

Abstract
Background
von Hippel–Lindau Disease (VHL) is an autosomal dominant inherited systemic cancer syndrome. Recently, many advances have contributed to the understanding of VHL pathophysiology.
Methods
In this article we review recent developments and summarize our findings in VHL molecular pathology related to retinal and optic nerve diseases.
Results
Loss of heterozygosity (LOH) within the VHL gene is detected in the stromal cells surrounding the capillary endothelial cells and admixed with glial cells in ocular hemangioblastomas. This finding is in line with similar findings in VHL-associated CNS hemangioblastoma and renal clear cell carcinomas. Increases of vascular endothelial growth factor (VEGF), hypoxia induced factor (HIF), and ubiquitin are found in ocular hemangioblastomas.
Interestingly, tumorlet cells, which are composed of poorly differentiated small cells with prominent dark nuclei and little cytoplasm, as well as several stem cell markers, such as erythropoietin (Epo), Epo receptor (EpoR), and CD133, are present in ocular VHL lesions. CXCR4, a CXC chemokine receptor is also expressed in retinal VHL hemangioblastomas.
Conclusions
These findings imply that VHL cells with LOH of the tumor suppressor gene, most likely originate from a hematopoietic/vascular lineage. Targeting these proteins and ischemic factors, not VEGF alone, may be a potential therapeutic approach for VHL-associated ocular hemangioblastomas.
Keywords: von Hippel Lindau (VHL) disease, retinal or optic nerve hemangioblastoma, loss of het-erozygosity, hypoxia-induced factor (HIF), vascular endothelial growth factor (VEGF), tumorlet, hemato-poietic/vascular stem cell
Von Hippel–Lindau (VHL) disease occurs in 1/36,000 live births per year.1 VHL has a remarkable penetrance of over 90% by 65 years of age.1 Of all VHL patients, 49–85% develop retinal hemangioblastomas, making this ocular sign the most common presentation of the disease.2,3 VHL-associated retinal hemangioblastomas usually develop at a mean age of 25 years. Recently, in a large series of 406 patients (199 families) with ocular VHL disease, Chew reported that approximately 8.4% of the eyes had a visual acuity of 20/200 or worse and that 8.1% of these eyes required enucleation.4 However, patients can maintain a relatively normal quality of life as well as life span if comprehensive screening methods and timely treatments are performed.
VHL disease is an autosomal, dominant inherited cancer syndrome. It is caused by a germline alteration of the VHL gene, a tumor suppressor gene located on chromosome 3p25.5.5,6 VHL-associated tumors have revealed an allelic deletion within the VHL gene locus. According to the “Knudson two-hit” model that one allele is constitutively inactivated while the other allele is subsequently inactivated (second hit) at the somatic level.7 Therefore, it is believed that the pathology is linked to the somatic inactivation or loss of the remaining wild-type VHL allele. The genetic inactivation leads to malignant tumors in multiple organs including the eye, CNS, kidney, inner ear, pancreas, broad ligament, epididymis, and rarely the liver, lung, and ovary.1,8
The VHL mRNA encodes a protein termed VHL protein (pVHL).8,9 A subdomain of pVHL binds to elongin C, a protein which nucleates a complex containing elongin B, cullin-2 (CUL2), and RBX1. This complex targets specific proteins for the polyubiquitylation of the α-subunits of hypoxia-inducible factor (HIF). A lack of pVHL induces profound intracellular metabolic changes that closely resemble changes observed under oxidative stress. In pVHL defective cells, there is a lack of HIF degradation causing the excessive accumulation of this protein. HIF is primarily used to signal hypoxia and further mediates the upregulation of hypoxia-induced genes such as vascular endothelial growth factor (VEGF), erythropoietin (Epo), platelet-derived growth factor (PDGF-β), and transforming growth factor (TGF- α).8 HIF also activates NF- κB and cycline A, thereby producing many cytokines and growth factors. In addition, HIF can cause the release of fibroblast growth factor (FGF) and epidermal growth factor (EGF).10 By blocking EGF, the growth of renal cell carcinoma can be inhibited; siRNA inhibition of FGF, on the other hand, has been shown to reduce the proliferation of pulmonary artery smooth muscle cells.10–12 In summary, the VHL protein stimulates a number of angiogenic factors capable of inducing and promoting the formation of various VHL-associated lesions such as hemangioblastomas, cysts, and other tumors.
Recent advances in the molecular genetics and pathology of VHL have furthered our understanding of the disease pathogenesis and have introduced new potential therapeutic strategies. In this review, we describe our findings on the molecular pathology of ocular VHL with a focus on the mechanisms behind tumorigenesis and on potential therapeutic strategies for this disease.
