

Abstract
Cancer epidemiology has undergone marked development since the nineteen-fifties. One of the most spectacular and specific contributions was the demonstration of the massive effect of smoking on the occurrence of lung, larynx and bladder cancer. Major chemical, physical and biological carcinogenic agents have been identified in the working environment and in the overall environment. The chain of events from environmental exposures to cancer requires hundreds of polymorphic genes coding for proteins involved in the transport and metabolism of xenobiotics, or in repair, or in an immune or inflammatory response. The multifactorial and multistage characteristics of cancer create the theoretical conditions for statistical interactions which have been exceptionnally detected. Over the last two decades, a considerable mass of data has been generated, mostly addressing the interactions between smoking and xenobiotic-metabolizing enzymes in smoking-related cancers. They are sometimes considered disappointing but they actually brought a lot of information and raised many methodological issues. In parallel, the number of polymorphisms which can be considered candidate per function increased so much that multiple testing has become a major issue, and genome wide screening approaches have more and more gained in interest.
Facing the resulting complexity, some instruments are being set up: our studies are now equipped with carefully sampled biological collections, high-throughput genotyping systems are becoming available, work on statistical methodologies is ongoing, bioinformatics databases are growing larger and access to them is becoming simpler; international consortiums are being organized. The roles of environmental and genetic factors are being jointly elucidated. The basic rules of epidemiology, which are demanding with respect to sampling, with respect to the histological and molecular criteria for cancer classification, with respect to the evaluation of environmental exposures, their timeframes, quantification and covariables, with respect to study size and with respect to the rigor of multivariate analyses, are more pertinent than ever before.
L’épidémiologie des cancers a connu un essor important à partir des années 1950s. L’un de ses apports les plus spectaculaires et les plus spécifiques a été la mise en évidence de l’effet massif de la consommation de tabac, avec un temps de latence important, sur la survenue des cancers du poumon, du larynx et de la vessie. Des carcinogènes majeurs, nombreux et de nature variée, ont été identifiés dans l’environnement de travail (fibres d’amiante, métaux lourds, suies et goudrons. arsenic, benzène, amines aromatiques, monochlorure de vinyle, radiations ionisantes), dans l’environnement général (radon résidentiel, UV, aflatoxines). Plusieurs variétés de virus (EBV. HPV, HBC, HCV, HTLV1, HHV8…) se sont révélés cancérogènes, de même que des bactéries (helicobacter pylori) et des parasites (bilharzie). Certains cancérogènes majeurs sont d’origine iatrogène, en particulier les traitements anticancéreux (radiations ionisantes, alkylants. epipodophyllotoxines) ou, à un moindre degré, certains traitements hormonaux. Jusqu’à présent, en dehors de la consommation d’alcool dans les cancers de l’úsophage, des voies aéro-digestives supérieures (VADS) et du foie, peu de facteurs liés à l’alimentation ont fait la preuve de leur cancérogénicité. Les expositions environnementales et les comportements possiblement ou probablement cancérogènes d’après la somme des recherches anterieures constituent une très longue liste, qui se clarifie progressivement. En parallèle des avancées sur les cancérogènes environnementaux, des progrès considérables ont permis de comprendre certains cancers héréditaires dus à des délétions transmissibles de gènes suppresseurs de tumeurs, comme le gène Rb dans le rétinoblastome ou le gène TP53 dans le syndrome de Li et Fraumeni, qui sont des gènes-clé du cycle cellulaire et de l’apoptose, ou les genes BRCA1 et BRCA2 dans le cancer du sein ou MSH2 et MLH1 dans le cancer du côlon non polypoïde, qui sont des gènes de la réparation de l’ADN impliqués dans le maintien de l’intégritè du génome. Il existe encore peu d’exemples dans lesquels des interactions entre prédisposition héréditaire au cancer et facteurs environnementaux ont été identifiés. C’est le cas dans le xeroderma pigmentosum où des anomalies des gènes XP empêchent la réparation des lésions de l’ADN induites par les UV, ce qui conduit à la transformation maligne des cellules cutanées.
Les modèles classiques de cancérogenèse supposent l’existence de plusieurs évènements mutationnels successifs sur un même clone cellulaire, qui le font progressivement échapper aux mécanismes assurant la régulation de la division cellulaire et le maintien de l’intégrité du génome. La chaîne d’évènements qui conduit d’une exposition environnementale au cancer fait potentiellement intervenir des centaines de génes polymorphes codant pour des protéines impliquées soil dans le transport et le métabolisme des xénobiotiques, soil dans la réparation, soil dans la réponse immunitaire ou l’inflammation, avec des fréquences d’allèles variants suffisantes pour que l’on puisse envisager de contraster des groupes de sujets ayant des prédispositions différentes au cancer en cas d’exposition. D’autres gènes également polymorphes peuvent intervenir dans la propension individuelle è développer un cancer dans certaines conditions d’exposition (gènes de la pigmentation, genes des recepteurs hormonaux…). Les caractères multifactoriel et multi–étape du cancer créent des conditions théoriques d’interaction statistique, puisque certains facteurs interviennent à des étapes complémentaires du processus cancéreux. La réalité est que ces interactions ne sont visibles que si les associations sont assez fortes, que les expositions sont bien mesurées en considérant des périodes d’exposition pertinentes, et que la multiplicité des facteurs n’est pas trop importante. De fait, la recherche d’une interaction statistique entre exposition et génes dans les cancers s’est généralement révélée infructueuse, malgré l’exemple inaugural confirmé de l’interaction entre NAT2 et amines aromatiques dans le cancer de la vessie.
