disease. Brain Res Rev. 2004;47:275–289. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 86.Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid β peptide (25–35) induces the inactivation of phosphatidylinositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3β. Neurosci Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- 87.Magrane J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal β-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005;25:10960–10969. doi: 10.1523/JNEUROSCI.1723-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312:922–929. doi: 10.1016/j.bbrc.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 89.Johnson GVW, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: an update. Alzheimer Dis Rev. 1998;3:125–141. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 90.Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- 91.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 92.Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S, Woodgett JR, Miller CCJ. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol. 1994;4:1077–1086. doi: 10.1016/s0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]
- 93.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 94.Cho JH, Johnson GVW. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 95.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 96.Hernandez F, Perez M, Lucas JJ, Mata AM, Bhat R, Avila J. Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J Biol Chem. 2004;279:3801–3806. doi: 10.1074/jbc.M311512200. [DOI] [PubMed] [Google Scholar]
- 97.Fath T, Eidenmuller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J Neurosci. 2002;22:9733–9741. doi: 10.1523/JNEUROSCI.22-22-09733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 100.Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear β-catenin, tau hyper-phosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 103.Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167–176. doi: 10.1016/j.ijdevneu.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 104.Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 105.Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2001;158:1345–1354. doi: 10.1016/S0002-9440(10)64085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bach JH, Chae HS, Rah JC, Lee MW, Park CH, Choi SH, Choi JK, Lee SH, Kim YS, Kim KY, Lee WB, Suh YH, Kim SS. C-terminal fragment of amyloid precursor protein induces astrocytosis. J Neurochem. 2001;78:109–120. doi: 10.1046/j.1471-4159.2001.00370.x. [DOI] [PubMed] [Google Scholar]
- 107.Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFα -dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4:247–256. doi: 10.2174/1568010053586237. [DOI] [PubMed] [Google Scholar]
- 109.Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- 110.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 111.Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:906–916. doi: 10.1093/jnen/60.9.906. [DOI] [PubMed] [Google Scholar]
- 112.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial–neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem. 1997;272:6641–6646. doi: 10.1074/jbc.272.10.6641. [DOI] [PubMed] [Google Scholar]
- 114.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 115.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 116.Gelinas DS, DaSilva K, Fenili D, St George-Hyslop P, McLaurin J. Immunotherapy for Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14657–14662. doi: 10.1073/pnas.0404866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.03.013. in press. [DOI] [PubMed] [Google Scholar]
- 118.Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 120.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 121.White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 122.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 123.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 124.Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 125.Eldar-Finkelman H, Schreyer S, Shinohara M, LeBoeuf R, Krebs E. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes. 1999;48:1662–1666. doi: 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- 126.Semiz S, Orvig C, McNeill J. Effects of diabetes, vanadium, and insulin on glycogen synthase activation in Wistar rats. Mol Cell Biochem. 2002;231:23–35. doi: 10.1023/a:1014437019586. [DOI] [PubMed] [Google Scholar]
- 127.Henriksen EJ, Kinnick TR, Teachey MK, O’Keefe MP, Ring D, Johnson KW. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am J Physiol Endocrinol Metab. 2003;284:892–900. doi: 10.1152/ajpendo.00346.2002. [DOI] [PubMed] [Google Scholar]
- 128.Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- 129.Laviola L, Belsanti G, Davalli AM, Napoli R, Perrini S, Weir GC, Giorgino R, Giorgino F. Effects of streptozotocin diabetes and diabetes treatment by islet transplantation on in vivo insulin signaling in rat heart. Diabetes. 2001;50:2709–2720. doi: 10.2337/diabetes.50.12.2709. [DOI] [PubMed] [Google Scholar]
- 130.Gispen WH, Biessels GJ. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000;23:542–549. doi: 10.1016/s0166-2236(00)01656-8. [DOI] [PubMed] [Google Scholar]
- 131.Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schubert M, Gautam D, Surjo D, Ueki K, Stephanie B, Schubert D, Kondo T, Alber J, Galldiks N, Kustermann E, Arndt S, Jacobs AH, Krone W, Kahn CR, Bruning JC. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci USA. 2004;101:3100–3105. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 134.Clodfelder-Miller B, De Sarno P, Zmijewska AA, Song L, Jope RS. Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J Biol Chem. 2005;280:39723–39731. doi: 10.1074/jbc.M508824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, Farhang-Fallah J, Dikkes P, Warot XM, Rio C, Corfas G, White MF. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23:7084–7092. doi: 10.1523/JNEUROSCI.23-18-07084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 137.Plotkin B, Kaidanovich O, Talior I, Eldar-Finkelman H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exp Ther. 2003;305:974–980. doi: 10.1124/jpet.102.047381. [DOI] [PubMed] [Google Scholar]
- 138.Smith SA, Porter LE, Biswas N, Freed MI. Rosiglitazone, but not glyburide, reduces circulating proinsulin and proinsulin:insulin ratio in type 2 diabetes. J Clin Endocrinol Metab. 2004;89:6048–6053. doi: 10.1210/jc.2004-0705. [DOI] [PubMed] [Google Scholar]
- 139.Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, Ohlstein EH, Jucker BM. Rosiglitazone treatment in Zucker diabetic fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes. 2005;54:554–562. doi: 10.2337/diabetes.54.2.554. [DOI] [PubMed] [Google Scholar]