Ocular Clinical Manifestations of VHL
The most common attributes of this disease are associated with maximum morbidity and mortality including retinal hemangioblastomas (angiomas or hemangiomas), brain and spinal hemangioblastomas, renal cell carcinomas and cysts, pancreatic cancers and cysts, and pheochromocytomas.13,14Germline VHL mutations have been identified that result in three distinct cancer phenotypes: 1) renal carcinoma without pheochromocytoma; 2) renal carcinoma with pheochromocytoma; and 3) pheochromocytoma alone.15,16 The mutations associated with the first group of cancer phenotypes, or Type 1, are more commonly deletions and truncating mutations which cause a predisposition to hemangioblastomas and renal cell carcinomas. The mutations in the second and third phenotype groups, or Type 2, are more commonly missense mutations. The cancer phenotypes which belong to Type 2 are further divided into three subtypes: type 2A, which is detected in hemangioblas-tomas or pheochromocytomas but is rarely observed in renal cell carcinomas; type 2B, which is detected in hemangioblastomas, renal cell carcinomas, and pheochromocytomas; and type 2C, which is detected in pheochromocytomas only. VHL-associated ocular lesions are retinal and optic nerve hemangioblastomas, and belong to types 1, 2A, and 2B.
Retinal hemangioblastomas are often the most common and earliest presentation of VHL disease.3 Usually the hemangioblastomas present as a solitary “vascular” tumor in the superior temporal region of the peripheral retina in a patient in their mid twenties (Figure 1).17,18 Upon ocular examination, the tumor is noted to be accompanied by exudation, subretinal fluid, and prominent feeder vessels. Vitreous or retinal hemorrhages are uncommon. Retinal tractional detachment may develop at the end stage of the disease. In Chew’s series, 205 of the 406 patients with VHL developed ocular diseases.4 In addition to the retinal hemangioblastomas, retinal neovascularization was described in 8.3% (17 cases). Intraorbital/intracranial hemangioblastomas were found in 5.4% (11 cases) of the patients with ocular manifestations. The differential diagnoses of VHL-associated retinal hemangioblastomas include Coats’ disease, racemose hemangioma (Wyburn-Mason disease), retinal cavernous hemangioma, retinal microaneurysm, and vasoprolif-erative retinal tumor.2,19
Fig. 1.
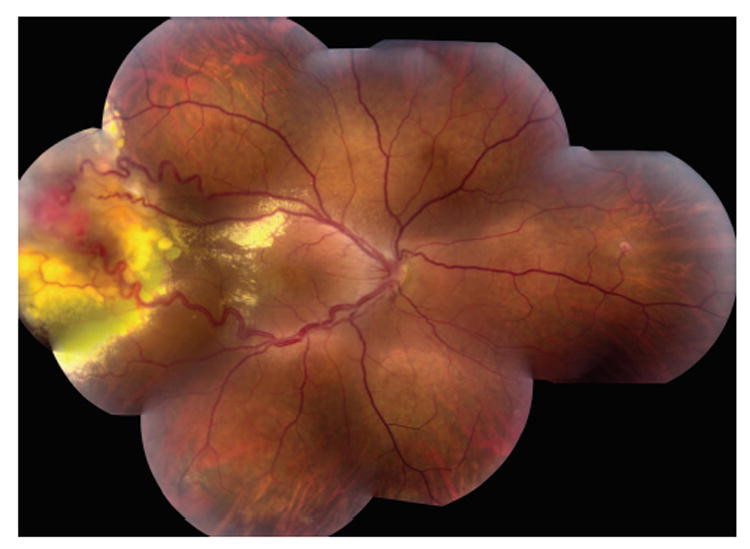
Funduscopic photograph showing a typical hemangioblastoma in the peripheral retina of a patient with von Hippel–Lindau disease.
Optic nerve capillary hemangioblastomas or juxtapapillary capillary hemangioblastomas associated with VHL are difficult to treat and the visual prognosis is often poor (Figure 2).20 In Chew’s report, optic nerve hemangioblastomas occurred in 23.4% of the patients with VHL eye disease and in 11.1% of the 406 patients with VHL.4 These tumors can first present as small lesions at the optic disk or in the peripapillary region. The optic nerve tumors may grow very slowly over a number of years. With increased growth and/or vascular leakage, visual acuity deteriorates with the development of retinal edema, hard exudates, epiretinal membranes, and even detachment of the macula.21Differential diagnoses of optic nerve hemangioblastoma include disk edema, papillitis, sarcoidosis, and anterior ischemic optic neuropathy.22
Fig. 2.