Les 2 dernières décennies ont produit une masse trés importante de données, pour la plupart en suivant une approche gène candidat, ciblant les polymorphismes connus situés dans un nombre limité de gènes. Des consortiums internationaux ont permis de réunir des effectifs de grande taille pour étudier des associations faibles et s’intéresser à des sous-groupes de cancer ou à des expositions rares. Plusieurs exemples de ces travaux sont présentés. Les publications les plus nombreuses concernent les interactions entre la consommation de tabac et les gènes des enzymes impliquées dans le métabolisme des xénobiotiques dans les cancers du poumon, de la vessie et des VADS. En effet, il s’agit de cancers fréquents, d’expositions prévalentes et assez facilement quantifiées, et les expositions aux hydrocarbures polycycliques aromatiques, aux nitrosamines et aux amines aromatiques générées par la consommation de tabac suivent des schémas métaboliques de mieux en mieux identifiés. Il existe, de plus, dans le cancer du poumon une composante familiale faible mais décelable qui contribue à accréditer l’idée d’une inégalité constitutionnelle vis-à-vis du cancer à exposition identique. Les génes impliqués dans les mécanismes de réparation de l’ADN sont également de plus en plus étudiés dans ces cancers liés aux radiations et au tabac. L’étude des interactions gènes-environnement concernant les autres types de cancers et/ou d’autres expositions environnementales a dans l’ensemble moins de recul.
L’un des enseignements, prévisible, de ces années d’investigation, est que l’étude des interactions gènes-environnement nécessite un effort important sur l’évaluation des expositions environnementales si l’on veut qu’elle contribue à améliorer la détection des faibles risques relatifs. Jusqu’à présent, à notre connaissance, aucun facteur de risque environnemental de cancer n’a été révélé par la prise en compte d’un génotype de susceptibilité. Un autre enseignement est la remise en cause progressive de la notion de gène candidat. Le nombre élevé des polymorphismes candidats par fonction tempère de fait l’opposition entre une démarche de type génération d’hypothèses par screening versus test d’hypothèse d’un gène particulier. Classiquement, la première serait une pêche à la ligne improductive alors que la seconde est supposée intelligente et productive. Les gènes candidats, déjà multiples, ne sont probablement qu’une partie des gènes que l’on pourra considérer comme candidats dans quelques mois ou années. Même en limitant notre intérêt aux gènes candidats, on ne peut plus échapper à la problématique des tests multiples, et que l’on choisisse cette approche ou l’approche génome entier, le développement de méthodes aidant à extraire de l’information du bruit au sein des données issues des plateformes de génotypage à haul-débit constitue un enjeu majeur.
En conclusion, les cancers résultent de processus complexes, qui font intervenir des gènes et des facteurs environnementaux, avec de multiples interactions entre polymorphismes génétiques, entre facteurs environnementaux et entre polymorphismes génétiques et facteurs environnementaux. C’est un constat et l’on ne peut pas échapper à cette complexité, que les prochaines années nous aideront à mieux déchiffrer. Les outils se mettent en place : nos enquêtes ont su se doter de collections biologiques bien échantillonnées, des plateformes de génotypage à haul débit deviennent accessibles, les travaux de méthodologie statistiques se développent, les bases bioinformatiques s’étoffent et leur accès se clarifie, les consortiums internationaux s’organisent. La compréhension du rôle des facteurs environnementaux et génétiques avancent conjointement. Les règles de base de l’épidémiologie, exigeantes à la fois sur l’échantillonnage et les critères histologiques et moléculaires de classement du cancer, sur l’evaluation des expositions environnementales, leur temporalité, leur quantification et leurs covariables, sur la taille des enquêtes, et sur la rigueur des analyses multivariées, sont plus que jamais d’actualité.
