

Abstract
Inflammation is associated with obesity and insulin resistance. Proinflammatory cytokines produced by adipose tissue in obesity could alter insulin signaling and action. Recent studies have shown a relationship between interleukin (IL)-1β amount and metabolic syndrome or type 2 diabetes. However, the ability of IL-1β to alter insulin signaling and action remains to be explored. We demonstrated that IL-1β sligthly increased Glut 1 translocation and basal glucose uptake in 3T3-L1 adipocytes. Importantly, we found that prolonged-IL-1β treatment reduced the insulin-induced glucose uptake whereas an acute treatment had no effect. Chronic treatment with IL-1β slightly decreased the expression of Glut 4 and markedly inhibited its translocation to the plasma membrane in response to insulin. This inhibitory effect was due to a decrease in the amount of IRS-1 but not IRS-2 expression both in 3T3-L1 and human adipocytes. The decrease in IRS-1 amount resulted in a reduction in its tyrosine phosphorylation and in the alteration of insulin-induced PKB activation and AS160 phosphorylation. Pharmacological inhibition of ERK totally inhibited IL-1β–induced down regulation of IRS-1 mRNA. Moreover, IRS-1 protein expression and insulin-induced PKB activation, AS160 phosphorylation and Glut 4 translocation were partially recovered following treatment with the ERK inhibitor. These results demonstrate that IL-1β reduces IRS-1 expression at a transcriptional level through a mechanism that is ERK dependent and at a posttranscriptional level independently of ERK activation. By targeting IRS-1, IL-1β is capable of impairing insulin signaling and action, and could thus participate in concert with other cytokines, in the development of insulin resistance in adipocytes.
Keywords: 3T3-L1 Cells; Adipocytes; drug effects; immunology; metabolism; Animals; Down-Regulation; drug effects; immunology; Extracellular Signal-Regulated MAP Kinases; metabolism; GTPase-Activating Proteins; metabolism; Gene Expression; drug effects; immunology; Glucose; metabolism; Glucose Transporter Type 1; genetics; metabolism; Glucose Transporter Type 4; genetics; metabolism; Inflammation; immunology; metabolism; Insulin Resistance; immunology; Interleukin-1beta; immunology; pharmacology; Interleukin-6; pharmacology; MAP Kinase Signaling System; drug effects; immunology; Mice; Mice, Obese; Phosphoproteins; genetics; Phosphorylation; drug effects; Proto-Oncogene Proteins c-akt; metabolism; Tyrosine; metabolism
INTRODUCTION
The prevalence of obesity and type 2 diabetes characterised by an insulin resistance has increased considerably in recent years (1). Although the molecular mechanisms leading to insulin resistance are not fully understood, accumulation of adipose tissue appears to be closely related to the development of insulin resistance (2). It is now clearly established that adipose tissue is not only a storage organ for excess calories but also an endocrine organ that secretes various factors collectively called adipokines that regulate glucose homeostasis, food intake, and energy expenditure (2). It has been recently determined that obesity and insulin resistance are associated with a low-grade chronic systemic inflammation. The origin of this inflammation could be related to adipose tissue expansion because the expression of pro-inflammatory cytokines including TNF-α, IL-1 and IL-6 is increased in this tissue in obese state (3, 4). Increased macrophage population in adipose tissue is thought to be responsible for this elevated production of cytokines (5, 6). Proinflammatory cytokines produced in adipose tissue could alter the endocrine function of the tissue and could impinge insulin signaling and action in adipocytes, liver and muscles leading to the development and/or the aggravation of insulin resistance (4).
Insulin regulates blood glucose level through suppression of hepatic endogenous glucose production and stimulation of glucose uptake in muscle and adipocyte. These biological responses require the tyrosine phosphorylation of the IRS-1 and/or IRS-2 proteins that in turn bind and activate PI 3-kinase. Downstream effectors of PI 3-kinase such as protein kinase B (PKB) are involved in insulin metabolic effects (7). Several reports have shown that TNF-α and IL-6 alter insulin signaling by targeting IRS-1 proteins. TNF-α increases the serine phosphorylation of IRS-1. This mechanism reduces its tyrosine phosphorylation by the insulin receptor (8–10) and several kinases including JNK (11), mTOR (12, 13) and ERK (13–15) have been implicated in the serine phosphorylation of IRS-1. TNF-α and IL-6 also enhance the expression of SOCS (Suppressor of Cytokine Signaling) proteins that can attenuate insulin signaling by binding to the insulin receptors and reducing their ability to phosphorylate IRS proteins (16, 17). Alternatively, SOCS proteins can bind directly to IRS proteins leading to their degradation (18, 19). Finally, TNF-α and IL-6 could inhibit IRS-1 expression at the transcriptional level (20–22). In obesity, alteration of IRS-1 tyrosine phosphorylation in muscle is not linked to a change in its expression. By contrast, a down-regulation of IRS-1 mRNA expression seems to be the major mechanism involved in alteration in IRS-1 tyrosine phosphorylation in adipocytes of obese rodents and in adipocytes from type 2 diabetic subjects, obese patients and relatives of diabetic subjects (23, 24).
Whereas the implication of TNF-α and IL-6 insulin resistance is well documented, little is known about a potential role of IL-1β. IL-1β is one of the major pro-inflammatory cytokines that is produced by monocytes and macrophages (25). IL-1β exerts its biological function by binding to IL-1 type I receptor and activates the IKK/NF-κB pathway and the three types of mitogen-activated protein (MAP) kinases ERK, JNK and p38MAPK (25). Recent studies suggest that IL-1β could also belong to the network of cytokines involved in insulin resistance. Indeed, in a case/control study, individuals with detectable circulating levels of IL-1β and elevated levels of IL-6 have an increased risk to develop type 2 diabetes compared with individuals with increased concentrations of IL-6 but undetectable levels of IL-1β (26). Further, IL-1β concentration is elevated in non-diabetic offspring of diabetic individuals and is correlated with the metabolic syndrome (27). Finally, expression of both IL-1β and its receptor is increased in visceral adipose tissue of obese subjects (28).
However, whether and how this overproduction of IL-1β could alter the metabolic function of insulin in adipocytes remains unclear. In the present study, we found that IL-1β markedly inhibits insulin-induced glucose transport in adipocytes by decreasing IRS-1 expression. Further, we demonstrate that activation of the ERK pathway is involved in the inhibitory action of IL-1β on insulin signaling.
MATERIAL AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum, and calf serum were obtained from Life Technologies (St Louis, MO, USA). Insulin was from Lilly (Paris, France). Recombinant murine IL-1β and IL-6 were from AbCys S.A (Paris, France). U0–126 and PD-169316 were from Calbiochem (La Jolla, CA, USA). Polyvinylidene difluoride (PVDF) membranes were purchased from Millipore (Bedford, MA, USA). BCA reagent was obtained from Pierce Biotechnology (Rockford, I11, USA). Protease inhibitors cocktails were obtained from Roche Diagnostics (Mannheim, Germany). Enhanced chemiluminescence reagent was purchased from PerkinElmer Life Sciences (Boston, MA, USA). All other chemical reagents were purchased from Sigma (St Louis, MO, USA).
Antibodies against IRS-2 and phosphotyrosine were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Polyclonal IRS-1 antibody used in immunoprecipitation experiments was raised against a peptide corresponding to the last 14 amino-acids of IRS-1 (Eurogentec, Seraing, Belgium). Monoclonal anti-IRS1 antibody used in immunoblotting experiments was purchased from BD Biosciences (Pharmingen, San Diego, CA). Antibodies against the β subunit of Insulin Receptor, Glut 4, Glut 1 and IκB were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phospho-PKB(Thr308), PKB, phospho-ERK, ERK, phospho-JNK1/2, JNK1/2, phospho-p38MAPK, and p38MAPK were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-phospho-AS160 (T642) was purchased from Biosource (Biosource International Inc, Camarillo, CA) and anti-AS160 (TBC1D4) antibody was from Abcam (Abcam Ltd, Cambridge, UK). Horseradish peroxydase-conjugated and FITC-coupled secondary antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove, PA).
Animals
ob/ob, db/db mice and their lean control littermates were purchased from Charles River Laboratories (St. Aubin les Elbeuf, France) and housed at the animal facility of the Faculty of Medicine (Nice, France). Mice were maintained on a 12h:12h light:dark cycle and were provided free access to water and standard rodent show. Mice were killed by cervical dislocation, and epididymal fat pads were removed and freeze-clamped in liquid nitrogen. Principles of laboratory animals care were followed and the Ethical Committee of the Faculty of Medicine approved the animal experiments.
Cells culture
3T3-L1 fibroblasts were grown at 7 % CO2 and 37 C in 35 or 100 mm dishes in DMEM, 25 mM glucose, 10% calf serum, and induced to differentiate in adipocytes as previously described (13). Briefly, 2 days after confluence, medium was changed for DMEM, 25 mM glucose, 10% fetal calf serum (FCS), supplemented with isobutylmethylxanthine (0.25 mM), dexamethasone (0.25 μM), insulin (5 μg/ml), and pioglitazone (10 μM). The medium was removed after 2 days and replaced with DMEM, 25 mM glucose, 10% FCS, supplemented with insulin (5 μg/ml) and pioglitazone (10 μM) for 2 days. Then, the cells were fed every 2 days with DMEM, 25 mM glucose, 10% FCS. 3T3-L1 adipocytes were used 8–15 days after the beginning of the differentiation protocol.