- 140.Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, Wagman AS, Hammond ME, Harrison SD. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- 141.Cline GW, Johnson K, Regittnig W, Perret P, Tozzo E, Xiao L, Damico C. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes. 2002;51:2903–2910. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- 142.Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA, Llano A, Moore GB, Murphy GJ, Smith SA, Taylor CM, Yates JW, Morrison AD, Harper AJ, Roxbee-Cox L, Abuin A, Wargent E, Holder JC. Development of glucose intolerance in male transgenic mice over-expressing human glycogen synthase kinase-3β on a muscle-specific promoter. Metabolism. 2004;53:1322–1330. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 143.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 144.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 145.Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25:4–7. doi: 10.1016/j.it.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 146.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 148.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 149.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-α, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278:45777–45784. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 150.Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 151.Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. doi: 10.1016/s0140-6736(99)01046-6. [DOI] [PubMed] [Google Scholar]
- 152.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6:351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 153.Polakis P. The oncogenic activation of β-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 154.Manoukian AS, Woodgett JR. Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv Cancer Res. 2002;84:203–229. doi: 10.1016/s0065-230x(02)84007-6. [DOI] [PubMed] [Google Scholar]
- 155.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 156.Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Boissan M, Beurel E, Wendum D, Rey C, Lecluse Y, Housset C, Lacombe ML, Desbois-Mouthon C. Overexpression of insulin receptor substrate-2 in human and murine hepatocellular carcinoma. Am J Pathol. 2005;167:869–877. doi: 10.1016/S0002-9440(10)62058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leis H, Segrelles C, Ruiz S, Santos M, Paramio JM. Expression, localization, and activity of glycogen synthase kinase 3β during mouse skin tumorigenesis. Mol Carcinog. 2002;35:180–185. doi: 10.1002/mc.10087. [DOI] [PubMed] [Google Scholar]
- 159.Ban KC, Singh H, Krishnan R, Seow HF. GSK-3β phosphorylation and alteration of β-catenin in hepatocel-lular carcinoma. Cancer Lett. 2003;199:201–208. doi: 10.1016/s0304-3835(03)00421-x. [DOI] [PubMed] [Google Scholar]
- 160.Desbois-Mouthon C, Blivet-Van Eggelpoel MJ, Beurel E, Boissan M, Delelo R, Cadoret A, Capeau J. Dysregulation of glycogen synthase kinase-3β signaling in hepatocellular carcinoma cells. Hepatology. 2002;36:1528–1536. doi: 10.1053/jhep.2002.37192. [DOI] [PubMed] [Google Scholar]
- 161.Goto H, Kawano K, Kobayashi I, Sakai H, Yanagisawa S. Expression of cyclin D1 and GSK-3β and their predictive value of prognosis in squamous cell carcinomas of the tongue. Oral Oncol. 2002;38:549–556. doi: 10.1016/s1368-8375(01)00121-x. [DOI] [PubMed] [Google Scholar]
- 162.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3β: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25:329–337. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 163.Gotoh J, Obata M, Yoshie M, Kasai S, Ogawa K. Cyclin D1 over-expression correlates with β-catenin activation, but not with H-ras mutations, and phosphorylation of Akt, GSK3β and ERK1/2 in mouse hepatic carcinogenesis. Carcinogenesis. 2003;24:435–442. doi: 10.1093/carcin/24.3.435. [DOI] [PubMed] [Google Scholar]
- 164.Shakoori A, Ougolkov A, Yu ZW, Zhang B, Modarressi MH, Billadeau DD, Mai M, Takahashi Y, Minamoto T. Deregulated GSK3β activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 165.Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD, Seldin DC. Kinase-inactive glycogen synthase kinase 3β promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65:5792–5801. doi: 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- 166.Fujimuro M, Hayward SD. Manipulation of glycogen-synthase kinase-3 activity in KSHV-associated cancers. J Mol Med. 2004;82:223–231. doi: 10.1007/s00109-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 167.Cohen Y, Chetrit A, Cohen Y, Sirota P, Modan B. Cancer morbidity in psychiatric patients: influence of lithium carbonate treatment. Med Oncol. 1998;15:32–36. doi: 10.1007/BF02787342. [DOI] [PubMed] [Google Scholar]
- 168.Gould TD, Gray NA, Manji HK. Effects of a glycogen synthase kinase-3 inhibitor, lithium, in adenomatous polyposis coli mutant mice. Pharmacol Res. 2003;48:49–53. [PubMed] [Google Scholar]
- 169.Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, Piette J, Siebenlist U, Bours V, Chariot A. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell. 2004;16:35–45. doi: 10.1016/j.molcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 170.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 171.Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3β. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 172.Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 174.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 175.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006 doi: 10.1016/j.pneurobio.2006.07.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GVW, Jope RS. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc Natl Acad Sci USA. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Ruiz-Ruiz C, Cadoret A, Capeau J, Desbois-Mouthon C. GSK3β inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Exp Cell Res. 2004;300:354–364. doi: 10.1016/j.yexcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 178.Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Cadoret A, Capeau J, Desbois-Mouthon C. GSK-3β reactivation with LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis. Int J Oncol. 2005;27:215–222. [PubMed] [Google Scholar]
- 179.Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. J Biol Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 181.Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14:433–439. doi: 10.1016/j.semcancer.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 182.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 183.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]