1. Introduction
Environment and cancer
Cancer epidemiology has undergone marked development since the nineteen-fifties. One of the most spectacular and specific contributions was the demonstration of the massive effect of smoking, with a long lag time, on the occurrence of lung, larynx and bladder cancer. Major carcinogens, numerous and of varied natures, have been identified in the working environment (asbestos fibers, heavy metals, soot and tar, arsenic, benzene, aromatic amines, vinyl monochloride, ionizing radiation, etc.), and in the overall environment (residential radon, UV radiation, aflatoxin). Several viruses (EBV, HPV, HBV, HCV, HTLV1, HHV8, etc.) have been shown to be carcinogenic as have bacteria (Helicobacter pylori) and parasites (Schistosoma). Certain major carcinogens are of iatrogenic origin, in particular cancer treatments (ionizing radiations, alkylating agents, epipodophyllotoxin), as are, to a lesser degree, some hormone treatments. To date, apart from alcohol intake with respect to esophageal, upper aerodigestive tract and liver cancer, few diet-related factors have been shown to be carcinogenic.
Factors influencing the occurrence of cancer without being directly carcinogenic have frequently been evidenced before the underlying cause was revealed. Thus, the influence of sexual relationships with multiple partners on cervical cancer was known long before the papilloma virus was implicated. Similarly, the role of the mode of food storage on stomach cancer was elucidated before the role of Helicobacter pylori was discovered. A certain number of dietary behavior patterns, a sedentary lifestyle and excess weight are currently known to be associated with the risk of various types of cancer, without their role in carcinogenesis having been clearly elucidated.
Generally speaking, environmental exposures and behavior patterns that may be considered possibly or probably carcinogenic on the basis of the sum of previous research constitute a very long list. But the list drawn up 20 years ago has been markedly clarified, first, because our know-how has improved (better designs, better questionnaires, large-scale studies, better analytical instruments) and, secondly, because we have gradually benefited from concomitant technological progress. The availability of sensitive and reliable dosimeters contributed to establishing the association between domestic radon and lung cancer; the existence of viral genome markers enabled demonstration of the involvement of HPV in cervical cancer and EBV in certain Hodgkin’s lymphomas; laboratory exposure markers have confirmed the association between aflatoxin and hepatic carcinoma. The same techniques will doubtless make new contributions in coming years.
Research on the environment and cancer has led to cancer being modeled as a multistage process during which the cell genome undergoes a succession of changes that are sufficient for the cell to escape from its control mechanisms but insufficient for it not to survive. Little by little, biological research has elucidated the main biological pathways through which a cell becomes malignant, the multiple factors involved, their interactions and, frequently, their pleiotropic nature. Recent concepts such as epigenetics and the role of micro-RNA in the regulation of gene expression will probably further modify our view of the role of the environment in the occurrence of cancer.
Heredity and cancer
In parallel, considerable progress has enabled elucidation of certain hereditary cancers due to inherited deletions of tumor-suppressor genes such as gene Rb in retinoblastoma and gene TP53 in Li-Fraumeni syndrome. The latter are key genes in the cell cycle and apoptosis. Further examples include genes BRCA1 and BRCA2 in breast cancer and MSH2 and MLH1 in non-polypoid colonic cancer. The genes are DNA repair genes involved in maintaining the integrity of the genome. Few examples of interactions between a hereditary predisposition to cancer and environmental factors have so far been identified. However, in xeroderma pigmentosum, anomalies of genes XP prevent the repair of UV-radiation-induced DNA lesions, resulting in the malignant transformation of skin cells.
‘-omics’
Study of the transcriptome has undergone explosive development over the last decade. The etiological research value is not yet clear but may become so through the introduction of tumor classifications that are more functional than histological and by revealing potential sources of between-individual response variability which could play a role before the tumor develops, during the cancerization process.
Study of the tumor genomic map in cancers of multifactorial origin has also raised the hope of evidencing specific signatures for particular exposures (e.g.: mutations of gene RET and ionizing radiation in thyroid cancer), or a sort of molecular synopsis of the pathways of cell cancerization (e.g.: pathways TP53, RB1, CDKN2A, etc.), and distinguishing subjects who are genetically predisposed to a given response. Currently, to the author’s knowledge, those studies have not yet shown gene-environment interactions.
Gene-environment interactions: the biological viewpoint
Cancers are chronic multifactorial diseases whose genesis occurs at a variable rate. For example, leukemia induced by epipodophyllotoxin treatment may develop in 6 months while the lag time for asbestos-related pleural mesothelioma emergence is of the order of 40 years.