Human preadipocytes (Biopredic, Rennes, France) were grown at 5% CO2 and 37 C in 12-wells collagen-coated plates in DMEM Ham’s F12 containing 15 mM Hepes, 2 mM L-Glutamine, 5% fetal calf serum, 1% antimycotic solution, ECGS/H-2, hEGF-5, and HC-500 from Supplement Pack Preadipocyte Growth Medium (Promocell). Differentiation in adipocytes was induced after confluence by changing the medium for DMEM Ham’s F12 15 mM Hepes, 2 mM L-glutamine, 3% fetal calf serum, supplemented with biotin (33 μM), insulin (100 nM), pantothenate (17 μM), isobutylmethylxanthine (0.2 mM), dexamethasone (1 μM), rosiglitazone (10 μM). The medium was removed after 3 days and replaced with Ham’s F12 containing 15 mM Hepes, 2 mM L-glutamine, 10% FCS, supplemented with biotin (33 μM), insulin (100 nM), pantothenate (17 μM) and dexamethasone (1 μM). Then, the cells were fed every 2 days with the same medium. Human adipocytes were used 15 days after the beginning of the differentiation protocol.
Immunoprecipitation and immunoblotting
3T3-L1 adipocytes were treated as indicated in figure legends for different periods of time at 37 C, 7% CO2 in DMEM 25 mM glucose, 10% fetal calf serum (FCS). The cells were washed with ice-cold buffer (20 mM Tris pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 10 mM pyrophosphate, 2 mM sodium orthovanadate) before solubilization for 1 hour at 4°C in lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 10 mM pyrophosphate, 2 mM sodium orthovanadate, 100 nM okadaic acid, protease inhibitors, and 1% Triton X-100 (v/v)). Following centrifugation at 14 000 g for 10 min at 4 C, the supernatant (cells lysates) was incubated for 4 hours at 4 C with antibody of interest (5–10 μg/sample) preadsorbed on protein-A-Sepharose beads. The beads were washed 3 times with the lysis buffer and boiled for 5 min in Laemmli buffer. The proteins were separated by SDS-PAGE using a 7.5 or 10% resolving gel. Proteins were transferred to PVDF membrane and the membrane was blocked with saline buffer (10 mM Tris pH 7.4, 320 mM NaCl, 0.1 % Tween) containing 5% (w/v) non-fat dry milk for 1 hour at room temperature and incubated overnight at 4 C with the indicated antibody. Following incubation with horseradish-peroxydase conjugated secondary antibodies, proteins were detected by enhanced chemiluminescence (ECL). Some membranes were subsequently incubated at 55 C for 30 min in stripping buffer (62 mM Tris pH 6.7, 100 mM 2-mercaptoethanol, and 2% SDS) and reprobed with the indicated antibody.
Glucose transport
3T3-L1 adipocytes were incubated or not with IL-1β at 20 ng/ml for 48 hours as indicated in the figure legends. Cells were then washed twice with Krebs-Ringer phosphate buffer (10 mM phosphate buffer, pH 7.4, 1.25 mM MgSO4, 1.25 mM CaCl2, 136 mM NaCl, 4.7 mM KCl) and incubated without or with 0.5 or 100 nM insulin for 20 min in Krebs-Ringer phosphate buffer supplemented with 0.2% bovine serum albumin. Glucose transport was determined by the addition of 2-[3H]deoxyglucose (0.1 mM, 0.5 μCi/ml) as described previously (29). The reaction was stopped after 3 min at 37 C by washing the cells four times with ice-cold PBS. Cells were lysed in lysis buffer, and glucose uptake was assessed by scintillation counting. Results were normalized for protein content measured by BCA assay.
Real-time quantitative PCR
Total RNAs from 3T3-L1 adipocytes or white adipose tissues were prepared using Trizol reagent (Life Technologies Inc, UK). The integrity of the RNA was confirmed by electrophoresis in ethidium bromide containing agarose gels and the RNA concentration was determined spectrophotometrically. cDNA was synthesized using MMLV transcriptase (Invitrogen) from 1 μg of total RNA. PCRs were performed using an AbiPrism 7700 Sequence Detection System instrument and software (Applied Biosystems). The PCR conditions were: 2 min at 50 C, 10 min at 95°C followed by forty cycles of a two-step PCR reaction denaturation at 95 C for 15 s and annealing extension at 60°C for 60 s. Each sample contained 0.5 to 5 ng cDNA in 1X SYBR®Green PCR Master Mix (Eurogentec, Seraing, Belgium) and 200 or 400 nM of each primer (Invitrogen) in a final volume of 25 μl. A control without cDNA was performed for each experiment. The number of cycles (CT) required for the fluorescence to reach a threshold limit was determined in duplicate for each sample. For each target an efficiency of the PCR method between 95 and 100%, a reproducibility of Ct values with a standard error less than 3% and a linear range of covering more at least 7 log units were obtained. To exclude the contamination of nonspecific PCR products such as primer dimers, melting curve analysis was applied to all final PCR products after the cycling protocols. The relative amounts of the different mRNAs were quantified by using the second derivative maximum method. 36B4 was used as an invariant control, and the relative quantification for a given gene was corrected to 36B4 mRNA values. The results were expressed relative to the control condition, which was arbitrary assigned a value of 1. Primers used for IRS-1 were (sense) 5′-GTGAACCTCAGTCCCAACCATAAC-3′, (anti sense) 5′-CCGGCACCCTTGAGTGTCT-3′; for IRS-2 (sense) 5′-TCCCACATCGGGCTTGAA-3′, (anti sense) 5′-CTGCACGGATGACCTTAGCA-3′; for PPARγ (sense) 5′-CTGTTTTATGCTGTTATGGGTGAAA-3′; (anti sense) 5′-CGACCATGCTCTGGGTCAA-3; for C/EBPα (sense) 5′-GACCATTAGCCTTGTGTGTACTGTATG-3′, (anti sense) 5′-TGGATCGATTGTGCTTCAAGTT-3′; for 36B4 (sense) 5′-TCCAGGCTTTGGGCATCA-3′, (anti sense) 5′-CTTTATCAGCTGCACATCACTCAGA-3.
Preparation of plasma membrane sheets and immunofluorescence labeling
3T3-L1 cells were grown on glass coverslips and differentiated into adipocytes as described above. Cells were treated without or with IL-1β at 20 ng/ml for 48 hours, and stimulated or not with insulin (100 nM) for 20 min. Plasma membrane sheets were prepared as previously described (30). Cells were washed twice with ice-cold PBS, and fixed with 0.55 mg/ml Poly- L-lysine for 1 min at 4°C and then swollen by three successive rinses with an hypotonic buffer (30 mM HEPES pH 7.5, 70 mM KCl, 5 mM MgCl2, 3 mM EGTA). The swollen cells were sonicated in hypotonic buffer containing 1 mM DTT and proteases inhibitors and the bound membrane sheets were fixed with 4% paraformaldehyde and blocked with PBS containing 1% BSA and 4% calf serum. Plasma membrane sheets were then incubated with anti-Glut 4 or anti-Glut 1 antibodies (5 μg/ml in blocking buffer) for 1 hour at room temperature and washed three times 10 min with blocking buffer. Following washes, lawns were incubated for 1 hour at room temperature with FITC-conjugated anti-goat antibodies and WGA-Texas Red to normalize, then rinsed out with three 10 min washes with blocking buffer. The coverslips were mounted in Mowiol onto glass slides. Plasma membrane sheets were analyzed with an Axiovert 200 microscope using a Plan-Neofluar 40 X 1.3 numeral aperture oil objective (Carl Zeiss, Göttingen, Germany). Images were acquired using a cooled digital camera (Coolsnap HQ, Roger Scientific Princeton Instruments, Evry, France) and quantification was made using Metamorph image analysis software (Universal Imaging Corporation, Downington, PA) with autothreshold detection of pixels as previously described (30).
Statistical Analysis
Statistical analysis was performed by Student’s t test or Mann-Whitney test. A p value <0.05 was considered significant
RESULTS
Effect of acute and chronic IL1-β treatments on insulin-induced glucose transport in 3T3-L1 adipocyte
The likelihood that 3T3-L1 adipocytes are target cells for IL-1β was investigated by incubating 3T3-L1 adipocytes with IL-1β for 20 min. IL-1β incubation induced the degradation of IκBα and the phosphorylation of the MAP kinases ERK, JNK and p38MAPK (Fig. 1A). These data indicate that 3T3-L1 adipocytes are responsive to IL-1β stimulation.
Figure 1.
