

Abstract
Bacterial virulence factors are typically surface-associated or secreted molecules that in Gram-negative bacteria must cross the outer membrane (OM). Protein translocation across the bacterial OM is not well understood. To elucidate this process we studied P pilus biogenesis in Escherichia coli. We present high-resolution electron micrographs of the OM usher PapC and show that it forms an oligomeric complex containing a channel approximately 2 nm in diameter. This is large enough to accommodate pilus subunits or the linear tip fibrillum of the pilus but not large enough to accommodate the final 6.8-nm-wide helical pilus rod. We show that P pilus rods can be unraveled into linear fibers by incubation in 50% glycerol. Thus, they are likely to pass through the usher in this unwound form. Packaging of these fibers into their final helical structure would only occur outside the cell, a process that may drive outward growth of the pilus organelles. The usher complex appears to be similar to complexes formed by members of the PulD/pIV family of OM proteins, and thus these two protein families, previously thought to be unrelated, may share structural and functional homologies.
Pilin, one of the most abundant class of proteins produced by many bacterial pathogens (1), must cross the outer membrane (OM) to be assembled into thread-like fibers called pili. Pili contain adhesins, typically located at their tips, that mediate the initial attachment of bacteria to their hosts (2), a key step in the pathogenesis of an organism. Translocation of a superfamily of over 27 bacterial adhesive organelles across the outer membrane depends on OM proteins called ushers that work in concert with periplasmic chaperones as part of the chaperone/usher pathway (2, 3). Assembly of P pili by uropathogenic Escherichia coli represents the prototype for the chaperone/usher pathway. P pili are composite structures consisting of a thin flexible tip fibrillum connected to a rigid helical rod (4, 5). The PapG adhesin is situated at the distal end of the flexible tip fibrillum where it is capable of binding to the globo series of glycolipids in the human kidney (6). PapG recognition of the galabiose receptor is thought to be a prerequisite for pyelonephritis.
Assembly of P pili requires the PapD chaperone, which facilitates release of pilus subunits from the cytoplasmic membrane and likely provides a template for their folding in the periplasm (7). The chaperone caps interactive surfaces on the subunits, preventing their premature aggregation (8, 9). Chaperone–subunit complexes are targeted to the PapC usher in the OM (10), where the chaperone dissociates from the subunits. Dissociation of the chaperone uncaps the interactive surfaces of the subunits, which drives their assembly into pili (8). In the absence of an usher, chaperone–subunit complexes accumulate in the periplasm but cannot translocate across the OM for assembly into pili on the bacterial surface (11–13).
Gram-negative bacteria have devised a number of methods for exporting proteins outside the cell (14–17), but many aspects of these pathways are poorly understood, especially secretion across the OM. For the present study, PapC was purified to address the mechanism by which ushers guide pilus proteins across the outer membrane for assembly into pili. High-resolution electron microscopy and biochemical analyses of the usher revealed that PapC assembles into a ring-shaped oligomeric complex possessing a pore on the order of 2 nm in diameter, which is large enough to accommodate a folded pilus subunit. We argue that pilus subunits are exported across the OM through the usher channel as linear fibers and only adopt their final quaternary structure upon reaching the surface of the bacteria.
MATERIALS AND METHODS
Plasmid Construction.
Plasmid pMJ3, containing papC under an arabinose-inducible promoter with a tag coding for six histidines inserted directly after the terminal lysine of the papC coding sequence, was constructed as follows. Plasmid pFJ20 was cleaved with PinAI and the large fragment was religated, leaving a unique PinAI site, to make plasmid pMJ1. Primers 5′-ACGGATGGTGTGGGAGGTGTACCGGTTGA and 5′-CGGGATCCTCAGTGATGGTGATGGTGATGTTTCTGAGGCGTACAGGGAAGCAATAA were used in the PCR to amplify the end of papC and add histidine codons. The PCR product was cleaved with PinAI and BamHI and ligated into pMJ1 digested with PinAI and BglII. The small PinAI fragment from pFJ20 was then cloned into the PinAI site of the new plasmid, recreating the entire papC gene and making pMJ3. Plasmid pMON6235 (11) is the parent vector for pFJ20 and pMJ3.
Plasmid pFJ29 (11) carries a wild-type pap operon under an isopropyl β-d-thiogalactoside-inducible promoter. Plasmid pMJ2, carrying a papC− pap operon, was made by cleaving plasmid pFJ29 with KpnI and RsrII and ligating in the equivalent fragment from plasmid pPAP33 (12), which contains a linker insertion mutation in papC.
Purification of PapC.
PapC was purified from E. coli W3110II5 (11) harboring plasmid pMJ3. W3110II5/pMJ3 was grown with aeration in LB broth containing ampicillin (100 μg/ml). At OD600 = 0.6 unit, PapC expression was induced with 0.1% l-arabinose for 1 h. Bacteria were harvested and resuspended into 20 mM Tris⋅HCl (pH 8), and OM was collected by disruption with a French press (SLM Instruments, Rochester, NY) and differential extraction with Sarkosyl (Sigma) as described (18). OM was solubilized by rocking (30 min, 25°C) with 1% Zwittergent 3–14 (Calbiochem)/0.15 M NaCl/20 mM Tris⋅HCl, pH 8. The majority of PapC was found in the soluble fraction after this step. After spinning out insoluble material (100,000 × g, 1 h, 4°C), the OM extract was diluted with 20 mM Tris⋅HCl, pH 8/0.15 M NaCl to reduce the Zwittergent 3–14 concentration to 0.1% and rocked with Ni-nitrilotriacetic acid resin (Qiagen, Chatsworth, CA; 2 h to overnight, 4°C) to bind the His-tagged PapC. The Ni-NTA resin was washed several times with buffer A (20 mM Tris⋅HCl, pH 8/0.1% Zwittergent 3–14/0.3 M NaCl) containing 0–20 mM imidazole to remove any contaminants, and then His-tagged PapC was eluted from the resin in buffer A containing 10 mM EDTA. When needed, purified PapC was concentrated with a Centricon 50 concentrator (Amicon), according to manufacturer’s instructions, and/or dialyzed to remove the EDTA and reduce the salt concentration (the protein tended to precipitate at NaCl concentrations of less than 0.1 M). His-tagged FimD (E.T.S., D.G.T., J. Pinkner & S.J.H., unpublished results) was purified by the same procedure.