The conventional models used to describe carcinogenesis suppose the existence of several successive mutation events impacting a given cell clone and inducing its gradual escape from the mechanisms ensuring regulation of cell division and the maintenance of genomic integrity (figure 1). Neighboring cells also exert a direct or indirect influence on the process. According to those models, the greater the number of stages, the greater the age of onset. The earlier a carcinogen intervenes in the process, the greater the tumor induction time (and hence lag time). In the chain of events from exposure to cancer, the chemical agent is transported and metabolized by a set of proteins. In most of the models of chemical carcinogenesis, the exposure, or more generally the metabolites, interact with DNA forming adducts that, if not eliminated, induce DNA mutation at their binding sites. Ionizing radiation, ultraviolet radiation and alkylating agents elicit different DNA repair pathways, while carcinogenesis with an infectious etiology elicits different immune response genes. Hundred of genes coding for proteins involved in the transport and metabolism of xenobiotics, or in repair, or in an immune or inflammatory response are polymorphic with variant allele frequencies sufficient for it to be possible to envisage contrasting groups of subjects with different predispositions to cancer in the event of exposure. In addition to those polymorphisms, there are other categories of genes which may intervene in the individual propensity to develop a cancer under certain exposure conditions (pigmentation genes, hormone-receptor genes, etc.).
Figure 1.
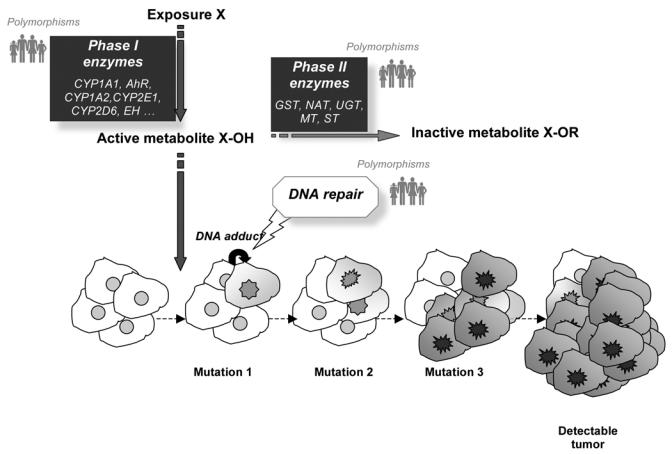
Simplified diagram of the chemical carcinogenesis model
There is no escape from the complexity revealed by awareness of the array of polymorphisms, but we may attempt to manage that complexity. Many studies aim to identify the sensitivity profiles for specific carcinogens by considering all the genes involved in the body's management of a given physical, chemical or biological agent.
These designs will probably be amended when we are able to incorporate the epigenetic mechanisms enabling response to the external environment and to potential carcinogens not just at cell or even body level, but on the scale of a population.
Gene-environment interactions: the statistical and epidemiological viewpoint
The biological interaction between the environment and the genome of a cell of an individual may exceptionally be reflected in statistical interactions that can be detected at population level. This is not surprising given the number and diversity of the parameters. In fact, the complexity of environmental exposures is additional to the complexity of the genetic predisposing factors cited above. Environmental exposure frequently consists in exposure to a mixture of potential carcinogens (e.g., smoking, paint, pesticides and air pollution) and various exposures are associated for a given individual, concomitantly or serially. The multifactorial and multistage characteristics of cancer create the theoretical conditions for statistical interaction. The cancer risk associated with early exposure to a carcinogen depends on the subsequent exposures of the impaired cell to carcinogens inducing pursuit of cell transformation. Conversely, the risk associated with carcinogen exposure at a late stage in carcinogenesis depends on the completion or non-completion of the previous carcinogenetic stages. The non-independence of the exposures occurring at complementary stages in the process should theoretically result in a statistical interaction. The relative risk for the joint effect of the exposures is then greater than the multiplication of the relative risks estimating the effects of each exposure. In reality, the interactions are only visible if the associations are fairly strong, the exposures are correctly measured during pertinent exposure periods and the multiplicity of factors is not too great. In fact, investigation for a statistical interaction between exposure and genes in cancer has generally proved fruitless, despite the confirmed inaugural example of the interaction between NAT2 and aromatic amines in bladder cancer (Cartwright et al., 1982).
Research on gene-environment interactions in cancer increasingly means studying the joint effects of genes and environmental exposures. The research has even been extended, in a certain manner, to investigating for the specific effect of polymorphisms in diseases strongly related to a particular exposure. Thus, the study of the role of polymorphisms of genes coding for the enzymes metabolizing benzo[a]pyrene in smoking-related cancers such as lung cancer is frequently presented as the study of gene-environment interactions.
2. Data on gene-environment interactions in cancer
Over the last two decades, a considerable mass of data has been generated. Most of the data were obtained using a candidate-gene approach targeting known polymorphisms situated in a limited number of genes. Very logically, the most numerous publications address the interactions between smoking and xenobiotic-metabolizing enzymes (XME) in lung cancer, bladder cancer and cancer of the upper aerodigestive tract. These are frequent cancers, the exposures are prevalent and relatively easily qualified and the exposures to aromatic polycyclic hydrocarbons, nitrosamines and aromatic amines generated by smoking follow metabolic pathways that are increasingly well known. In addition, in lung cancer there is a familial component that is weak but detectable and has contributed to accrediting the idea of a constitutional inequality with respect to cancer for a given exposure level. Obviously, smoking is also typically a mixture of chronic exposures accompanied by varied co-exposures and excessively simple designs obligatorily yield disappointing results. XME intervene in a major manner and some of their polymorphisms are relatively prevalent and markedly modify their effects. The polymorphisms of XME genes are naturally candidates for modulation of the clearly established relationship between cancers and complex chemical exposures. However, the redundancy of the pathways in which XME are involved may reduce the impact of those polymorphisms. The genes involved in DNA repair mechanisms are also being increasingly studied in cancers related to radiation and smoking. Less experience has been accumulated in the study of gene-environment interactions for other types of cancer and/or other environmental exposures.