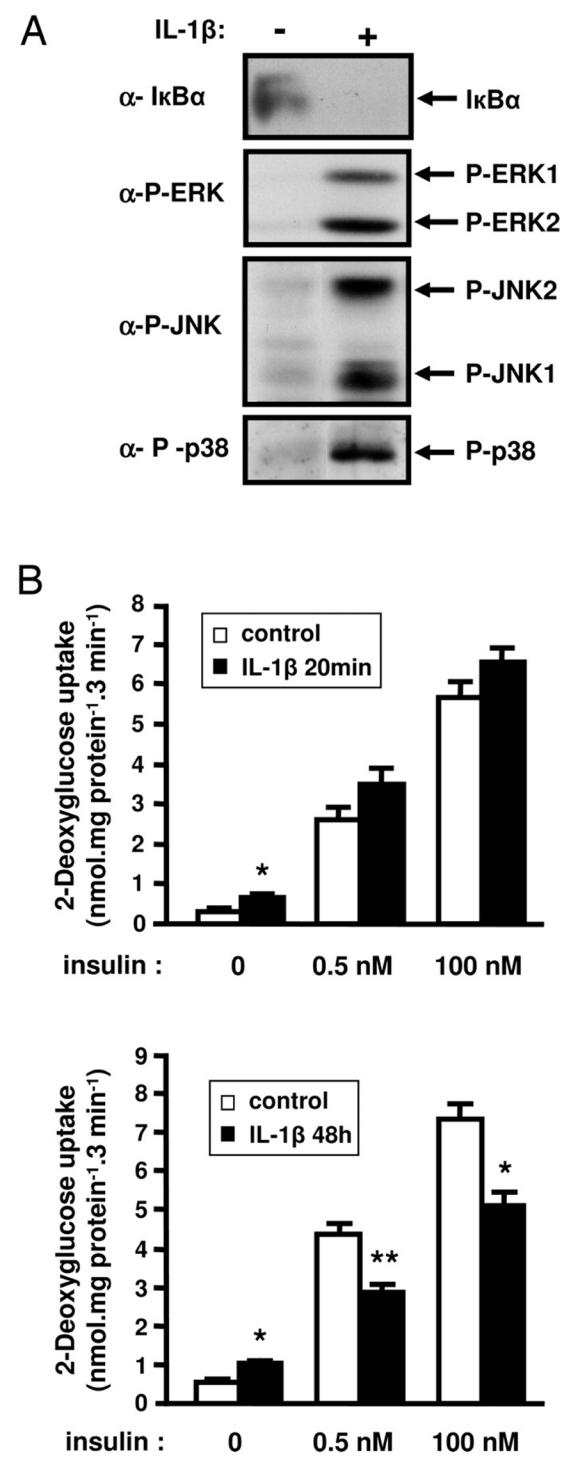
Effect of short or long-term IL-1β treatment on insulin-induced glucose transport in 3T3-L1 adipocytes. A, 3T3-L1 adipocytes were treated with IL-1β for 20 min or left untreated and proteins from cell lysates were prepared as described in “Material and Methods”. IkBα and the phosphorylated forms of ERK, JNK and p38MAPK were detected using specific antibodies. Typical autoradiographs representative of three independent experiments are shown. B, 3T3-L1 adipocytes were treated without (empty bars) or with (black bars) IL-1β (20 ng/ml) for 20 min (upper panel) or 48 h (lower panel) and then cells were incubated without or with insulin (0.5 or 100 nM) for 20 min at 37°C. Uptake of [2-3H]deoxyglucose was measured during a 3-min period as described under “Materials and Methods”. Means ± SEM of five independent experiments are shown. *, IL-1β effect significant with p < 0.01. **, IL-1β effect significant with p < 0.05.
We then determined whether IL-1β was able to induce an insulin-resistant state for glucose transport in 3T3-L1 adipocytes. Cells were incubated with IL-1β (20 ng/ml) and then incubated without or with insulin (0.5 or 100 nM). Treatment of 3T3-L1 adipoytes with IL-1β for 20 min slightly increased the basal rate of glucose uptake but did not modify insulin-induced glucose uptake (Fig. 1B, upper panel). Importantly, IL-1β treatment of 3T3-L1 adipocytes for 48 h reduced the absolute response at both insulin concentration (Fig. 1B, lower panel). Thus, the increment in glucose uptake at 0.5 nM insulin was 3.91 ± 0.21 and 1.71 ± 0.12 nmol·mg−1· 3 min−1 for control or IL-1β treated cells respectively and at 100 nM insulin, this increment was 6.90 ± 0.48 and 4.01 ± 0.42 nmol·mg−1· 3 min−1 for the same conditions. Further, 0.5 nM insulin induced 56.6 ± 0.8 % versus 42.6 ± 0.4 % of the maximal insulin effect in the absence or presence of IL-1β respectively, indicating that the insulin sensitivity was decreased.
Effect of IL-1β on glucose transporters expression and insulin-induced glucose transporters translocation
We first examined whether the inhibitory effect of IL-1β on glucose uptake was due to a change in glucose transporters expression. 3T3-L1 adipocytes were treated without or with IL-1β for 48 h and the amount of Glut 1 and Glut 4 in cell lysate was quantified by immunoblotting with specific antibodies. As shown in Fig. 2A, IL-1β treatment increased the amount of Glut 1 and slightly decreased Glut 4 expression but did not modify the differentiation state of adipocyte because C/EBPα, PPARγ, and aP2 mRNAs expression was not altered (Fig. 2B). The increased Glut 1 expression induced by IL-1β was associated with an increase in the amount of the transporter at the plasma membrane (Fig. 2C, left panel) and could explain the enhancement of glucose uptake induced by IL-1β alone. Importantly, prolonged IL-1β treatment did not modify the basal amount of Glut 4 at the plasma membrane but inhibited by 50 % the amount of Glut 4 at the plasma membrane following insulin stimulation. (Fig. 2C, right panel). These results indicated that IL-1β can trigger an insulin resistant state for glucose transport by altering insulin-induced Glut 4 translocation.
Figure 2.
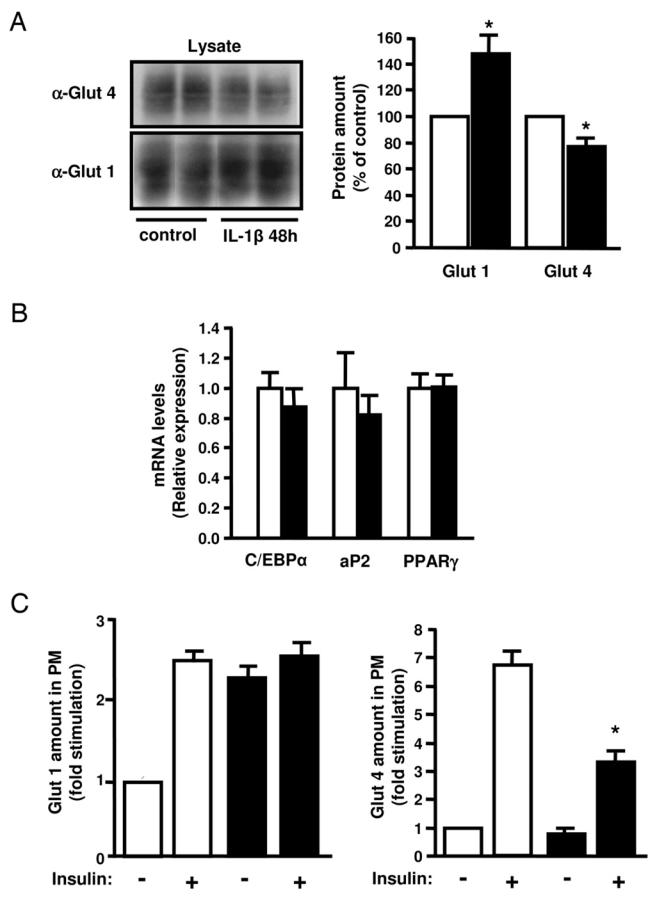
Effect of IL-1β treatment on Glut 4 and Glut 1 expression and insulin-induced Glut 4 and Glut 1 translocation to the plasma membrane in 3T3-L1 adipocytes. A, 3T3-L1 adipocytes were incubated without (empty bars) or with (black bars) IL-1β (20 ng/ml) for 48 h. A, Glut 4 and Glut 1 were detected in cell lysates by immunoblotting with anti-Glut4 or anti-Glut 1 antibodies (α-Glut 4, α-Glut 1). Glut 4 and Glut 1 amounts were quantified by densitometry scanning analysis. Data are expressed as percentage of Glut 4 and Glut 1 amounts in untreated cells and presented as the means ± SEM of four independent experiments. (* p < 0.01). B, total RNAs from cells treated as described in A were extracted and the amount of C/EBPα, aP2 and PPARγ mRNA were quantified by real-time quantitative PCR. mRNA expression was normalized using 36B4 RNA levels and expressed in arbitrary units, with the control cells taken as 1. Results are expressed as the means ± SEM of three independent experiments. C, Plasma membrane sheets were prepared from 3T3-L1 adipocytes incubated without (empty bars) or with IL-1β (20 ng/ml) (black bars) for 48 h and then without or with insulin (100 nM) for 20 min. Glut 1 (Left) and Glut 4 (Right) were detected in plasma membrane (PM) by immunofluorescence using goat anti-Glut 1 or anti-Glut 4 antibodies followed by incubation with FITC-coupled anti-goat antibodies. Quantification of fluorescence level was performed with MetaMorph software as described in “Materials and Methods”. Results are expressed as fold stimulation of control cells and presented as the means ± SEM of three independent experiments. *, IL-1β effect significant with p < 0.05.