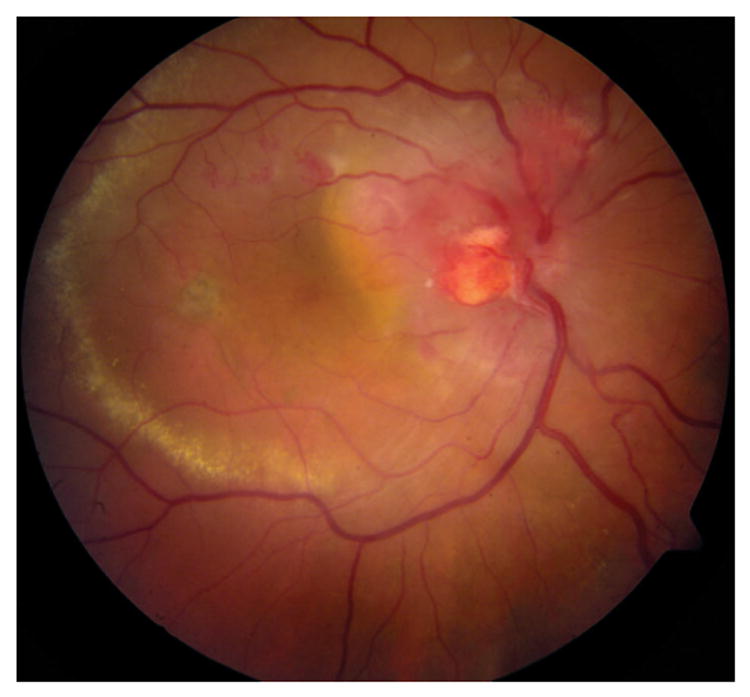
Funduscopic photograph showing a hemangioblastoma in the optic nerve head of a patient with von Hippel–Lindau disease.
Ophthalmic Pathology of VHL
Histopathologically, retinal and optic nerve hemangioblastomas are similar to CNS hemangioblastomas (Figure 3). The tumors can replace the entire layers of the neuroretina and are composed of many abnormal capillarylike fenestrated channels surrounded by vacuolated foamy cells.3,23–29The vacuolated foamy cells are easily identified in the larger lesions. These foamy cells contain large intracytoplasmic lipid inclusions that are homogeneous with medium electron density (Figure 4A).27The tumors lack endothelial cells and some contain reactive glial cells (Figure 4B).3,23–29Reactive glial proliferation occurs most often at the margins of relatively large hemangioblastomas.28,30
Fig. 3.
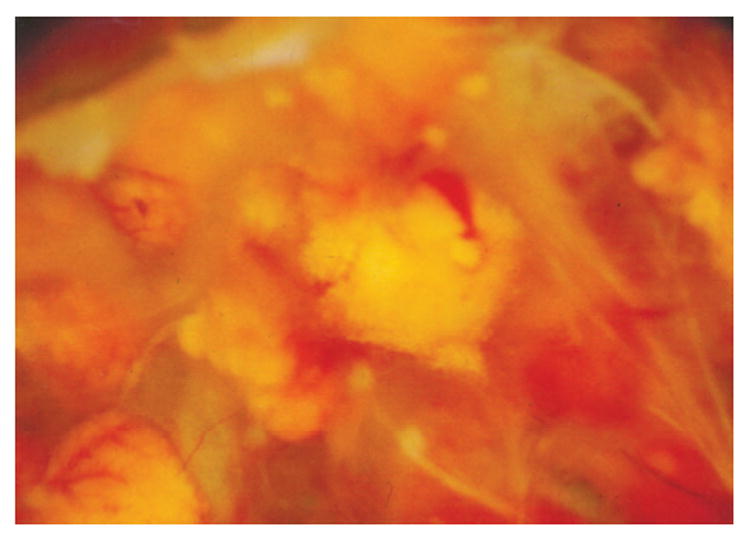
Macroscopic photograph showing multiple retinal hemangio-blastomas in the eye of a patient with von Hippel–Lindau disease.
Fig. 4.
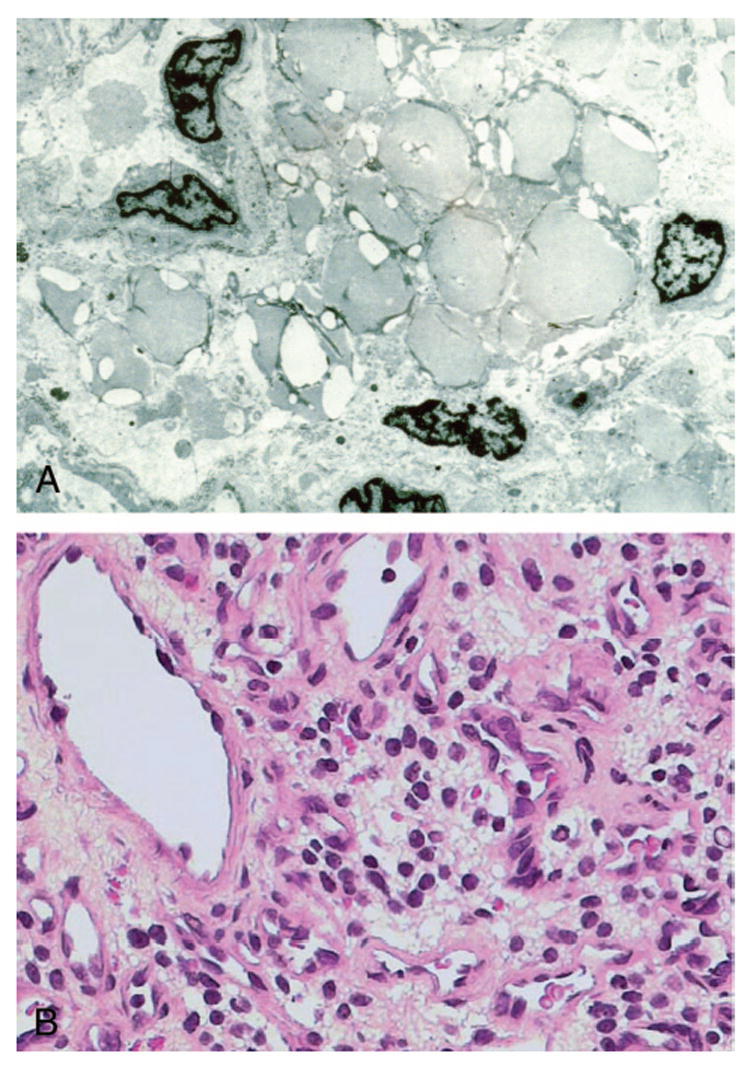
A, Transmission electron micrograph showing many electron homogenous globules in the cytoplasm of a stomal cell (x30,000). B, Microphotograph showing many foamy cells composing the stroma of a von Hippel–Lindau-associated retinal hemangioblastoma (hematoxylin & eosin, x400).