2.1. Xenobiotic-metabolizing enzyme genes
The international Genetic Susceptibility to Environmental Carcinogenesis (GSEC) group (Gaspari et al., 2001) retrospectively pooled the data generated by the contributors to 152 studies: about 24,000 cases of different cancers and 28,000 controls. The analyses of the pooled data enabled indispensable synthetic findings but nonetheless showed certain limitations, inconsistencies and sources of bias, which were amplified by the scale. In addition, several meta-analyses contributed to further elucidation. The results for CYP1A1, GSTM1 and NAT2 clearly illustrate the issue as a whole.
2.1.1. Smoking, benzopyrene and CYP1A1 and GSTM1 polymorphisms
Benzopyrene is a combustion product present, in particular, in cigarette smoke, exhaust fumes and soot and tar. Benzopyrene metabolism particularly involves the enzymes P4501A1 and GSTM1. With marked simplification, it may be considered that the former intervenes in phase 1 yielding a diol epoxide able to bind to DNA to form adducts, while the latter intervenes in phase 2. detoxifying the oxygen derivatives. The polymorphisms of the genes for those enzymes have a variable distribution depending on race. The frequency of allele CYP1A1*2A, which increases the activity of the enzyme, was estimated to be 6, 15 and 22% in Caucasians, Asians and Africans, respectively, while the frequency of GSTM1 deletion was estimated to be 53% for Caucasians and Asians and 27% for Africans (Garte et al., 2001).
Table I summarizes the results of the recent meta-analyses and pooled analyses of the relationships between those polymorphisms and the three smoking-related cancers. Overall, GSTM1 deletion appears to be associated with the three disease sites and the association is stronger for Asians. The raw results for CYP1A1 are more difficult to interpret. At the very least, on the basis of table I, the possibility of a link between CYP1A1 polymorphisms and lung and upper aerodigestive tract cancers cannot be ruled out.
Table I.
Published meta-analyses and pooled analyses on the association between CYP1A1 (msp1) and GSTM1 null and smoking-related cancers (adjusted for age, gender and study)
| CYP1A1 | GSTM1 (null vs. present) | NAT23 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Caucasian | Asian | Caucasian | Asian | Caucasian | Asian | ||||||||
| OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | ||
| Lung cancer | |||||||||||||
| (Vineis et al., 2003) | 2451 cases | 2.4 | [1.2–4.8] | 1.1 | [0.8–1.7] | ||||||||
| 22 pooled studies 1 (GSEC) | 3358 controls | ||||||||||||
| (Le Marchand et al., 2003) | 1950 cases | 1.3 | [0.4–4.7] | 1.4 | [0.8–2.4] | ||||||||
| 11 pooled studies2 (GSEC) | 2617 controls | ||||||||||||
| (Benhamou et al., 2002) | 3940 cases | 1.0 | [0.9–1.1] | 1.1 | [0.8–1.5] | ||||||||
| 21 pooled studies (GSEC) | 5515 controls | ||||||||||||
| (Benhamou et al., 2002) | 7463 cases | 1.1 | [1.0–1.2] | 1.3 | [1.1–1.7] | ||||||||
| Meta-analysis - 43 studies | 10789 controls | ||||||||||||
| (Ye et al., 2006) | 19729 cases | 1.0 | [1.0–1.1] | 1.5 | [1.4–1.5] | ||||||||
| Meta-analysis - 1 19 studies | 25931 controls | ||||||||||||
| Upper aerodigestive tract cancers | |||||||||||||
| (Hashibe et al., 2003) | 2334 cases | 1.0 | [0.6–1.4] | 1.2 | [0.9–1.5] | ||||||||
| 11 pooled studies (GSEC) | 2766 controls | ||||||||||||
| (Hashibe et al., 2003) | 4635 cases | 1.1 | [0.8–1.5] | 1.7 | [0.9–3.2] | 1.0 | [0.9–1.1] | 1.6 | [1.2–2.1] | ||||
| Meta-analysis - 30 studies | 5770 controls | ||||||||||||
| Bladder cancer | |||||||||||||
| (Vineis et al., 2003) | 1530 cases | 1.4 | [1.1–1.8] | ||||||||||
| 8 pooled studies (GSEC) | 731 controls | ||||||||||||
| (Engel et al., 2002) | 1935 cases | 1.3 | [1.1–1.6] | 1.7 | [1.2–2.4] | ||||||||
| 17 pooled studies (GSEC) | 1444 controls | ||||||||||||
| (García-Closas et al., 2005) | 5091 cases | 1.4 | [1.2–1.6] | 1.4 | [1.2–1.6] | 1.4 | [1.3–1.5] | 1.5 | [0.8–2.6] | ||||
| Meta-analysis - 22 studies | 6501 controls | ||||||||||||
polymorphism msp1 (mutant homozygotes vs. non-mutant homozygotes)
polymorphism exon 7 (mutant homozygotes vs. non-mutant homozygotes)
polymorphism NAT2*5 (homozygote + heterozygote variants vs. reference homozygotes)
The association between variant CYP1A1 and lung cancer appears to be restricted to non-smokers and more particularly Caucasian non-smokers (table II) in the GSEC pooled studies. The associations with GSTM1 do not appear to be influenced by smoker status.