Hemagglutination.
Hemagglutination assays were performed by serial dilution of strain W3110II5, containing the indicated plasmids, in microtiter plates as described (19). The pap operon was induced from pFJ29 or pMJ2 with 0.1 mM isopropyl β-d-thiogalactoside, and papC was induced from pMJ3 with 0.4% l-arabinose.
Liposome Swelling Assay.
Swelling assays were carried out as in ref. 20. Liposomes were made from 2.4 μmol of egg phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) and 0.2 μmol of dicetyl phosphate (Sigma). PapC to be incorporated into the liposomes was generally concentrated as described above and dialyzed into 20 mM Tris⋅HCl, pH 8/0.1 M NaCl/0.1% Zwittergent 3–14. Final resuspension of the proteoliposomes was in 10 mM Tris⋅HCl (pH 8) containing 15% (wt/vol) Dextran T-40 (Pharmacia). OM used in the swelling assay was isolated from strain JM101 (21) as described above. When used, lipopolysaccharide (LPS; from E. coli Ra mutant, Sigma) was added at the reconstitution step along with the protein. Swelling was monitored on a Beckman DU-650 spectrophotometer. Incorporation of PapC into the liposomes was determined by sucrose density gradient flotation (22), followed by inspection of Coomassie-stained SDS/PAGE gels.
Electron Microscopy.
Samples were adsorbed to mica chips, which were quick-frozen, then freeze-fractured, and deep-etched, followed by rotary replication with platinum (23). Glutaraldehyde, when used, was added to 0.2% and the mixture was incubated for 10 min at room temperature before freezing. Purified P pili (5) were unraveled by incubation in 50% glycerol (24).
Rotary replication results in an artifactual enlargement of the size of the objects imaged due to the deposition of platinum. Furthermore, this enlargement is not linear with respect to the size of the objects imaged. For example, the apparent diameter of an isolated protein will be equal to its true diameter, d, plus two times the thickness of the platinum replica, t, or d + 2t (i.e., the platinum coats both sides of the subunit). The apparent diameter of two proteins packed side by side will be equal to twice the true diameter of the protein plus twice the thickness of the platinum, or 2d + 2t (i.e., the platinum coats the outer surfaces of each subunit but not the subunit–subunit interface). Note that this is less than would be obtained by adding the apparent diameter of two isolated proteins together, which would equal 2d + 4t. For this reason, an assembled complex often appears to be smaller than the sum of its subunits. Note also that the true diameter of a subunit can be obtained by subtraction of the apparent diameter of the isolated subunit from the apparent diameter of two subunits packed side by side or (2d + 2t) − (d + 2t) = d (for an in-depth discussion of this and other aspects of the deep-etch technique, see ref. 23).
Cross-Linking.
Purified PapC (10–20 μg) was dialyzed into 20 mM Hepes, pH 8/0.1 M NaCl/0.1% Zwittergent 3–14 and incubated with 1 mM bis(sulfosuccinimidyl)suberate (Pierce) for 30 min at 25°C. The reaction was quenched with 0.1 M glycine, followed by heating in SDS/PAGE sample buffer (95°C, 5 min). Other cross-linkers used were dithiobis(succinimidyl propionate) and 3,3′-dithiobis(sulfosuccinimidyl propionate) (Pierce) and glutaraldehyde (Sigma). Cross-linking was also performed on OM isolated from W3110II5/pMJ3, followed by purification of the cross-linked PapC as described above.
Immunoblotting.
OM, isolated from strains W3110II5/pMON6235 and W3110II5/pMJ3 as described above, was subjected to SDS/PAGE, transferred to Westran poly(vinylidene difluoride) membrane (Schleicher & Schuell), and probed with anti-PapC antibody (provided by MedImmune, Gaithersburg, MD). The blot was developed with alkaline phosphatase-linked secondary antibodies (Sigma).
Blue Native PAGE.
Blue native PAGE was performed as described (25). Coomassie blue G was added to 0.05% to all samples before electrophoresis (PapC samples also contained 0.1% Zwittergent 3–14). Samples were electrophoresed on a 5–11% gradient gel, overnight at 4°C and 100 V, with 0.01% Coomassie blue G in the cathode buffer. Protein standards were from Pharmacia.
RESULTS
Purification and Initial Characterization of PapC.
A six-His tag was engineered onto the carboxyl terminus of PapC to facilitate its purification. His-tagged PapC was found to be fully functional in vivo, as judged by its ability to complement a papC− pap operon to assemble adhesive pili (Table 1). PapC cofractionated with the OM and behaved as an integral OM protein, as expected (10, 12, 13, 26, 27). PapC was extracted from OM preparations with the detergent Zwittergent 3–14 (Calbiochem) and purified by nondenaturing nickel chromatography (Fig. 1).
Table 1.