Recently, Chan and associates have identified sporadic tumorlet cells characterized by small cellular clusters, which form angiomesenchymal islands in ocular VHL-associated lesions (Figure 5).31Tumorlet cells have also been described in VHL-associated CNS hemangioblastomas as well as in other systemic lesions.32–35Unlike the solitary tumorlets observed in the CNS, ocular tumorlet cells are frequently located adjacent to either small retinal vessels or admixed within the inner retinal layers. Isolated tumorlet cells are also detected inside or near the retinal and optic nerve lesions.31
Fig. 5.
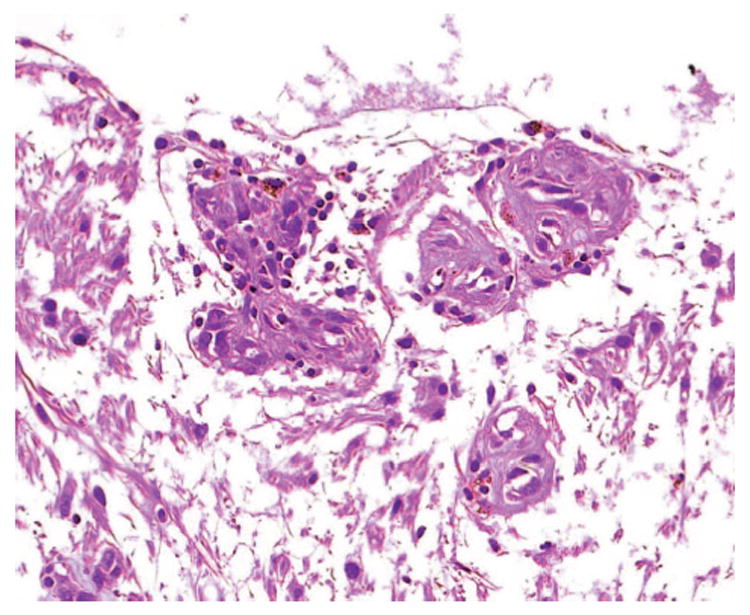
Microphotograph showing small clusters of tumorlet like cells in a von Hippel–Lindau-associated optic nerve head hemangioblastoma (hematoxylin & eosin, x400).
All ocular hemangioblastomas express high levels of VEGF, HIF, ubiquitin, Epo, and EpoR.23,24,31VEGF is detected in the neovasculature of fibrovascular epiretinal membranes, but is not seen in the vitreous of an eye with VHL disease (Figure 6, A and B).36VEGF, HIF, ubiquitin, and Epo are closely related to hypervascularization, angiogenesis, or both.37–40pVHL plays a critical role in the transduc-tion of signals generated by changes in ambient oxygen tension.37In a normal cell, the pVHL forms stable pVHL-elongin-CUL2 complexes. These complexes regulate ubiquitin and HIF production and accumulation and thus repress many proteins such as VEGF, Epo, PDGF, FGF, and EGF. When pVHL is defective, HIF will not be regulated properly and will act as a transcription factor for downstream target genes including VEGF and Epo, which are both activated during hypoxia. Upregulation of VEGF and its receptors have also been reported in VHL-associated and sporadic CNS hemangioblastomas.41
Fig. 6.