Table II.
Smoking, polymorphisms of CYP1A1 (msp1) and GSTM1 null and lung cancer - Pooled analyses (adjustment for age, gender and study)
| CYP1A1 | GSTM1 (null vs. present) | NAT22 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Non-smokers | Smokers | Non-smokers | Smokers | Non-smokers | Smokers | |||||||||
| OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | |||
| Lung cancer | ||||||||||||||
| (Vineis et al., 2003) | 2451 cases | Caucasian | 2.1 | [1.0–4.6] | 1.0 | [0.8–1.4] | ||||||||
| 22 pooled studies1 (GSEC) | 3358 controls | Asian | 1.1 | [0.5–2.3] | 1.3 | [0.6–3.1] | ||||||||
| (Raimondi et al., 2005) | 555 cases | Caucasian | 1.7 | [1.0–2.8] | 1.0 | [0.8–1.3] | ||||||||
| 21 pooled studies1 (GSEC) | 2209 controls | Asian | 1.0 | [0.5–2.3] | 1.0 | [0.6–1.7] | ||||||||
| (Le Marchand et al., 2003) | 1950 cases | 2.1 | [1.2–3.1] | ≈ 1.1 | ||||||||||
| 11 pooled studies2 (GSEC) | 2617 controls | |||||||||||||
| (Benhamou et al., 2002) | 3940 cases | 1.1 | [0.8–1.4] | 1.1 | [1.0–1.2] | |||||||||
| 21 pooled studies (GSEC) | 5515 controls | |||||||||||||
| Upper aerodigestive tract cancers | ||||||||||||||
| (Hashibe et al., 2003) | 2334 cases | 1.0 | [0.6–1.5] | 0.9 | [0.5–1.5] | 1.6 | [1.1–2.2] | 1.3 | [1.0–1.7] | |||||
| 11 pooled studies (GSEC) | 2766 controls | |||||||||||||
| Bladder cancer | ||||||||||||||
| (Vineise et al., 2003) | 1530 cases | Caucasian | 1.1 | [0.7–1.8] | 1.6 | [1.3–2.1] | ||||||||
| 8 pooled studies (GSEC) | 731 controls | |||||||||||||
| (Engel et al., 2002) | 1935 cases | 1.2 | [0.9–1.7] | 1.4 | [1.1–1.6] | |||||||||
| 17 pooled studies (GSEC) | 1444 controls | |||||||||||||
| (García-Closas et al., 2005)4 | 1935 cases | 1.0 | [0.9–1.2] | 1.2 | [1.1–1.5] | |||||||||
| 17 pooled studies (GSEC) | 1444 controls | |||||||||||||
polymorphism msp 1 (homozygote + heterozygote variants vs. reference homozygotes)
polymorphism exon 7 (homozygote + heterozygote variants vs. reference homozygotes)
polymorphism NAT2*5 (homozygote + heterozygote variants vs. reference homozygotes)
OR for gene-smoking interaction estimated in the case-only study
There are certain inconsistencies in the results of the GSEC analyses. Thus, the Le Marchand pooled analyses (Le Marchand et al., 2003) appear to indicate that the association between CYP1A1 (exon 7) and lung cancer is more marked for women than for men and more marked for squamous-cell carcinoma than for adenocarcinoma. In contrast, Vineis’ analysis (Vineis et al., 2003) (mspl). which was stratified on ethnic origin, showed associations that were systematically more marked for men with, curiously, a significantly lower OR (0.3 [0.1–1.0]) for Asian women. The association was of the same order for the two main histological types.