Hemagglutination assays: His-tagged PapC is fully functional in vivo
| Plasmids | Relevant genotype | Titer |
|---|---|---|
| pFJ29, pMON6235 | Wild-type pap operon, vector | 32 |
| pMJ2, pMON6235 | papC− pap operon, vector | 0 |
| pMJ2, pMJ3 | papC− pap operon, his-tag papC+ | 32 |
Host strain for the plasmids was W3110II5. Titer is the highest fold dilution of bacteria that still gives hemagglutination.
Figure 1.
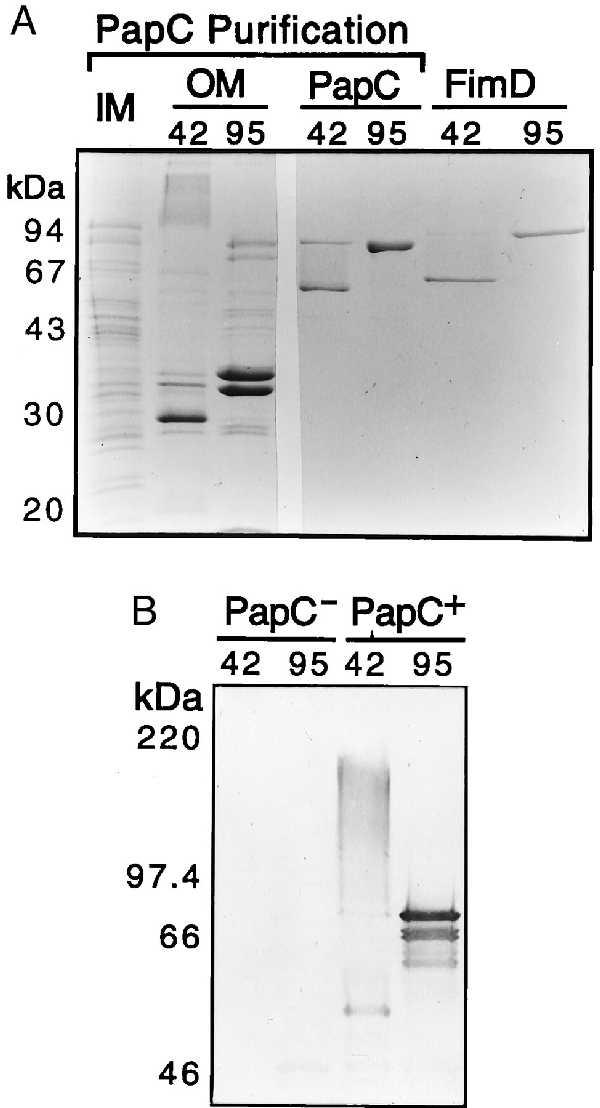
Purification and heat-modifiable mobility of PapC and FimD. (A) Coomassie blue-stained SDS/PAGE showing W3110II5/pMJ3 inner (lane IM) and outer (lanes OM) membrane fractions isolated during PapC purification, and PapC and FimD after purification from the OM by nickel chromatography. OM, PapC, and FimD samples were heated to 42°C or 95°C in sample buffer for 5 min before loading. IM was treated at 95°C. (B) Anti-PapC immunoblot of OM from strains W3110II5/pMON6235 (PapC−) or W3110II5/pMJ3 (PapC+). Samples were heated to 42°C or 95°C in sample buffer for 5 min before SDS/PAGE and transfer to a poly(vinylidene difluoride) membrane.
PapC was found to have heat-modifiable mobility on SDS/PAGE. When heated to 95°C in SDS/PAGE sample buffer, purified PapC migrated at its calculated molecular mass of 88 kDa, but when treated at 42°C, it migrated mostly as a ∼50-kDa protein (Fig. 1A). This suggests that a large portion of the usher adopts a β-barrel conformation, which is stable to SDS unless heated (28). Immunoblot analysis revealed that PapC in outer membrane preparations also exhibited heat-modifiable mobility (Fig. 1B). In addition, when treated at 42°C much of the PapC in the OM migrated as a high molecular weight smear centered at approximately 180 kDa (Fig. 1B). This suggests that PapC assembles into an oligomeric complex in the OM. This high molecular weight species was also detected in immunoblots of the purified PapC but was much less abundant (data not shown), suggesting that the PapC complex becomes (partially) destabilized when extracted out of the OM. FimD, the usher for E. coli type 1 pili (13), exhibited similar behavior (Fig. 1A), indicating that this is likely a general property of the usher family. Heat-modifiable mobility on SDS/PAGE is characteristic of the bacterial OM pore-forming proteins known as porins (29).
Pore-Forming Activity of PapC.
The hypothesis that PapC forms a channel was tested by using a proteoliposome swelling assay that has been used extensively in the past to study the pore-forming properties of bacterial porins (20). Purified PapC was reconstituted into multilamellar proteoliposomes and the osmotic swelling of the proteoliposomes was monitored in the presence of various molecular weight solutes. When arabinose was used as the solute, pore-forming activity was detected that was directly proportional to the amount of PapC incorporated into the liposomes (Fig. 2A). Proteoliposomes made with purified FimD also exhibited pore-forming activity (data not shown). Therefore, pore formation appears to be a property of the usher family. As a control, liposomes reconstituted with PapD–PapG complex (10 μg) or BSA (30 μg) showed no significant pore-forming activity (data not shown).
Figure 2.