Differential effect of IL-1β on insulin-induced tyrosine phosphorylation of IRS proteins
Because IL-1β altered insulin-induced Glut4 translocation, we studied the effect of the cytokine on proximal insulin signaling steps involved in this process. We first analyzed the effect of IL-1β on the insulin receptor tyrosine phosphorylation. 3T3-L1 adipocytes were pretreated or not with 20 ng/ml IL-1β for 48 h and then stimulated or not with insulin. The insulin-induced tyrosine phosphorylation of the insulin receptor, which was analyzed by immunoblotting using anti-phosphotyrosine antibodies, was not altered following IL-1β treatment (Fig. 3). We then determined whether IL-1β could alter the insulin-induced IRS tyrosine phosphorylation. IRS-1 or IRS-2 were immunoprecipitated from 3T3-L1 adipocytes with specific antibodies and their tyrosine phosphorylation was quantified by immunoblotting with anti-phosphotyrosine antibody. As shown in Fig. 3, IL-1β alone did not induce IRS-1 or IRS-2 tyrosine phosphorylation. Importantly, IL-1β inhibited insulin-induced IRS-1 tyrosine phosphorylation by 60% but did not modify insulin-induced IRS-2 tyrosine phosphorylation. In parallel to the decrease in IRS-1 tyrosine phosphorylation, less IRS-1 was immunoprecipitated in IL-1β-treated cells suggesting that IL-1β could alter IRS-1 expression. To directly assess this possibility, we analyzed IRS-1 expression in homogenates prepared from 3T3-L1 adipocytes treated for 24 or 48h with IL-1β. IL-1β decreased IRS-1 protein expression by 50% and 60% at 24 and 48 h, respectively (Fig. 4A) whereas IRS-2 amount was unchanged (data not shown).
Figure 3.
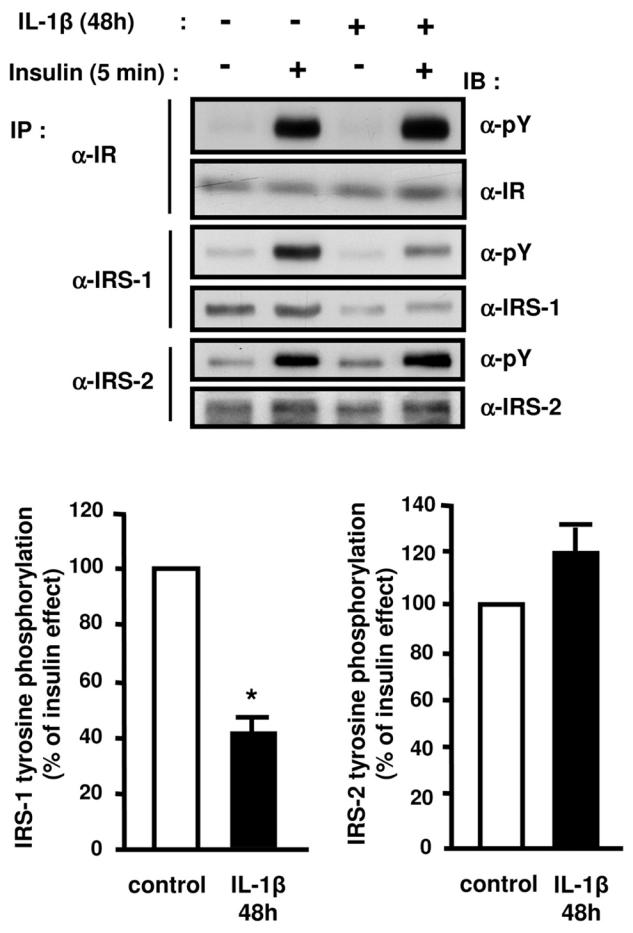
Differential effects of IL-1β treatment on insulin-stimulated tyrosine phosphorylation of the insulin receptor, IRS-1 and IRS-2. 3T3-L1 adipocytes were treated without or with IL-1β (20 ng/ml) for 48 h and then without or with insulin for 5 min. Proteins were immunoprecipitated (IP) using anti-insulin receptor (α-IR) antibodies, anti-IRS1 (α-IRS1) or anti-IRS2 (α-IRS2) antibodies and immunoprecipitated proteins were resolved by SDS-PAGE and blotted (IB) using anti-phosphotyrosine (α-pY) antibodies. The membrane was then stripped and probed using α-IR, α-IRS1, or α-IRS2 antibodies. Typical autoradiographs representative of four to five experiments are shown. The absolute Tyr-phosphorylation of IRS-1 or IRS-2 was quantified by densitometry scanning analysis. Results are expressed as percentage of insulin effect in control cells and presented as the means ± SEM of five independent experiments. *, IL-1β effect significant with p < 0.01.
Figure 4.
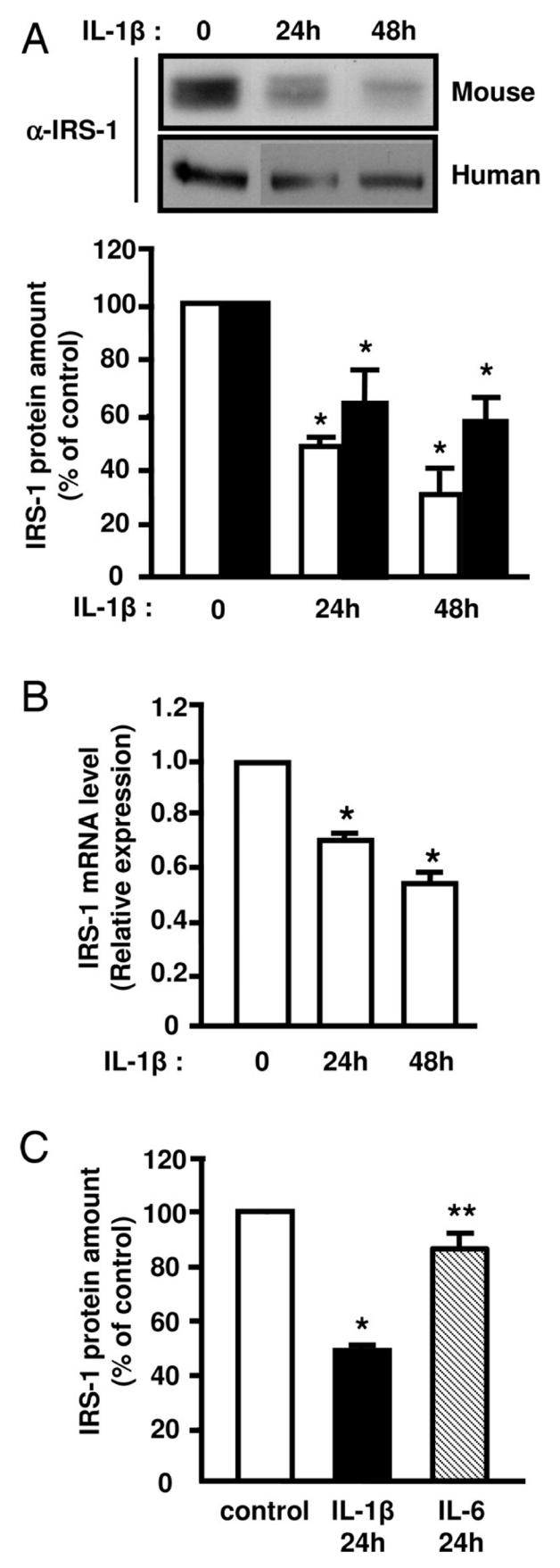
IL-1β treatment decreases IRS-1 expression in 3T3-L1 adipocytes and in human adipocytes. Mouse 3T3-L1 adipocytes (empty bars) or human preadipocytes-derived adipocytes (black bars) were incubated without or with IL-1β (20 ng/ml) for 24 or 48 hours. Proteins from cell lysates were blotted using anti-IRS1 (α-IRS1) antibodies. A, Representative autoradiograph is shown, and IRS-1 amount was quantified by densitometry scanning analysis. Results are expressed as percentage of IRS-1 protein amount in control cells and presented as the means ± SEM of four independent experiments. *, IL-1β effect significant for 3T3-L1 adipocytes or human adipocytes respectively with p < 0.01. B, Total RNAs were extracted and the relative amounts of IRS-1 mRNA were determined by real-time PCR. mRNA expression was normalized using 36B4 RNA levels. Results are expressed in arbitrary units, with the control values taken as 1 and are the means ± SEM of three independent experiments. *, IL-1β effect significant with p < 0.01. C, 3T3-L1 adipocytes were left untreated (empty bar) or incubated with IL-1β (20 ng/ml) (black bar) or IL-6 (20 ng/ml) (hatched bar) for 24 hours and IRS-1 amount was determined as described in A. Results are expressed as percentage of IRS-1 protein amount in control cells and presented as the means ± SEM of four independent experiments. * p < 0.05 control versus IL-1β, ** p <0.05 IL-1βversus IL-6.
Because IRS-1 amount is dramatically decreased in adipocytes of obese patients, we investigated whether IL-1β could also down-regulate IRS-1 expression in human adipocytes. As shown in Fig. 4A, IL-1β markedly reduced IRS-1 amount in human adipocytes suggesting that IL-1β could participate in the negative regulation of IRS-1 in adipocyte of obese subjects.
We then found, by using real-time PCR, that IL-1β treatment decreased IRS-1 mRNA amount by 40% (Fig 4B) indicating that the decrease in IRS-1 protein was partly linked to a reduced gene expression. In agreement with these in vitro finding, IL-1β mRNA expression was increased in two models of obese mice and IRS-1 mRNA was decreased whereas IRS-2 mRNA level was not markedly reduced (Fig. 5).
Figure 5.