2.1.2. Aromatic amines and NAT2 polymorphisms in bladder cancer
Aromatic amines were particularly widely used in the dye industry before the potent carcinogenicity to the bladder of benzidine, 4-aminobiphenyl and auramine and magenta manufacturing had been demonstrated. Certain aromatic amines remain present in occupational settings and 4-aminobiphenyl was recently identified in brown tobacco smoke. Briefly, NAT2 acetylates the oxygen derivatives and reduces the formation of DNA adducts. The NAT2 polymorphisms which slow that metabolism thus increase the risk of adduct formation. The NAT2*5 polymorphism, responsible for a slow acetylator phenotype, is present in about half of Caucasians (Garte et al., 2001). The pooled analysis conducted by Vineis (Vineis et al., 2001) on the GSEC data generated an overall OR estimated of 1.4 [1.1–1.8] associated with the slow acetylator genotype in Caucasians. The association appears restricted to smokers (table II).
2.1.3. Other examples
Many other polymorphisms of the genes coding for XME have been studied, sometimes extensively, using the candidate gene approach. Thus, the risk of upper aerodigestive tract cancer is strongly associated with alcohol intake and polymorphisms of the genes coding for ADH and ALDH, which modify alcohol metabolism, may modulate the risk. The pooled analysis conducted by the IARC on 7 studies that collected data on alcohol intake did not however demonstrate an influence of polymorphism on one of the ADH, ADH1C*1 (Brennan et al., 2004). In contrast, in a multicenter Central European study coordinated by the IARC, the expected interactions were observed: the tumors were negatively associated with the variant allele for ADH1B and positively with the variant alleles for ALDH2. The associations were markedly stronger for intermediate and high alcohol intakes (Hashibe et al., 2006). The international consortium, Inhance, will afford the opportunity of replicating those findings.
2.2. Repair genes
Repair genes are broadly divided into 4 groups on the basis of whether their products ensure DNA single-strand break repair by base excision repair (BER), double-strand break repair by homologous recombination (HR) or end-joining (EJ), repair of insertions, deletions and mismatches by mismatch repair (MMR) or repair of pyrimidine adducts and dimers by nucleotide excision repair (NER). Certain genes involved in DNA repair also directly affect cell cycle homeostasis. Some ten familial cancers are due to hereditary deficiency in repair genes. Those cancers include hereditary non-polyposis colon cancer, due to constitutional mutation of genes of the MMR type, and ataxia-telangiectasia (malignant blood diseases and breast cancer) and xeroderma pigmentosum (skin cancer) due to mutations of HR and NER genes, respectively. Xeroderma pigmentosum is a textbook example of gene-environment interaction, since it results from association of exposure to UV radiation and the presence of a variant allele (seven NER genes may be involved). The 100% prevalence of the exposure masks the interaction.
BER, HR and EJ enzymes are involved in the repair of the lesions induced by ionizing radiation and chemotherapy (e.g.: cisplatin) and NER enzymes in the repair of the lesions induced by UV radiation and adducts generated by chemical exposures. The polymorphisms of the genes coding for those enzymes are thus candidates for modulating the risk of cancer associated with both physical and chemical environmental exposures.
Lung cancer, smoking and repair gene polymorphisms
The most widely studied polymorphisms in the cancers related to chemical exposures are those of genes XP. The recent meta-analyses, pooling 6 to 8 studies, do not rule out a moderate association between lung cancer and the frequent polymorphisms of genes ERCC2/XPD, XPD-751 and XPD-312 (Benhamou and Sarasin, 2005; Manuguerra et al., 2006). Their interaction with smoking could not be estimated. Variant alleles XRCC1 3194Trp and Arg280His may be negatively associated with the risk of lung cancer in heavy smokers but this has only been reported by a single large-scale multicenter study conducted in Central Europe (Hung et al., 2005).
2.3. Cell cycle genes
Malignant cutaneous melanoma provides an indirect example of interaction between the genes involved in regulating the cell cycle and environmental factors, in this case UV radiation. Certain mutations of genes CDKN2A and CDK4 predispose toward the risk of malignant cutaneous melanoma (Hayward, 2003) with high penetrance. However, on the basis of the data generated by a consortium pooling Australian, European and North-American data, the penetrance of gene CDKN2A varies by country (Bishop et al., 2002). The variants of gene MC1R, involved in skin pigmentation, and a high number of nevi would appear to account for the influence of a history of sunburn on the penetrance of CDKN2A (Chaudru et al., 2005).
2.4. Assessment: contribution to the elucidation of carcinogenesis and limitations
The incidence of cancers, their multifactorial nature and the absence of intermediate phenotypes in most cases justify the fact that the case-control approach is usually preferred, together with familial approaches in the event of familial clusters. Certain cohorts, such as the EPIC international cohort to which E3N constitutes a major French contribution, are sufficiently large to enable powerful case-control studies to be conducted. A specific interest of cohorts is the fact that the specimens are obtained prior to the disease. This sometimes enables use of biological exposure markers and study of the DNA adducts which form during the very early and reversible stages of carcinogenesis. The data on gene-environment interactions in those contexts remain disparate. TDT studies using index case-father-mother triads, which are possible at least for childhood and young adulthood cancers, are increasing. While such studies do not enable direct study of gene-environment interactions, they are complementary to the case-control and case-only approaches and have the advantage of overcoming potential population sampling bias with respect to ethnic origin (stratification bias) in the study of the association between polymorphism and disease.