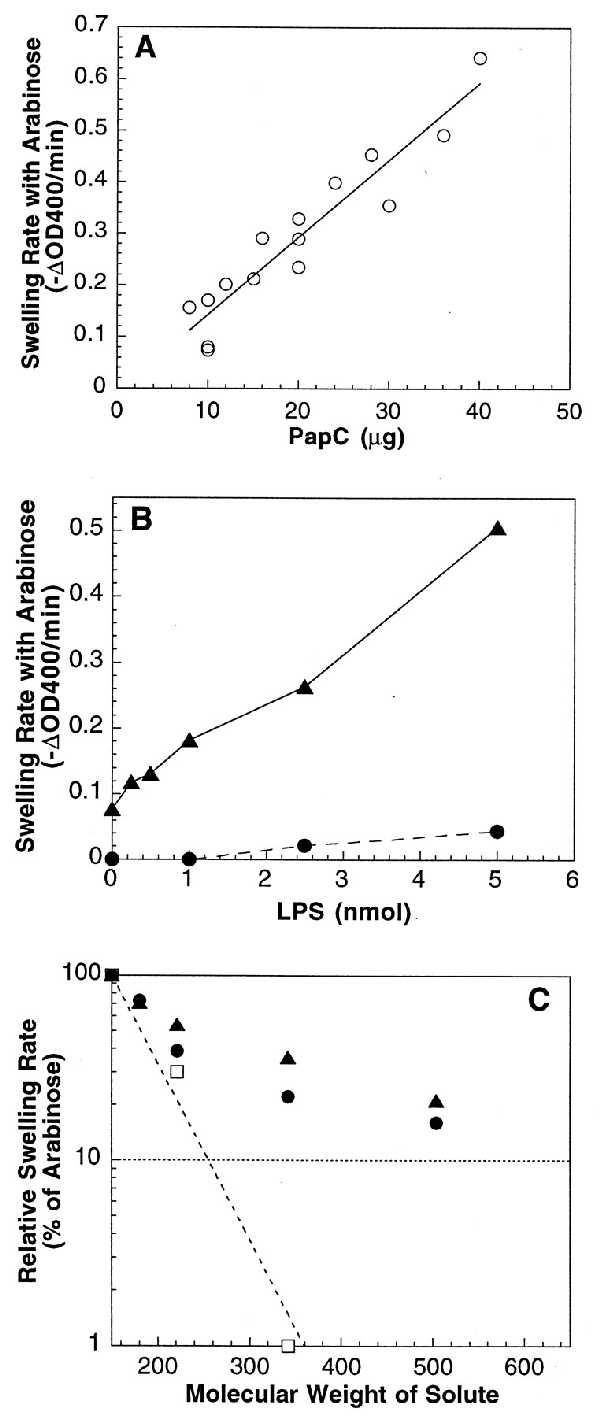
Liposome swelling assay. Liposomes (18-μl aliquots) were diluted into 0.6 ml of an isoosmotic solution containing the indicated solute, and initial rates of decrease at OD400 were determined. (A) Swelling rates of PapC proteoliposomes with l-arabinose as the solute. The swelling rate is directly proportional to the amount of PapC incorporated into the liposomes. (B) LPS stimulates the swelling activity of PapC-containing proteoliposomes. LPS (0–5 nmol) was reconstituted along with 0 (•) or 10 (▴) μg of PapC into liposomes and swelling rates were measured with l-arabinose as the solute. (C) Swelling rates in the presence of various molecular weight solutes. Swelling rates of proteoliposomes reconstituted with 20–36 μg of PapC (•), 10 μg of PapC plus 1.0–2.5 nmol of LPS (▴), or 0.5 μg of isolated OM (□) were determined in the presence of l-arabinose (150 Da), d-galactose (180 Da), N-acetyl-d-glucosamine (221 Da), sucrose (342 Da), or d-raffinose (504 Da). Rates were plotted as the percent of the arabinose swelling rate for each proteoliposome preparation.
The specific activity of the PapC proteoliposomes was 0.016 ± 0.001 ΔOD400 per min per μg for arabinose, about 20 times lower than the specific activity of the classical porin OmpF (30). This low activity may indicate that PapC exists in the liposomes predominantly in a “closed,” incorrectly inserted, or nonassembled state. The nonnative environment of the liposomes, which consisted mainly of egg phosphatidylcholine, may have hindered the ability of PapC to attain an active conformation. Indeed, swelling of the PapC-containing proteoliposomes increased up to fivefold upon the addition of the OM lipid LPS (Fig. 2B). Because the addition of LPS did not increase incorporation of PapC into the liposomes (data not shown), LPS must have affected the conformation of PapC in the liposome membrane. LPS may help to stabilize the oligomeric state of PapC, similar to the effect observed for PapC in the outer membrane (Fig. 1B).
The size of the PapC channel was estimated by plotting the relative swelling rates of PapC proteoliposomes in the presence of various molecular weight sugars (20) (Fig. 2C). The relative swelling rates of proteoliposomes made with isolated E. coli outer membrane (not expressing PapC) were plotted alongside for comparison. Large molecular weight sugars caused a much greater decrease in the swelling rates of the OM proteoliposomes than the PapC proteoliposomes, indicating that the PapC pore is larger than the porins in the OM, the largest of which is OmpF (29). A measure of pore size is the molecular weight of the sugar that would produce 10% of the swelling activity of arabinose [Mr (0.1 Ara)] (20). The Mr (0.1 Ara) for the OM proteoliposomes was 255 Da (Fig. 2C), which is close to that found for OmpF (280 Da) (20). The Mr (0.1 Ara) for the PapC proteoliposomes could not be determined exactly, because the data did not fit well to a single slope but, instead, appeared biphasic in nature (Fig. 2C). This might be a reflection of the fact that PapC functions as a specific transporter of pilus subunit proteins, whereas porins such as OmpF serve as nonspecific diffusion channels. Regardless, the PapC pore is clearly larger than that of OmpF, with the best fit for the PapC Mr (0.1 Ara) equal to 620 Da. This would place the PapC pore at roughly twice the size of the OmpF pore. The diameter of the OmpF pore is known from crystallography to be 1.1 nm (31), thus the diameter of the PapC pore would be on the order of 2 nm. Note that addition of LPS did not significantly affect the pore size of PapC (Fig. 2C). This argues that the LPS-stimulated increase in the swelling rate of the PapC proteoliposomes (Fig. 2B) was due to a greater amount of PapC attaining a pore-forming conformation and not to an increase in the PapC pore size.