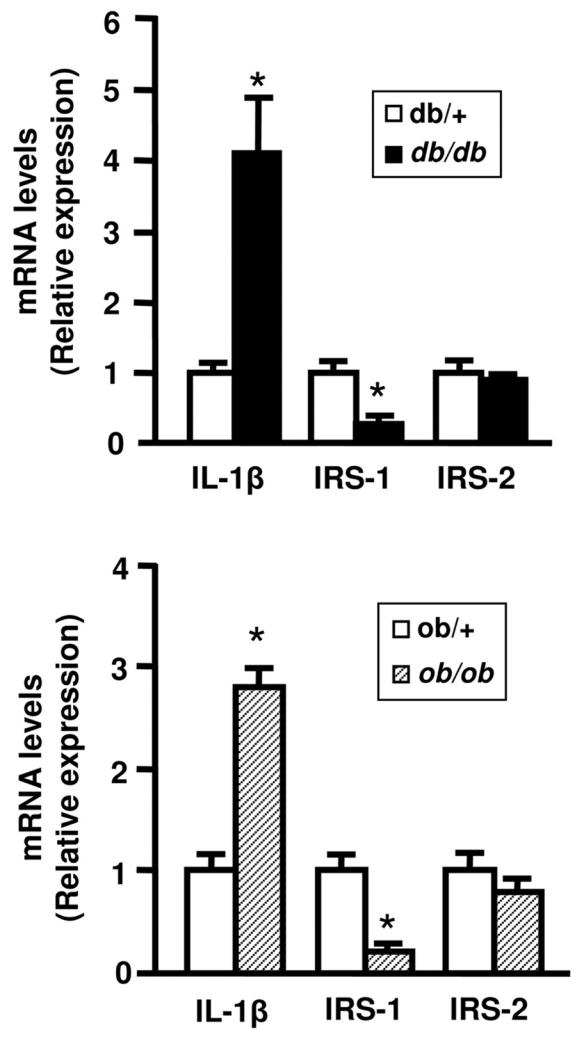
mRNA expression of IL-1β, IRS-1 and IRS-2 in adipose tissue of obese mice. Total RNA were prepared from adipose tissue of lean (white bars) and obese mice (db/db, black bars or ob/ob, hatched bars). The expression level of mRNA were analyzed by real-time quantitative PCR, normalized to the level 36B4 mRNA and expressed in arbritary units, with the control values taken as 1. Results are the means ± SEM of n=6/group. *, Significantly different from lean mice with p < 0.05.
Taken together, these results indicate that IL-1β specifically reduced the amount of IRS-1 leading to a decrease in the amount of tyrosine phosphorylated IRS-1 following insulin stimulation.
IL-1β is more potent than IL-6 to inhibit IRS-1 expression
Because IL-1β increases IL-6 production in fat cells (31) and because IL-6 was shown to down-regulate IRS-1 expression (20), we compared the ability of IL-1β and IL-6 to regulate IRS-1 expression. Treatment of 3T3-L1 adipocytes with IL-6 (20 ng/ml) for 24 h slightly decreased IRS-1 amount whereas in the same conditions, IL-1β induced a 50% inhibition in IRS-1 expression (Fig. 4C). These data indicate that IL-1β is more potent than IL-6 to down-regulate IRS-1 expression and from these data, it is unlikely that the observed effect on IRS-1 expression is mediated by an increase in IL-6 production.
Insulin-induced PKB activation and AS160 phosphorylation are reduced in IL-1β treated 3T3-L1 adipocytes
Activation of PKB following tyrosine phosphorylation of IRS-1 or IRS-2 is a critical step for insulin-induced glucose transport and Glut 4 translocation (7). Because IL-1β differentially regulated the expression and the tyrosine phosphorylation of these proteins, we aimed at determining the effect of IL-1β treatment on insulin-induced PKB activation. PKB activation was monitored by immunoblotting with a phosphospecific antibody against Threonine 308 located in the activation loop of PKB, phosphorylation which correlates with PKB activation. Importantly, following IL-1β treatment, the insulin-induced phosphorylation of PKB was reduced by 50% without any change in the total amount of PKB (Fig. 6). We then assessed the effect of IL-1β on phosphorylation of the PKB substrate AS160 (Akt substrate 160). Indeed, PKB-induced phosphorylation of AS160, a protein containing a Rab GAP (GTPase activating protein) domain promotes Glut 4 translocation to the plasma membrane (32, 33). Using a phosphospecific antibody against the PKB phosphorylation site on AS160, we found that IL-1β treatment markedly reduced insulin-induced AS160 phosphorylation (Fig. 6). This data confirmed that PKB activity was reduced following IL-1β treatment, and the reduced AS160 phosphorylation may provide a mechanism for IL-1β–induced insulin resistance on Glut 4 translocation and glucose uptake.
Figure 6.
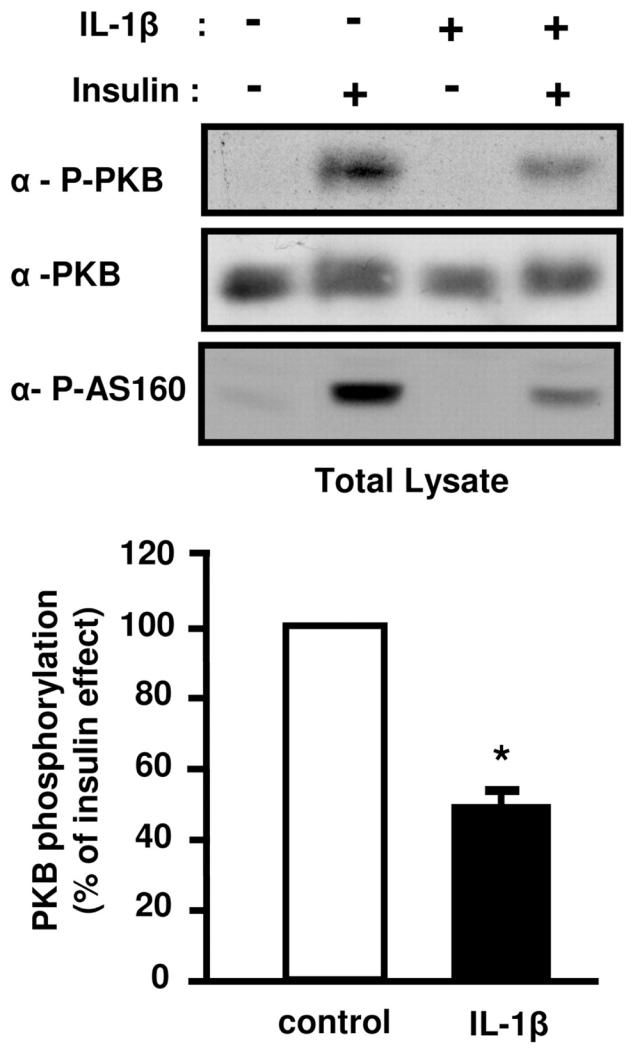
IL-1β decreases insulin-induced PKB and AS160 phosphorylation. 3T3-L1 adipocytes pretreated or not with IL-1β (20 ng/ml) for 48 h were stimulated or not with insulin (0.5 nM) for 5 min. Cells were then lysed. Top, Phosphorylation of PKB and AS160 were monitored by immunoblotting with anti-phospho-PKB (Thr308) (α-P-PKB) or with anti-phospho-AS160 (Thr642) (α-P-AS160) antibodies respectively. Representative autoradiographs are shown. Bottom, PKB phosphorylation was normalized for the total amount of PKB and results are expressed as the means ± SEM of four independent experiments. *, IL-1β effect significant with p < 0.01.
ERK pathway is involved in IL-1β induced alteration in insulin signaling
The activity of the MAPK kinases family members ERK, JNK and p38MAPK is increased in adipose tissue of insulin resistant type 2 diabetic patients or in obese animals. Further, several studies have evidenced down-regulatory effect of the MAP kinases on insulin signaling (8). Because IL-1β activates the MAP kinases family (25), we investigated whether their activation could be involved in the inhibition of insulin signaling. Amount of phosphorylated ERK-1 and -2 or p38MAPK was significantly higher in 3T3-L1 adipocytes treated with IL-1β compared to control cells (3 ± 0.8 fold; p = 0.03 for ERK−1/−2 and 4.4 ± 1.5 fold; p = 0.02 for p38MAPK, Fig. 7), whereas phosphorylated JNK was not detected (data not shown). For directly assessing the role of ERK and p38MAPK in IL-1β-induced down-regulation of IRS-1 expression and PKB activation, 3T3-L1 adipocytes were treated with 10 μM U0126 or PD169316, potent inhibitors of ERK or p38MAPK pathways respectively, and incubated without or with IL-1β for 48 h. Incubation with PD169316 did not prevent the down-regulation of IRS-1 expression induced by IL-1β (data not shown). By contrast, U0126 that totally inhibited ERK activation (Fig. 8A) fully prevented the decrease in IRS-1 mRNA amount induced by IL-1β treatment (Fig. 8B) but only partially prevented the decrease in IRS-1 protein expression (Fig. 8C). These data indicate that ERK activation plays a major role in the negative regulation of IRS-1 expression induced by IL-1β. To determine whether insulin-induced PKB activation could be rescued by blocking ERK pathway, adipocytes were treated as described above and stimulated without or with insulin for 5 min. Inhibition of ERK with U0126 partially overcame the inhibitory effect of IL-1β. (Fig. 9A, B) on PKB and AS160 phosphorylation. Further, inhibition of ERK with U0126 also partially prevented the inhibitory effect of IL-1β on insulin-induced Glut 4 translocation to the plasma membrane (Fig. 9C). These data are in favour of the importance of ERK activation by IL-1β in the inhibition of insulin signaling and Glut 4 translocation.