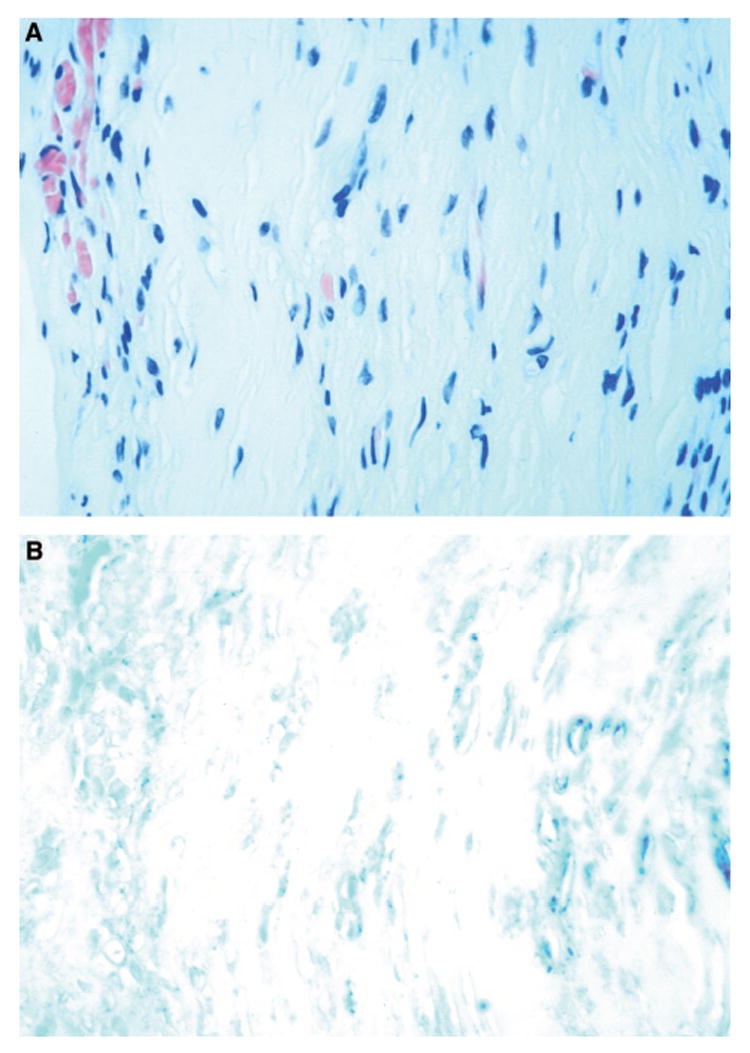
Microphotographs showing (A) a preretinal neovascular membrane removed from a patient with von Hippel–Lindau disease (hematoxylin & eosin, x400) and (B) expression of vascular endothelial growth factor (dark black-bluish color) in the membrane (avidin-biotin-complex immunoperoxidase, x400).
Several stem cell markers including CD133, Epo, and EpoR have been detected in ocular hemangioblastomas associated with VHL.31CD133 is a surface molecule expressed on progenitors from hematopoietic, endothelial, and neural lineages.42This marker is also specific for embryonic stem cell-derived progenitor cells.43CD133 positive cells can be distributed either diffusely or sparsely within the hemangioblastoma. These CD133 positive cells are mostly the vacuolated “stromal” cells and tumorlet cells. Immunoreactivity against CD117, the stem cell factor receptor,44is rarely positive in ocular hemangioblastomas.31
Epo has been reported to regulate various human malignancies via its involvement in tumor growth, viability, and angiogenesis.45Expression of EpoR has been well documented in embryonic stem cells and tumors within the CNS, uterus, and ovaries.46Coexpressed Epo and EpoR have been identified in various VHL-associated lesions including CNS hemangioblastomas.32–34However, expression of EpoR is more prominent than Epo in ocular VHL-associated hemangioblastomas. Moreover, these two proteins are not coexpressed ubiquitously.31
Molecular Pathology of Ocular VHL Hemangioblastomas
Using microdissection and molecular analysis, we have successfully analyzed ocular hemangioblastomas of 20 patients with VHL, including 16 retinal and 4 optic nerve lesions. Loss of heterozygosity (LOH) of the VHL gene was clearly identified in the vacuolated “stromal” cells, but not in the vascular endothelial or reactive glial cells of the 15 retinal and 4 optic nerve hemangioblastomas.23,24,30,31Only one of the 20 cases was noninformative.23These findings demonstrate that the tumor cells (vacuolated foamy “stromal” cells) contain a deletion of one VHL gene wild-type allele. This supports the theory that it is the “stromal” cells that are genetically altered, whereas the vascular components retain heterozygosity of the VHL gene locus, in CNS hemangiobalstomas.25Thus, these vacuolated, foamy “stromal” cells are the true tumor cells of the disease.
In accordance with the findings on HIF and independent protein expression in ocular hemangioblastomas, the transcripts of HIF target genes such as VEGF and Epo are also detected in ocular VHL lesions.23,31 Interestingly, we have found an increased expression of VEGF mRNA correlates with a greater quantity of tumors within the eye as well as with an increased disease activity through quantitative reverse-transcriptase polymerase chain reaction (RT-PCR). The high expression of VEGF transcripts produces higher levels of VEGF and, therefore, may be responsible in part for abundant neovascularization and subsequent tumor growth. In addition to VEGF, other HIF-induced molecules that are involved in VHL also secrete a number of cytokines and growth factors such as TGF, PDGF, FGF, and EGF. Many of these factors are also related to angiogenesis. These factors may promote the proliferation of vessels within the tumefactions in the eye and the production of various systemic tumors in other tissues with VHL disease. Therefore, anti-VEGF therapy alone will not be efficient or sufficient to treat all VHL-associated lesions in the eye.