High-Resolution Ultrastructure of the Usher.
To facilitate pilus assembly, the usher must be able to translocate pili across the OM. P pilus tips are thin fibers formed by a linear array of subunits (5, 32). Tips are 2 nm in diameter; hence, the diameter of a subunit must also be in the range of 2 nm. Such structures would be able to pass through the usher channel. On the other hand, the pilus rod is composed of PapA subunits arranged in a helical cylinder with 3.28 subunits per turn and a diameter of 6.8 nm (4, 32). This would be too large to fit through the PapC channel. A mechanism by which PapA rods could be translocated across the OM was suggested by our finding that incubation of pilus rods in 50% glycerol caused local unwinding of the helix into linear fibers as determined by deep-etch electron microscopy (23) (Fig. 3A). Glycerol has been demonstrated to unravel type 1 pili (24) and local unwinding of P pili has been shown to result from mechanical stress (4, 32). The unraveled pilus fibers presumably consist of PapA subunits held together by head to tail interactions (4, 8), and their diameter would, therefore, be 2 nm. The diameter of the unraveled pilus shown in Fig. 3A was artifactually enlarged by residual glycerol in the sample, in addition to the platinum used for replication (see Materials and Methods). Studies of P pili unraveled by mechanical stress have confirmed the diameter of the unraveled regions to be 2 nm (4, 32). These unraveled pilus rods would, therefore, be narrow enough to pass through the usher pore. We propose that these linear fibers would then package into helical cylinders once outside the cell, a process that may facilitate their transport and outward growth.
Figure 3.
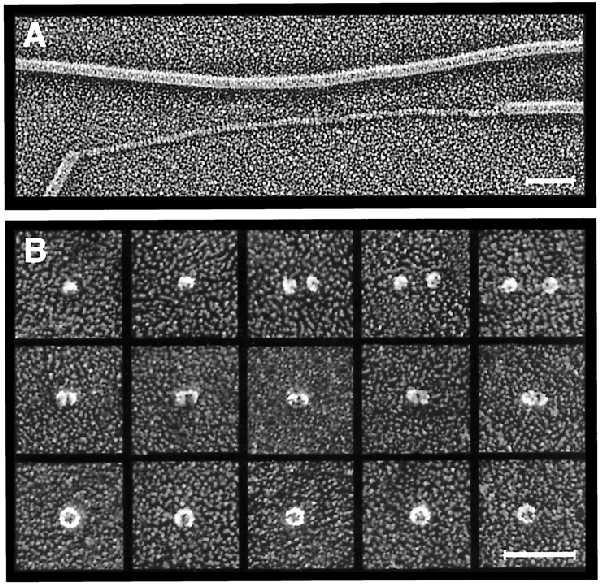
Deep-etch electron microscopy. (A) Platinum replica of purified P pili incubated in 50% glycerol showing unraveling of one of the pilus rods. (Bar = 50 nm.) (B) Montage of platinum replicas of purified PapC showing putative PapC monomers (top row), dimers (middle row), and ring-shaped complexes (bottom row). The ring-shaped complexes contain central pores 2–3 nm in diameter. (Bar = 50 nm.)
Deep-etch electron microscopy was also done to determine the fine structure of the PapC usher itself. Purified PapC was found to exist in a range of oligomeric states, which is not surprising considering the destabilization of PapC complexes that have been extracted out of the OM as noted above. Fig. 3B, top row, shows small globules measuring 9 nm in diameter that most likely represent PapC monomers. Fig. 3B, middle row, illustrates what appear to be dimers of this species. The diameter of the proteins was enlarged by the deposition of platinum, but the true diameter for the monomer can be obtained by subtraction of the apparent diameter of the monomer from that of the dimer, to give ∼6 nm (see Materials and Methods). Fig. 3B, bottom row, shows larger ring-shaped complexes measuring 15 nm in diameter also found in the PapC preparations. Samples pretreated with glutaraldehyde just before freezing yielded increased proportions of this larger form. These ring-shaped complexes have hexagonal symmetry and invariably display a central pore 2–3 nm in diameter. This pore size is in good agreement with the swelling assay results and would be large enough to allow an extended chain of pilus subunits to pass through it.
Oligomeric State of PapC.