Figure 7.
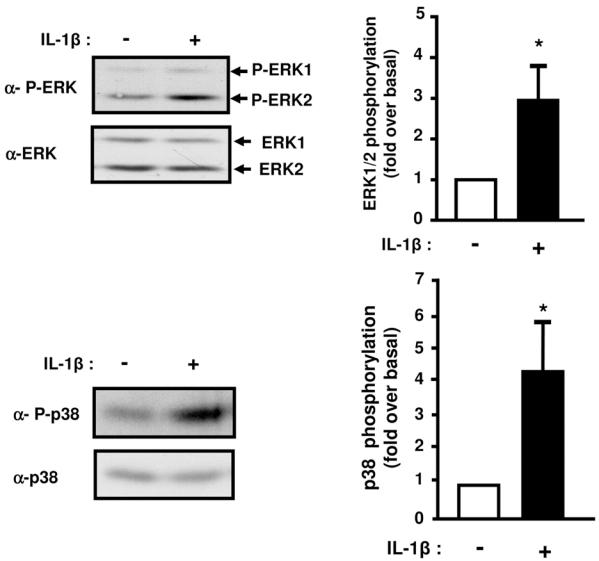
Prolonged IL-1β treatment induces ERK1/2 and p38MAP Kinases phosphorylation. 3T3-L1 adipocytes were treated without or with IL-1β (20 ng/ml) for 48 hours. Proteins from cell lysates were blotted using anti-phospho ERK1/2 (α-P-ERK) or anti-phospho-p38MAPK (α-P-p38) antibodies. The membranes were then stripped and probed using anti-ERK1/2 (α-ERK1/2) or anti-p38 (α-p38) antibodies. Representative immunoblots and means ± SEM of four independent experiments are shown. Results are expressed as fold phosphorylation over basal set to 1 in untreated cells (* p < 0.05).
Figure 8.
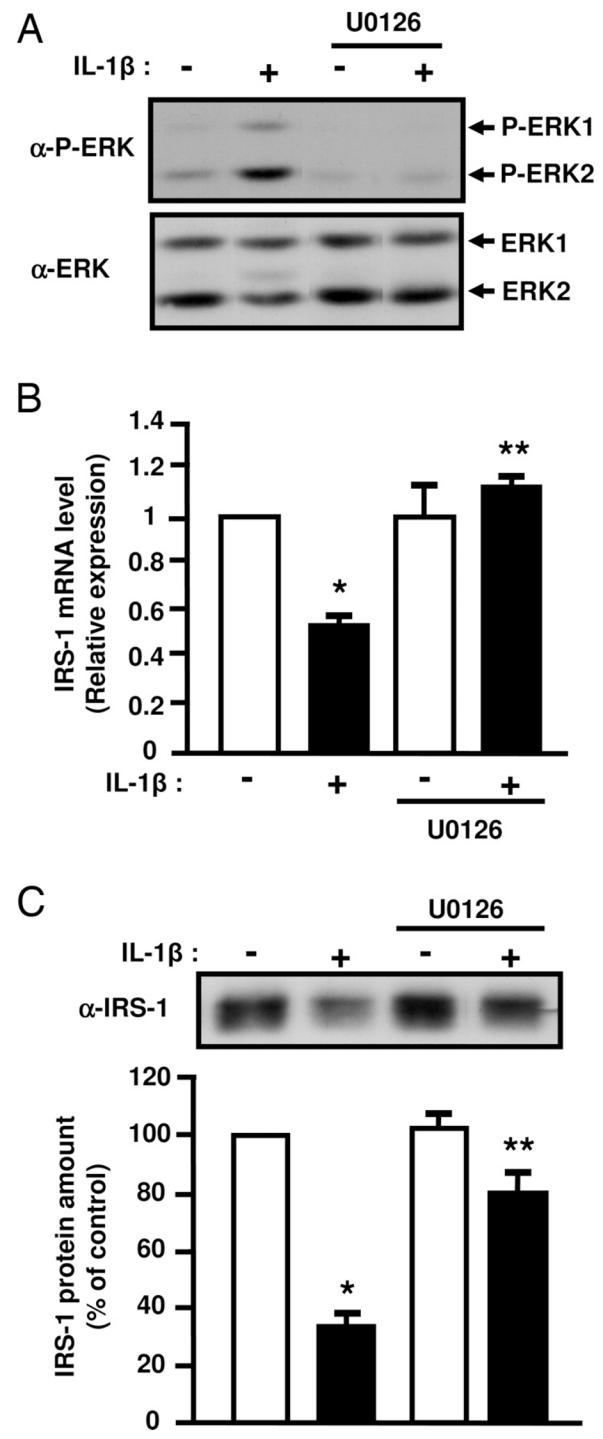
Inhibition of ERK1/2 activity prevents the inhibitory effect of IL-1β on IRS-1 expression. 3T3-L1 adipocytes were treated with vehicle (0.1 % DMSO) or U0126 (10 μM) and without or with IL-1β (20 ng/ml) for 48 hours. A, ERK activation was assessed by immunoblotting with anti-phospho ERK1/2 (α-P-ERK) antibodies. Typical autoradiographs representative of three experiments are shown. B, IRS-1 mRNA level was determined by real-time quantitative PCR. mRNA expression was normalized using 36B4 RNA levels. Results are expressed in arbitrary units, with the control values taken as 1 and are the means ± SEM from three independent experiments. C, IRS-1 amount in cell lysate was determined using anti-IRS1 (α-IRS1) antibodies. Typical autoradiograph is presented and graph shows the means ± SEM of four independent experiments. *, significantly different between control and IL-1β with p < 0.01, ** significantly different between IL-1β and IL-1β + U0126 with p < 0.01.
Figure 9.
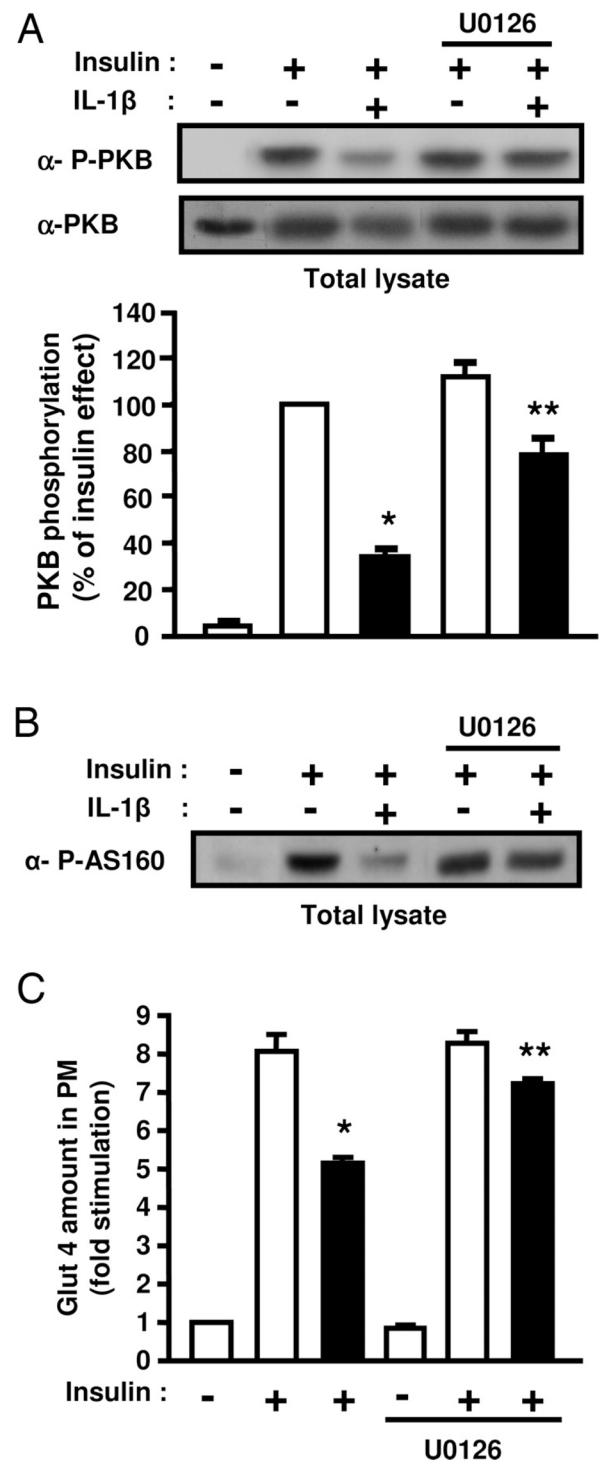
Inhibition of ERK1/2 activity partially prevents the inhibitory effect of IL-1β on insulin-induced PKB and AS160 phosphorylation and on insulin-induced Glut 4 translocation. A, 3T3-L1 adipocytes were treated with vehicle (0.1 % DMSO) or U0126 (10 μM) and without or with IL-1β (20 ng/ml) for 48 hours. Then cells were stimulated or not with insulin for 5 min. Phosphorylation of PKB was monitored by immunoblotting with anti-phospho-PKB (Thr308) (α-P-PKB) antibodies. The membrane was then stripped and probed using anti-PKB (α-PKB) antibodies. Representative autoradiographs are presented, PKB phosphorylation was normalized for the total amount of PKB and graph shows the means ± SEM of four independent experiments. *, significant different between control and IL-1β with p < 0.05, **, significant different between IL-1β and IL-1β + U0126 with p < 0.05. B, Level of AS160 phosphorylation in cell lysates was determined following immunoblotting with anti-phospho-AS160 (Thr642) (α-P-AS160) antibodies in cells treated as described above. A typical autoradiograph representative of four independent experiments is shown. C, 3T3-L1 adipocytes were incubated with vehicle (0.1 % DMSO) or U0126 (10 μM) in absence (empty bars) or presence of IL-1β (20 ng/ml) (black bars) for 48 hours, and then incubated without or with insulin (100 nM) for 20 min. Plasma membrane sheets were prepared and Glut 4 was detected in plasma membrane (PM) as described in Fig. 2. Quantifications of fluorescence level was performed with MetaMorph software as described in “Materials and Methods”. Results are the means ± SEM of three independent experiments. *, significantly different between insulin and IL-1β + insulin with p < 0.01, ** significantly different between IL-1β + insulin and IL-1β + insulin + U0126 with p < 0.01.