In an evaluation of six ocular hemangioblastomas, Epo and EpoR were coexpressed mainly within the tumorlet cells of VHL-associated lesions.31Coexpression of Epo and EpoR may reflect a developmental arrest in CNS angioblasts and in renal immature mesenchymal cells. This finding implicates progenitor cellular involvement in VHL-associated lesions.35,47Based on our findings, these “vascular” lesions are composed of vacuolated “stromal” and tumorlet cells with LOH at the VHL genes. These cells may represent potentially immature tumor VHL cells. Furthermore, there is lack of apoptosis or in situ DNA fragmentation in ocular VHL cells, which supports the notion that the true neoplastic cells are the vacuolated “stromal” cells.30
Recently, we have detected expression of both CXCR4, a chemokine receptor that is involved in the trafficking of hematopoietic progenitors and stem cells, transcript, and protein in VHL-associated retinal hemangioblastomas.48CXCR4 is another HIF-induced product and is reported to be downregulated by pVHL under normoxic conditions.49Under hypoxic condition, there is HIF-dependent CXCR4 activation. Expression of CXCR4, therefore, may play a role in the attraction, migration, development, and regulation of embryonic VHL cells in the eye.
New Therapeutic Approaches for VHL-Associated Ocular Hemangioblastomas
The management of patients with VHL is complex because of the progressive and variable nature of multiple neoplasms in various organ systems. For ocular hemangioblastomas, conventional treatments include observation, laser photocoagulation, and cryotherapies.1,2With continued growth of the ocular lesions, either with or without treatment, the retina may detach and develop proliferative neovascular membranes.4,36The neovascular membrane can be removed surgically, especially in those cases with tractional retinal detachment or those affecting the macula. Nevertheless, the underlying retinal hemangioblastomas still continue to produce VEGF and other neovascular growth stimulators.
Knowledge of the molecular genetics behind VHL disease has led to various pharmacologic agents that block different steps (antiangiogenesis, cycline-depending kinase inhibitor) in the pathway of pVHL.50–52Systemic or intravitreal injection of anti-VEGF drugs for ocular VHL has recently been subscribed.4,53,54Although macular edema and the retinal hard exudates are decreased in some cases, ocular hemangioblastomas are not affected by the anti-VEGF therapies alone.4,54In one study, SU5416, a VEGF receptor inhibitor, failed to reduce the size of VHL retinal hemangioblastoma in spite of intravenous administration for 7 months.53In another study, intravitreal injections (every 6 weeks for six doses) of Macugen, a VEGF inhibitor, did not alter clinical appearances of retinal hemangioblastomas but decreased retinal exudates and edema in two patients with VHL.4Since VHL cells are responsible for the production of many other cytokines that cause angiogenesis, a better therapeutic approach may be to target these cells.
Based on the above molecular and pathologic findings, therapeutic strategies for ocular VHL may require targeting the tumor cells directly. Inhibition of CXCR4 and Epo/EpoR has recently been considered in the treatment of some malignancies. Targeting CXCR4 is currently being tested in a clinical trial for breast carcinoma.55Epo has been implicated as a neurocognitive protectant used for the treatment of patients with lung cancer.56Stem cell transplantation and tumor vaccine may be another approach for the treatment of VHL disease.51,52Specific genetic and molecular analyses of ocular VHL-associated lesions will further our understanding of the mechanisms behind tumor growth and reinforce our understanding of normal retinal and optic nerve development as well as tumorigenesis in the eye. VEGF inhibition alone will likely not be adequate to treat VHL-associated hemangioblastomas in the eye.
Acknowledgments
The authors thank Ms. Christine M. Bojanowski for her editing review.
Supported by the NEI Intramural Research Program, NIH, Bethesda, Maryland.