To determine the number of PapC subunits present in the ring-shaped complexes, pure or OM-associated PapC was incubated with various cross-linking agents, including bis(sulfosuccinimidyl)suberate (Pierce), glutaraldehyde, and others. Cross-linking of both pure and OM-associated PapC produced a ladder of high molecular weight bands as visualized by SDS/PAGE (data not shown). These bands were rather diffuse, likely due to intramolecular as well as intermolecular cross-links, and determination of the number of cross-linked products and their relative mobilities was not possible. To circumvent this problem, purified usher was subjected to Blue native PAGE, a procedure that allows membrane proteins to be electrophoresed in the absence of cross-linker or detergent by using Coomassie blue G dye to keep the proteins soluble without disrupting complexes (25). Purified PapC ran as a series of high molecular weight oligomeric species on Blue native PAGE (Fig. 4). The band of slowest mobility had a molecular mass of ∼550 kDa, consistent with a hexameric complex. However, a ladder of faint bands could sometimes be seen extending above the 670-kDa thyroglobulin standard, suggesting the possibility of a dodecameric complex (data not shown). PapC boiled in SDS and 2-mercaptoethanol before Blue native PAGE yielded bands with mobilities of 100 and 150 kDa (Fig. 4). These are most likely a mixture of monomeric (88 kDa) and dimeric (176 kDa) forms, suggesting partial reassembly of the oligomer during electrophoresis. Thus, the ring-shaped PapC complex is at least a hexamer and may be as large as a dodecamer.
Figure 4.
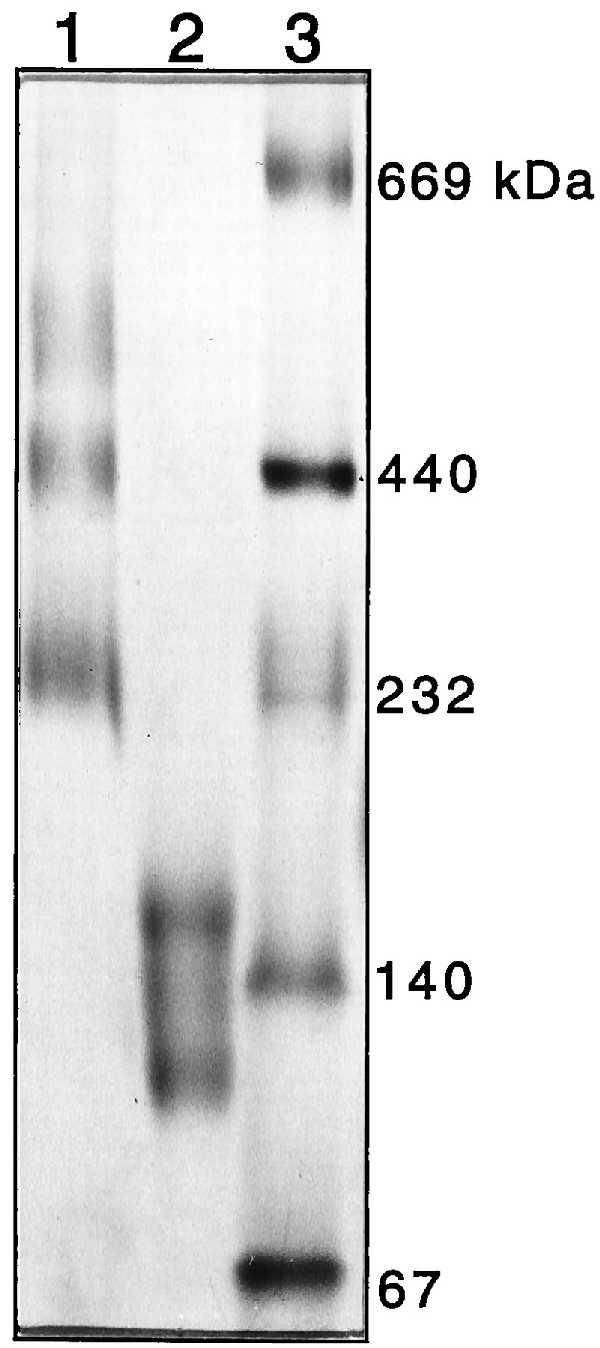
Oligomeric state of PapC determined by Blue native PAGE. Coomassie blue G (0.05%) was added to purified PapC, which was loaded directly (lane 1) or heated to 95°C for 5 min in the presence of 1.0% SDS and 2.5% 2-mercaptoethanol (lane 2) before loading. Protein standards (lane 3) were treated as in lane 1. The standards were thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), lactate dehydrogenase (140 kDa), and albumin (67 kDa). The band of slowest mobility in lane 1 is consistent with a hexameric PapC complex.
DISCUSSION
Recently, much attention has been focused on the secretion systems used by Gram-negative pathogenic bacteria to secrete, assemble, or inject virulence factors into host cells. Bacteria accomplish this with various machinery, including the ATP-binding cassette (ABC or type I) secretion system, the main terminal branch of the general secretory pathway (type II), and the contact-dependent (type III) secretion system (16, 33, 34). An underlying principle in all of these systems is the presence of an outer membrane protein that facilitates the secretion and/or assembly of the relevant virulence factor(s). However, little is known regarding the structure of these OM proteins or their mechanisms of action. In this article, we have used P pilus biogenesis as a model to elucidate the mechanism by which proteins are translocated across the bacterial outer membrane.
Our results are consistent with the view that PapC is a pore-forming protein that facilitates pilus assembly by creating a channel across the OM. Modeling of PapC (C. Stathopoulos, D.G.T., R. Curtiss III, and S.J.H., unpublished data) and other usher family members (26, 27) predicts largely β-stranded structures, and the heat-modifiable mobility of PapC and FimD suggests that large portions of the ushers fold with a β-barrel motif. PapC assembles into a ring-shaped oligomeric complex likely composed of six subunits. A number of OM proteins from the main terminal branch (type II) and contact-dependent (type III) secretion systems have also been shown to form large oligomeric assemblies, consisting of 10–12 subunits (35–38). These OM proteins, members of the PulD/pIV or secretin family, are thought to act as gated secretion channels (39, 40). Included in this family are the PilQ proteins required for assembly of type 4 pili by Psuedomonas aeruginosa and Neisseria gonorrhoeae (41, 42). Interestingly, four members of this family, YscC from Yersinia enterocolitica, pIV from filamentous phage f1, and XcpQ and PilQ from P. aeruginosa recently were purified and shown by electron microscopy to form ring-shaped complexes with apparent central pores (43–45). These complexes appear similar to the PapC complexes shown herein. Thus, these two protein families, which have been thought to be unrelated, may assemble into homologous structures in the OM. Further research may reveal that these two protein families use similar secretion mechanisms as well.