DISCUSSION
Recent studies demonstrate that expression of IL-1β is increased in adipose tissue of both obese rodents and humans (28), an observation that we confirmed in this study. However whereas the involvement of TNFα and IL-6 in insulin resistance is well documented, the potential role of IL-1β in the alteration of insulin signaling and metabolic effects is poorly documented.
In the present study, no acute inhibitory effect of IL-1β was observed on insulin-induced glucose uptake or on insulin signaling in accordance with a recent report with IL-1α (34). By contrast, in the β pancreatic cell line RINm5F, IL-1β was shown to rapidly induce SOCS-3 expression that evoked a decrease in insulin receptor phosphorylation and IRS phosphorylation (35). Moreover, the effect of IL-1β is clearly different from the TNFα effect that promotes a rapid inhibition of IRS-1 tyrosine phosphorylation through its serine phosphorylation (11, 36).
We found that prolonged IL-1β treatment induces an inhibition of insulin effect on glucose uptake as also recently published (37) during the revision process of this manuscript. A sustained increase in the expression of IL-1β in adipose tissue during obesity could thus participate in the development of the insulin resistance. This inhibitory effect was mainly due to the down-regulation of the expression of IRS-1 and to a lesser degree of Glut 4. TNFα and IL-6 have also been shown to negatively regulate the expression of these two proteins (20–22). However, whereas TNFα markedly suppressed Glut 4 expression (20), we found that IL-1β had only a modest effect. Further, upon prolonged exposure to TNFα, mature adipocytes loose their terminally differentiated phenotype whereas we found no effect of IL-1β on PPARγ or C/EBPα mRNAs expression in our experimental conditions indicating that IL-1β did not induce a change in the differentiation state of the 3T3-L1 adipocytes. In agreement with these findings, it was recently reported that treatment of mature 3T3-L1 adipocytes with IL-1β for 6 days did not modify their differention state (37) whereas addition of the cytokine during the differentiation process markedly altered the adipocyte phenotype (37, 38).
IL-1β increases the expression of IL-6 in fat cells (31) but it seems unlikely that the inhibition of IRS-1 expression was totally mediated by IL-6. Indeed, we found that a high level of IL-6 modestly decreased IRS-1 expression compared to IL-1β. By contrast, IL-6 was more potent than IL-1β to decrease Glut 4 and PPARγ expression (20, 21) suggesting that IL-1β could regulate IRS-1 expression independently of its effect on IL-6 expression.
Whereas, IRS-1 amount was decreased following IL-1β treatment, the expression of IRS-2 was unchanged. Despite normal IRS-2 amount and tyrosine phosphorylation, we found that insulin-induced PKB activation was markedly altered leading to a decreased phosphorylation of its substrate AS160, a Rab GTPase-activating protein recently described to play a role in insulin-stimulated Glut 4 translocation (32, 33). These data support that IRS-1 rather IRS-2 is involved in insulin-induced glucose uptake. In agreement with this finding, previous studies have shown that insulin-induced glucose uptake is altered in adipocytes from IRS-1 deficient mice (39). Further, in L6 myotube, knockdown of IRS-1 by siRNA strategy markedly impaired glucose transport, and knockdown of IRS-2 was without effect (40).
Elevated activities of the MAP kinases ERK, JNK and p38MAPK are found in adipocytes or muscles of obese and insulin resistant rodent and humans (8) and IL-1β is known to activate these protein kinases (25). We found that prolonged IL-1β treatment induced a sustained activation of ERK and p38MAPK but not JNK suggesting that an increase in IL-1β expression in adipose tissue could thus participate in the elevated activity of these kinases in obesity. Such activation of these kinases could be involved in the inhibitory effect of IL-1β on insulin-induced glucose transport. Indeed, activation of p38MAPK was shown to be involved in the down regulation of Glut 4 expression (41). Thus, the small decrease in Glut 4 amount in IL-1β treated cells could be due to the sustained activation of p38MAPK. In the other hand, we found that pharmacological inhibition of ERK pathway totally prevented the decrease in IRS-1 mRNA and partially prevented the inhibition of IRS-1 protein expression induced by IL-1β treatment. In parallel, the insulin-induced activation of PKB, phosphorylation of AS160 and Glut 4 translocation were improved. These results indicate that activation of ERK pathway by IL-1β negatively regulates IRS-1 mRNA transcription and underscore an important role of ERK pathway activation in IL-1β-induced down regulation of the IRS-1/PI 3-kinase/PKB signaling pathway necessary for insulin-induced Glut 4 translocation and glucose transport. A negative cross-talk between these two pathways was evidenced by different studies. For example, activation of ERK through expression of constitutively MEK inhibits IRS-1 expression to a greater extent than activation of p38MAPK or JNK (42). Other studies, using pharmacological inhibitors, demonstrate that ERK activation altered IRS-1 function by promoting its serine phosphorylation and that inhibition of ERK pathway could improved insulin resistance (13–15, 43–45). In agreement with an important role of ERK in insulin resistance, a recent report demonstrates that ERK1-deficient mice are more sensitive to insulin on a high fat diet regimen (46). Thus, ERK activation in response to different stimuli could be one important event to impair normal insulin metabolic effect by altering IRS-1 function.
The fact that ERK inhibition only partially prevents the decrease in IRS-1 expression induced by IL-1β indicates that another ERK-independent pathway is involved at a posttranscriptional level to regulate IRS-1 protein amount. Such a mechanism could be a regulated degradation of IRS-1 that has been shown to be a long term inhibitory mechanism involved in insulin resistance (8). For example, prolonged insulin treatment reduces the level of IRS-1 through proteasome dependent process (47, 48) whereas osmotic (49) or oxidative (50) stresses induce IRS-1 degradation through a proteasome-independent pathway. Some studies have proposed that SOCS-3 proteins by binding to IRS proteins could promote their ubiquitin-mediated degradation (18) but this finding is controversial (51). We found that IL-1β was able to induce SOCS-3 in 3T3-L1 adipocytes (data not shown) as previously shown in some other cell types (35, 52). Thus, it is plausible that SOCS-3 are involved, at a posttranscriptional level, in IL-1β-induced down-regulation of IRS-1 protein amount in adipocytes.
In conclusion, our data show that IL-1β decreases insulin-induced glucose transport in adipocytes mainly by inhibiting IRS-1 expression through a reduction in IRS-1 mRNA amount that is dependent on ERK pathway activation and by a posttranscriptional mechanism that is independent of ERK. Thus, IL-1β that is produced by resident macrophages in adipose tissue could act in synergy with TNFα and IL-6 to impair adipocytes biology that could be an important event for the development of the insulin resistance. Indeed, an attenuation of insulin signaling in adipocytes can impaired the lipid buffering capacity of the adipocytes that could favour accumulation of lipids in muscle and liver with deleterious effects on insulin action. Moreover, modification of glucose transport in adipocytes could alter the secretory function of adipocyte in a way that is detrimental to insulin action in muscle and liver.
Acknowledgments
This work was supported by grants from INSERM (France), the University of Nice, the Fondation Bettencourt-Schueller, the Fondation pour la Recherche Médicale, the Région Provence-Alpes Côte d’Azur, the Conseil Général des Alpes Maritimes, and the Association pour la Recherche Contre le Cancer (Grant 7449). J-F Tanti is supported by grant from CNRS (France). This work is part of the project “Hepatic and adipose tissue and functions in the metabolic syndrome” (HEPADIP, see http://www.hepadip.org/), which is supported by the European Commission as an Integrated Project under the 6th Framework Programme (Contract LSHM-CT-2005-018734). We thank T Gonzalez for her technical help for the culture and differentiation of human adipocytes.