References
- 1.Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 2.Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117–142. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- 3.Welsh RB. Von-Hippel-Lindau disease: the recognition and treatment of early angiomatosis retinae and the use of cryo-surgery as an adjunct to therapy. Trans Am Ophthalmol Soc. 1970;68:367–424. [PMC free article] [PubMed] [Google Scholar]
- 4.Chew EY. Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc. 2005;103:495–511. [PMC free article] [PubMed] [Google Scholar]
- 5.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 6.Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268–269. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 7.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14:123–130. doi: 10.1016/j.semcancer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1{alpha} promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2528–2534. doi: 10.1152/ajpheart.01077.2005. [DOI] [PubMed] [Google Scholar]
- 11.Cenni E, Perut F, Zuntini M, et al. Inhibition of angiogenic activity of renal carcinoma by an antisense oligonucleotide targeting fibroblast growth factor-2. Anticancer Res. 2005;25:1109–1113. [PubMed] [Google Scholar]
- 12.Cao H, Dronadula N, Rao GN. Thrombin induces expression of FGF-2 via activation of PI3K-Akt-Fra-1 signaling axis leading to DNA synthesis and motility in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;290:C172–182. doi: 10.1152/ajpcell.00284.2005. [DOI] [PubMed] [Google Scholar]
- 13.Zbar B, Kaelin W, Maher E, Richard S. Third International Meeting on von Hippel-Lindau disease. Cancer Res. 1999;59:2251–2253. [PubMed] [Google Scholar]
- 14.Maher ER, Kaelin WG., Jr von Hippel-Lindau disease. Medicine (Baltimore) 1997;76:381–391. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Zbar B, Kishida T, Chen F, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.Clifford SC, Maher ER. Von Hippel-Lindau disease: clinical and molecular perspectives. Adv Cancer Res. 2001;82:85–105. doi: 10.1016/s0065-230x(01)82003-0. [DOI] [PubMed] [Google Scholar]
- 17.Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371–378. doi: 10.1001/archopht.117.3.371. [DOI] [PubMed] [Google Scholar]
- 18.Dollfus H, Massin P, Taupin P, et al. Retinal hemangioblas-toma in von Hippel-Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci. 2002;43:3067–3074. [PubMed] [Google Scholar]
- 19.Shields JA, Decker WL, Sanborn GE, Augsburger JJ, Gold-berg RE. Presumed acquired retinal hemangiomas. Ophthalmology. 1983;90:1292–1300. doi: 10.1016/s0161-6420(83)34389-x. [DOI] [PubMed] [Google Scholar]
- 20.McCabe CM, Flynn HW, Jr, Shields CL, et al. Juxtapapillary capillary hemangiomas. Clinical features and visual acuity outcomes. Ophthalmology. 2000;107:2240–2248. doi: 10.1016/s0161-6420(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 21.Inoue M, Yamazaki K, Shinoda K, et al. A clinicopathologic case report on macular hole associated with von Hippel-Lindau disease: a novel ultrastructural finding of wormlike, wavy tangles of filaments. Graefes Arch Clin Exp Ophthal-mol. 2004;242:881–886. doi: 10.1007/s00417-004-0908-9. [DOI] [PubMed] [Google Scholar]
- 22.Aumiller MS. Juxtapapillary hemangioma: a case report and review of clinical features and management of von Hippel-Lindau disease. Optometry. 2005;76:442–449. doi: 10.1016/j.optm.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117:625–630. doi: 10.1001/archopht.117.5.625. [DOI] [PubMed] [Google Scholar]
- 24.Chan CC, Lee YS, Zhuang Z, Hackett J, Chew EY. von Hippel-Lindau gene deletion and expression of hypoxia-inducible factor and ubiquitin in optic nerve hemangioma. Trans Am Ophthalmol Soc. 2004;102:75–81. [PMC free article] [PubMed] [Google Scholar]
- 25.Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28:540–543. doi: 10.1016/s0046-8177(97)90075-7. [DOI] [PubMed] [Google Scholar]
- 26.Rubio A, Meyers SP, Powers JM, Nelson CN, de Papp EW. Hemangioblastoma of the optic nerve. Hum Pathol. 1994;25:1249–1251. doi: 10.1016/0046-8177(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 27.Nicholson DH, Green WR, Kenyon KR. Light and electron microscopic study of early lesions in angiomatosis retinae. Am J Ophthalmol. 1976;82:193–204. doi: 10.1016/0002-9394(76)90418-9. [DOI] [PubMed] [Google Scholar]
- 28.Grossniklaus HE, Thomas JW, Vigneswaran N, Jarret WHI. Retinal hemangioblastoma: a histologic, immunohistochem-ical, and ultrastructural evaluation. Ophthalmology. 1992;99:140–145. doi: 10.1016/s0161-6420(92)32024-x. [DOI] [PubMed] [Google Scholar]
- 29.Goldberg MF, Duke JR. Von Hippel-Lindau disease. His-topathologic findings in a treated and an untreated eye. Am J Ophthalmol. 1968;66:693–705. [PubMed] [Google Scholar]
- 30.Vortmeyer AO, Chan CC, Chew EY, et al. Morphologic and genetic analysis of retinal angioma associated with massive gliosis in a patient with von Hippel-Lindau disease. Graefes Arch Clin Exp Ophthalmol. 1999;237:513–517. doi: 10.1007/s004170050271. [DOI] [PubMed] [Google Scholar]