The PapC complex contains a central pore on the order of 2 nm in diameter, which is large enough to allow passage of folded pilus subunits. A pore this size in the OM would not necessarily be deleterious to the bacterium. PapC was found to have relatively low pore-forming activity when compared with OmpF, even though it produced a larger size pore. A similar result was found for the P. aeruginosa porin protein F (20). PapC may exist in open and closed forms or possess some sort of gating mechanism. Indeed, recent work on the FimD usher suggests that it undergoes a conformational change upon binding of a chaperone–adhesin complex from the periplasm (E.T.S., D.G.T., J. Pinkner, and S.J.H., unpublished results). Alternatively, the low pore-forming activity of PapC may simply be a reflection of the artificial lipid environment that was used for the swelling assays, because addition of the OM lipid LPS stimulated the swelling activity of PapC proteoliposomes (Fig. 2B). It should also be noted that PapC is normally only expressed in the context of the pap operon, and therefore “empty” PapC channels may essentially never exist, because the usher would always be occupied by pilus proteins.
The 15-nm-diameter ring-shaped PapC complex would present a large face to the periplasm for interaction with chaperone–subunit complexes. Chaperone–subunit complexes have been shown to target to the usher with affinities reflecting the subunit’s order of incorporation into the pilus (ref. 10 and E.T.S., D.G.T., J. Pinkner, and S.J.H., unpublished results). Each subunit also makes specific interactions with its appropriate neighbor subunits in the final pilus structure (8, 46). Chaperone dissociation from a subunit at the usher would allow that subunit to make the appropriate interaction with an incoming subunit, leading to the formation of a linear pilus fiber. We propose that pilus subunits are assembled into open helical fibers concomitant with their translocation through the usher channel. The tip fibrillum remains as an open helical fiber, but the pilus rod folds into a final 6.8-nm-wide helical cylinder (4). The physical constraints of the usher would maintain the nascently assembled pilus rod in a linear conformation. These constraints would then be removed upon its translocation to the outside surface of the cell, allowing the rod to snap into its final helical conformation. Pilus assembly is independent of an energized inner membrane (11). Winding of the PapA fiber into a helix on the external surface of the cell may provide a driving force for the translocation of pili across the OM, perhaps acting as a sort of ratcheting mechanism to force the pilus to grow outwards. This, coupled with the binding and targeting specificities of the subunits, may allow assembly of the pilus organelle in the absence of an external energy source.
Acknowledgments
We thank Hiroshi Nikaido and members of his lab, Etsuko Sugawara and Emiko Rosenberg, for help with the liposome swelling assay. This work was supported by National Institutes of Health Grants R01AI29549 and R01DK51406. D.G.T. was supported by National Institutes of Heath Fellowship GM18201.
ABBREVIATIONS
- OM
outer membrane
- LPS
lipopolysaccharide
References
- 1.Klemm P. Rev Infect Dis. 1985;7:321–340. doi: 10.1093/clinids/7.3.321. [DOI] [PubMed] [Google Scholar]
- 2.Hultgren S J, Jones C H, Normark S N. In: Escherichia Coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2730–2756. [Google Scholar]
- 3.Hung D L, Knight S D, Woods R M, Pinkner J S, Hultgren S J. EMBO J. 1996;15:3792–3805. [PMC free article] [PubMed] [Google Scholar]
- 4.Bullitt E, Makowski L. Nature (London) 1995;373:164–167. doi: 10.1038/373164a0. [DOI] [PubMed] [Google Scholar]
- 5.Kuehn M J, Heuser J, Normark S, Hultgren S J. Nature (London) 1992;356:252–255. doi: 10.1038/356252a0. [DOI] [PubMed] [Google Scholar]
- 6.Striker R, Nilsson U, Stonecipher A, Magnusson G, Hultgren S J. Mol Microbiol. 1995;16:1021–1030. doi: 10.1111/j.1365-2958.1995.tb02327.x. [DOI] [PubMed] [Google Scholar]
- 7.Jones C H, Danese P N, Pinkner J S, Silhavy T J, Hultgren S J. EMBO J. 1997;16:6394–6406. doi: 10.1093/emboj/16.21.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullitt E, Jones C H, Striker R, Soto G, Jacob-Dubuisson F, Pinkner J, Wick M J, Makowski L, Hultgren S J. Proc Natl Acad Sci USA. 1996;93:12890–12895. doi: 10.1073/pnas.93.23.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuehn M J, Normark S, Hultgren S J. Proc Natl Acad Sci USA. 1991;88:10586–10590. doi: 10.1073/pnas.88.23.10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodson K W, Jacob-Dubuisson F, Striker R T, Hultgren S J. Proc Natl Acad Sci USA. 1993;90:3670–3674. doi: 10.1073/pnas.90.8.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob-Dubuisson F, Striker R, Hultgren S J. J Biol Chem. 1994;269:12447–12455. [PubMed] [Google Scholar]