The abbreviation used are
- DMEM
Dulbecco’s modified Eagle’s medium
- PBS
phosphate buffered saline
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun NH2-terminal kinase
- PKB
protein kinase B
- AS160
Akt substrate of 160 k
- IRS
insulin receptor substrate
- PI 3-kinase
phosphoinositide 3-kinase
- TNFα
tumor necrosis-α
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Hauner H. The new concept of adipose tissue function. Physiol Behav. 2004;83:653–658. doi: 10.1016/j.physbeh.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 3.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marette A. Molecular mechanisms of inflammation in obesity-linked insulin resistance. Int J Obes Relat Metab Disord. 2003;27:S46–48. doi: 10.1038/sj.ijo.0802500. [DOI] [PubMed] [Google Scholar]
- 5.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 8.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001;11:437–441. doi: 10.1016/s0962-8924(01)02129-8. [DOI] [PubMed] [Google Scholar]
- 10.Tanti J-F, Grémeaux T, Van Obberghen E, Le Marchand-Brustel Y. Serine/threonine phosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling. J Biol Chem. 1994;269:6051–6057. [PubMed] [Google Scholar]
- 11.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307. J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 12.Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci USA. 2001;98:4640–4645. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gual P, Gremeaux T, Gonzalez T, Le Marchand-Brustel Y, Tanti JF. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia. 2003;46:1532–1542. doi: 10.1007/s00125-003-1223-4. [DOI] [PubMed] [Google Scholar]
- 14.Bouzakri K, Roques M, Gual P, Espinosa S, Guebre-Egziabher F, Riou JP, Laville M, Le Marchand-Brustel Y, Tanti JF, Vidal H. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes. 2003;52:1319–1325. doi: 10.2337/diabetes.52.6.1319. [DOI] [PubMed] [Google Scholar]
- 15.Engelman JA, Berg AH, Lewis RY, Lisanti MP, Scherer PE. Tumor necrosis factor alpha-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3-L1 adipocytes. Mol Endocrinol. 2000;14:1557–1569. doi: 10.1210/mend.14.10.0542. [DOI] [PubMed] [Google Scholar]
- 16.Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 17.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 19.Shi H, Cave B, Inouye K, Bjorbaek C, Flier JS. Overexpression of Suppressor of Cytokine Signaling 3 in Adipose Tissue Causes Local but Not Systemic Insulin Resistance. Diabetes. 2006;55:699–707. doi: 10.2337/diabetes.55.03.06.db05-0841. [DOI] [PubMed] [Google Scholar]
- 20.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278:45777–45784. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 21.Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun. 2003;311:372–379. doi: 10.1016/j.bbrc.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Warne JP. Tumour necrosis factor alpha: a key regulator of adipose tissue mass. J Endocrinol. 2003;177:351–355. doi: 10.1677/joe.0.1770351. [DOI] [PubMed] [Google Scholar]
- 23.White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 24.Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab. 2006;17:72–78. doi: 10.1016/j.tem.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592:265–280. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- 26.Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 27.Salmenniemi U, Ruotsalainen E, Pihlajamaki J, Vauhkonen I, Kainulainen S, Punnonen K, Vanninen E, Laakso M. Multiple abnormalities in glucose and energy metabolism and coordinated changes in levels of adiponectin, cytokines, and adhesion molecules in subjects with metabolic syndrome. Circulation. 2004;110:3842–3848. doi: 10.1161/01.CIR.0000150391.38660.9B. [DOI] [PubMed] [Google Scholar]
- 28.Juge-Aubry CE, Somm E, Chicheportiche R, Burger D, Pernin A, Cuenod-Pittet B, Quinodoz P, Giusti V, Dayer JM, Meier CA. Regulatory effects of interleukin (IL)-1, interferon-beta, and IL-4 on the production of IL-1 receptor antagonist by human adipose tissue. J Clin Endocrinol Metab. 2004;89:2652–2658. doi: 10.1210/jc.2003-031219. [DOI] [PubMed] [Google Scholar]
- 29.Gual P, Shigematsu S, Kanzali M, Grémeaux T, Gonzalez T, Pessin JE, Le Marchand-Brustel Y, Tanti J-F. A Crk-II/TC10 signaling pathway is required for osmotic shock-stimulated glucose transport. J Biol Chem. 2002;277:43980–43986. doi: 10.1074/jbc.M203042200. [DOI] [PubMed] [Google Scholar]
- 30.Mari M, Monzo P, Kaddai V, Keslair F, Gonzalez T, Le Marchand-Brustel Y, Cormont M. The Rab4 effector Rabip4 plays a role in the endocytotic trafficking of Glut 4 in 3T3-L1 adipocytes. J Cell Sci. 2006;119:1297–1306. doi: 10.1242/jcs.02850. [DOI] [PubMed] [Google Scholar]
- 31.Flower L, Gray R, Pinkney J, Mohamed-Ali V. Stimulation of interleukin-6 release by interleukin-1beta from isolated human adipocytes. Cytokine. 2003;21:32–37. doi: 10.1016/s1043-4666(02)00495-7. [DOI] [PubMed] [Google Scholar]
- 32.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 34.He J, Usui I, Ishizuka K, Kanatani Y, Hiratani K, Iwata M, Bukhari A, Haruta T, Sasaoka T, Kobayashi M. Interleukin-1alpha inhibits insulin signaling with phosphorylating insulin receptor substrate-1 on serine residues in 3T3-L1 adipocytes. Mol Endocrinol. 2006;20:114–124. doi: 10.1210/me.2005-0107. [DOI] [PubMed] [Google Scholar]
- 35.Emanuelli B, Glondu M, Filloux C, Peraldi P, Van Obberghen E. The potential role of SOCS-3 in the interleukin-1beta-induced desensitization of insulin signaling in pancreatic beta-cells. Diabetes. 2004;53:S97–S103. doi: 10.2337/diabetes.53.suppl_3.s97. [DOI] [PubMed] [Google Scholar]
- 36.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 37.Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, Caron M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 2006;49:2162–2173. doi: 10.1007/s00125-006-0335-z. [DOI] [PubMed] [Google Scholar]
- 38.Ohsumi J, Sakakibara S, Yamaguchi J, Miyadai K, Yoshioka S, Fujiwara T, Horikoshi H, Serizawa N. Troglitazone prevents the inhibitory effects of inflammatory cytokines on insulin-induced adipocyte differentiation in 3T3-L1 cells. Endocrinology. 1994;135:2279–2282. doi: 10.1210/endo.135.5.7956951. [DOI] [PubMed] [Google Scholar]
- 39.Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275:38990–38994. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- 40.Huang C, Thirone AC, Huang X, Klip A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem. 2005;280:19426–19435. doi: 10.1074/jbc.M412317200. [DOI] [PubMed] [Google Scholar]
- 41.Carlson CJ, Koterski S, Sciotti RJ, Poccard GB, Rondinone CM. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: potential role of p38 in the downregulation of GLUT4 expression. Diabetes. 2003;52:634–641. doi: 10.2337/diabetes.52.3.634. [DOI] [PubMed] [Google Scholar]
- 42.Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, Onishi Y, Ono H, Abe M, Shojima N, Fukushima Y, Kikuchi M, Oka Y, Asano T. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol Endocrinol. 2003;17:487–497. doi: 10.1210/me.2002-0131. [DOI] [PubMed] [Google Scholar]
- 43.Bouzakri K, Karlsson HK, Vestergaard H, Madsbad S, Christiansen E, Zierath JR. IRS-1 Serine Phosphorylation and Insulin Resistance in Skeletal Muscle From Pancreas Transplant Recipients. Diabetes. 2006;55:785–791. doi: 10.2337/diabetes.55.03.06.db05-0796. [DOI] [PubMed] [Google Scholar]
- 44.Hernandez R, Teruel T, de Alvaro C, Lorenzo M. Rosiglitazone ameliorates insulin resistance in brown adipocytes of Wistar rats by impairing TNF-alpha induction of p38 and p42/p44 mitogen-activated protein kinases. Diabetologia. 2004;47:1615–1624. doi: 10.1007/s00125-004-1503-7. [DOI] [PubMed] [Google Scholar]
- 45.Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54:2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- 46.Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, Dani C, Hofman P, Pages G, Pouyssegur J, Le Marchand-Brustel Y, Binetruy B. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54:402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 47.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1 [In Process Citation] Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 48.Takano A, Usui I, Haruta T, Kawahara J, Uno T, Iwata M, Kobayashi M. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol Cell Biol. 2001;21:5050–5062. doi: 10.1128/MCB.21.15.5050-5062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gual P, Gonzalez T, Gremeaux T, Barres R, Le Marchand-Brustel Y, Tanti JF. Hyperosmotic stress inhibits insulin receptor substrate-1 function by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem. 2003;278:26550–26557. doi: 10.1074/jbc.M212273200. [DOI] [PubMed] [Google Scholar]
- 50.Potashnik R, Bloch-Damti A, Bashan N, Rudich A. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia. 2003;46:639–648. doi: 10.1007/s00125-003-1097-5. [DOI] [PubMed] [Google Scholar]
- 51.He F, Stephens JM. Induction of SOCS-3 is insufficient to confer IRS-1 protein degradation in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2006;344:95–98. doi: 10.1016/j.bbrc.2006.03.142. [DOI] [PubMed] [Google Scholar]
- 52.Boisclair YR, Wang J, Shi J, Hurst KR, Ooi GT. Role of the suppressor of cytokine signaling-3 in mediating the inhibitory effects of interleukin-1beta on the growth hormone-dependent transcription of the acid-labile subunit gene in liver cells. J Biol Chem. 2000;275:3841–3847. doi: 10.1074/jbc.275.6.3841. [DOI] [PubMed] [Google Scholar]