- 31.Chan CC, Chew EY, Shen D, Hackett J, Zhuang Z. Expression of stem cells markers in ocular hemangioblastoma associated with von Hippel-Lindau (VHL) disease. Mol Vis. 2005;11:697–704. [PMC free article] [PubMed] [Google Scholar]
- 32.Vortmeyer AO, Frank S, Jeong SY, et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel-Lindau disease. Cancer Res. 2003;63:7051–7055. [PubMed] [Google Scholar]
- 33.Kim HJ, Butman JA, Brewer C, et al. Tumors of the en-dolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg. 2005;102:503–512. doi: 10.3171/jns.2005.102.3.0503. [DOI] [PubMed] [Google Scholar]
- 34.Vogel TW, Brouwers FM, Lubensky IA, et al. Differential expression of erythropoietin and its receptor in von Hippel-Lindau-associated and MEN type 2-associated pheochromo-cytomas. J Clin Endocrinol Metab. 2005;90:3747–3751. doi: 10.1210/jc.2004-1899. [DOI] [PubMed] [Google Scholar]
- 35.Vortmeyer AO, Yuan Q, Lee YS, Zhuang Z, Oldfield EH. Developmental effects of von Hippel-Lindau gene deficiency. Ann Neurol. 2004;55:721–728. doi: 10.1002/ana.20090. [DOI] [PubMed] [Google Scholar]
- 36.Kinyoun JL, Kalina RE, Chew EY, Chan CC. Neovascular-ization in Von Hippel-Lindau disease: Absence of vascular endothelial growth factor in vitreous specimens. Invest Ophthalmol Vis Sci. 2000;41:S358. [Google Scholar]
- 37.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci USA. 1996;93:10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krieg M, Marti HH, Plate KH. Coexpression of erythropoi-etin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood. 1998;92:3388–3393. [PubMed] [Google Scholar]
- 39.Li Z, Wang D, Messing EM, Wu G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep. 2005;6:373–378. doi: 10.1038/sj.embor.7400377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S, Sukhatme VP. The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol Cell Biol. 1997;17:5629–5639. doi: 10.1128/mcb.17.9.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wizigmann-Voos S, Breier G, Risau W, Plate KH. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res. 1995;55:1358–1364. [PubMed] [Google Scholar]
- 42.Hess DA, Wirthlin L, Craft TP, et al. Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood. 2006;107:2162–2169. doi: 10.1182/blood-2005-06-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kania G, Corbeil D, Fuchs J, et al. Somatic stem cell marker prominin-1/CD133 is expressed in embryonic stem cell-derived progenitors. Stem Cells. 2005;23:791–804. doi: 10.1634/stemcells.2004-0232. [DOI] [PubMed] [Google Scholar]
- 44.Escribano L, Ocqueteau M, Almeida J, Orfao A, San Miguel JF. Expression of the c-kit (CD117) molecule in normal and malignant hematopoiesis. Leuk Lymphoma. 1998;30:459–466. doi: 10.3109/10428199809057558. [DOI] [PubMed] [Google Scholar]
- 45.Youssoufian H, Longmore G, Neumann D, Yoshimura A, Lodish HF. Structure, function, and activation of the eryth-ropoietin receptor. Blood. 1993;81:2223–2236. [PubMed] [Google Scholar]
- 46.Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis. 2003;24:1021–1029. doi: 10.1093/carcin/bgg060. [DOI] [PubMed] [Google Scholar]
- 47.Lee YS, Vortmeyer AO, Lubensky IA, et al. Coexpression of erythropoietin and erythropoietin receptor in von Hippel-Lindau disease-associated renal cysts and renal cell carcinoma. Clin Cancer Res. 2005;11:1059–1064. [PubMed] [Google Scholar]
- 48.Liang X, Shen D, Huang Y, et al. Molecular pathology and CXCR4 expression in surgically excised retinal hemangio-blastomas associated with von Hippel-Lindau disease. Ophthalmology. 2006 doi: 10.1016/j.ophtha.2006.05.068. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hip-pel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 50.Madhusudan S, Deplanque G, Braybrooke JP, et al. Antian-giogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291:943–944. doi: 10.1001/jama.291.8.943. [DOI] [PubMed] [Google Scholar]
- 51.Sosman JA. Targeting of the VHL-hypoxia-inducible factor-hypoxia-induced gene pathway for renal cell carcinoma therapy. J Am Soc Nephrol. 2003;14:2695–2702. doi: 10.1097/01.asn.0000091589.10594.66. [DOI] [PubMed] [Google Scholar]
- 52.Martel CL, Lara PN. Renal cell carcinoma: current status and future directions. Crit Rev Oncol Hematol. 2003;45:177–190. doi: 10.1016/s1040-8428(02)00076-8. [DOI] [PubMed] [Google Scholar]
- 53.Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endo-thelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109:1745–1751. doi: 10.1016/s0161-6420(02)01159-4. [DOI] [PubMed] [Google Scholar]
- 54.Girmens JF, Erginay A, Massin P, Scigalla P, Gaudric A, Richard S. Treatment of von Hippel-Lindau retinal heman-gioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136:194–196. doi: 10.1016/s0002-9394(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 55.Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Mol Endocrinol. 2003;17:792–803. doi: 10.1210/me.2002-0438. [DOI] [PubMed] [Google Scholar]
- 56.Senzer N. Rationale for a phase III study of erythropoietin as a neurocognitive protectant in patients with lung cancer receiving prophylactic cranial irradiation. Semin Oncol. 2002;29:47–52. doi: 10.1053/sonc.2002.37362. [DOI] [PubMed] [Google Scholar]