- 12.Norgren M, Baga M, Tennent J M, Normark S. Mol Microbiol. 1987;1:169–178. doi: 10.1111/j.1365-2958.1987.tb00509.x. [DOI] [PubMed] [Google Scholar]
- 13.Klemm P, Christiansen G. Mol Gen Genet. 1990;220:334–38. doi: 10.1007/BF00260505. [DOI] [PubMed] [Google Scholar]
- 14.Wandersman C. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 955–966. [Google Scholar]
- 15.Salmond G P C, Reeves P J. Trends Biochem Sci. 1993;18:7–12. doi: 10.1016/0968-0004(93)90080-7. [DOI] [PubMed] [Google Scholar]
- 16.Pugsley A. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russel M. Science. 1994;265:612–614. doi: 10.1126/science.8036510. [DOI] [PubMed] [Google Scholar]
- 18.Nikaido H. Methods Enzymol. 1994;235:225–234. doi: 10.1016/0076-6879(94)35143-0. [DOI] [PubMed] [Google Scholar]
- 19.Slonim L N, Pinkner J S, Branden C I, Hultgren S J. EMBO J. 1992;11:4747–4756. doi: 10.1002/j.1460-2075.1992.tb05580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nikaido H, Nikaido K, Harayama S. J Biol Chem. 1991;266:770–779. [PubMed] [Google Scholar]
- 21.Messing J. Methods Enzymol. 1983;101:20–78. doi: 10.1016/0076-6879(83)01005-8. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura F, Zalman L S, Nikaido H. J Biol Chem. 1983;258:2308–2314. [PubMed] [Google Scholar]
- 23.Heuser J. J Electron Microsc Technol. 1989;13:244–263. doi: 10.1002/jemt.1060130310. [DOI] [PubMed] [Google Scholar]
- 24.Abraham S N, Land M, Ponniah S, Endres R, Hasty D L, Babu J P. J Bacteriol. 1992;174:5145–5148. doi: 10.1128/jb.174.15.5145-5148.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schägger H, von Jagow G. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 26.Valent Q A, Zaal J, de Graaf F K, Oudega B. Mol Microbiol. 1995;16:1243–1257. doi: 10.1111/j.1365-2958.1995.tb02346.x. [DOI] [PubMed] [Google Scholar]
- 27.Schifferli D M, Alrutz M A. J Bacteriol. 1994;176:1099–1110. doi: 10.1128/jb.176.4.1099-1110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugawara E, Steiert M, Rouhani S, Nikaido H. J Bacteriol. 1996;178:6067–6069. doi: 10.1128/jb.178.20.6067-6069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikaido H. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 29–47. [Google Scholar]
- 30.Sugawara E, Nikaido H. J Biol Chem. 1992;267:2507–2511. [PubMed] [Google Scholar]
- 31.Cowan S W, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit R A, Jansonius J N, Rosenbusch J P. Nature (London) 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 32.Gong M, Makowski L. J Mol Biol. 1992;228:735–742. doi: 10.1016/0022-2836(92)90860-m. [DOI] [PubMed] [Google Scholar]
- 33.Fath M J, Kolter R. Microbiol Rev. 1993;57:995–1017. doi: 10.1128/mr.57.4.995-1017.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee C A. Trends Microbiol. 1997;5:148–156. doi: 10.1016/S0966-842X(97)01029-9. [DOI] [PubMed] [Google Scholar]
- 35.Newhall V W J, Wilde C E, III, Sawyer W D, Haak R A. Infect Immun. 1980;27:475–482. doi: 10.1128/iai.27.2.475-482.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie K R, Lory S, Pugsley A P. EMBO J. 1996;15:978–988. [PMC free article] [PubMed] [Google Scholar]
- 37.Linderoth N A, Model P, Russel M. J Bacteriol. 1996;178:1962–1970. doi: 10.1128/jb.178.7.1962-1970.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L-Y, Chen D-Y, Miaw J, Hu N-T. J Biol Chem. 1996;271:2703–2708. doi: 10.1074/jbc.271.5.2703. [DOI] [PubMed] [Google Scholar]
- 39.Genin S, Boucher C A. Mol Gen Genet. 1994;243:112–118. doi: 10.1007/BF00283883. [DOI] [PubMed] [Google Scholar]
- 40.Russel M. Trends Microbiol. 1995;3:223–228. doi: 10.1016/s0966-842x(00)88929-5. [DOI] [PubMed] [Google Scholar]
- 41.Martin P R, Hobbs M, Free P D, Jeske Y, Mattick J S. Mol Microbiol. 1993;9:857–868. doi: 10.1111/j.1365-2958.1993.tb01744.x. [DOI] [PubMed] [Google Scholar]
- 42.Drake S L, Koomey M. Mol Microbiol. 1995;18:975–986. doi: 10.1111/j.1365-2958.1995.18050975.x. [DOI] [PubMed] [Google Scholar]
- 43.Koster M, Bitter W, de Cock H, Allaoui A, Cornelis G R, Tommassen J. Mol Microbiol. 1997;26:789–797. doi: 10.1046/j.1365-2958.1997.6141981.x. [DOI] [PubMed] [Google Scholar]
- 44.Linderoth N A, Simon M N, Russel M. Science. 1997;278:1635–1638. doi: 10.1126/science.278.5343.1635. [DOI] [PubMed] [Google Scholar]
- 45.Bitter W, Koster M, Latijnhouwers M, de Cock H, Tommassen J. Mol Microbiol. 1998;27:209–219. doi: 10.1046/j.1365-2958.1998.00677.x. [DOI] [PubMed] [Google Scholar]
- 46.Jacob-Dubuisson F, Heuser J, Dodson K, Normark S, Hultgren S J. EMBO J. 1993;12:837–847. doi: 10.1002/j.1460-2075.1993.tb05724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]