The considerable data compilation work implemented by GSEC has enabled large populations for the study of weak associations and an approach to cancer subgroups and rare exposures. This work constitutes a first stage that is ongoing today in the context of numerous consortiums and large-scale multicenter studies.
Population heterogeneity with respect to the risk of cancer associated with environmental exposures clearly exists. However, the influence of a given variant, considered in isolation, does not appear major and few studies have addressed several variants concomitantly. Indubitably, the identification of subpopulations that are more sensitive to certain carcinogens necessitates taking into account a set of polymorphisms and their joint role, but study of the interaction between genes is still at an early stage (Vineis et al., 2004). The low relative risks associated with variants liable to modulate the exposure-cancer relationship are only detectable in large-scale samples which, in the majority of cases, were obtained by pooling studies designed separately and conducted in different populations. Table III shows an example of the heterogeneity of the studies pooled by GSEC to study the interactions between genes and smoking in upper aerodigestive tract cancers (Hashibe et al., 2003). In that example, the associations between upper aerodigestive tract cancers and polymorphisms of CYP1A1 and GSTM1 were greatly influenced by the study designs. The biases of the individual studies are not resolved when the studies are pooled and the value of such studies in terms of the gain in power is relative. Sampling needs to be designed to prevent both bias due to differential stratification on ethnic origin and selection bias on environmental exposures. This rigor has sometimes become secondary in studies of genetic polymorphisms. The Spanish case-control study of bladder cancer (García-Closas et al, 2005) is exemplary from this point of view: first because of its large size, which confers strong power, but, above all, because all the stages of sampling and exposure evaluation were very carefully implemented, thus minimizing not only bias but losses of power.
Smoking is such an important risk factor that its association with certain cancers remains detectable, although poorly estimated, in the presence of numerous confounding factors and selection biases. However, this is a particular case and the study of gene-environment interactions requires a special effort to evaluate environmental exposures, particularly since the information on which evaluation is based is frequently retrospective and the lag times are long. However, gene-environment interaction studies are expected to contribute to improving the detection of low relative risks, which requires the most reliable estimates of the exposures. To the author’s knowledge, no environmental risk factor for cancer has been detected by taking into account a susceptibility genotype to date.
3. The debated candidate-gene approach
The number of genes that may reasonably be considered candidates on the dual basis of their probable functional involvement in the carcinogenetic process and the sufficient allelic frequency of their polymorphisms in the population has considerably increased over the last 10 years. Several hundred genes coding for XME, DNA repair enzymes, cell-cycle enzymes and immune response have become candidates. Depending on the case, other polymorphisms have also become candidates. In parallel, the advent of whole-genome arrays has also led to a review of the candidate-gene strategy and adoption of a screening strategy when the quantities of DNA available enable that approach. The cost remains extremely high but is not proportional to the number of polymorphisms studied. Thus, the cost differential between covering the main genes of interest and covering the whole genome is tending to become smaller. However, the question of managing the information generated is becoming increasingly complex. The proposed approaches involve stages using exploratory samples, followed by more refined stages during which one attempts to incorporate the information available on haplotypes and the functional pathways between genes in the model.
The high number of candidate polymorphisms per function effectively softens the opposition between the approach consisting in hypothesis generation by screening versus hypothesis testing for a given gene. Conventionally, the first method is considered to be unproductive fishing expedition while the second is considered intelligent and productive. The candidate genes, and there are already many of them, are probably only a fraction of the genes that may be considered candidates in a few months’ or years’ time. Even if we restrict our interest to the candidate genes, we cannot escape the issue of multiple testing. If this approach or the whole-genome approach is adopted, the development of methods enabling the extraction of information from the noise in the data derived from high-throughput genotyping systems constitutes a major challenge.
4. Conclusion
Cancers result from complex processes in which genes and environmental factors intervene with multiple interactions between genetic polymorphisms, between environmental factors and between genetic polymorphisms and environmental factors. The finding is now a given and we cannot escape the resulting complexity, but, with time, our ability to elucidate that complexity will improve. The instruments are being set up: our studies are now equipped with carefully sampled biological collections, high-throughput genotyping systems are becoming available, work on statistical methodologies is ongoing, bioinformatics databases are growing larger and access to them is becoming simpler; international consortiums are being organized. The roles of environmental and genetic factors are being jointly elucidated. The basic rules of epidemiology, which are demanding with respect to sampling, with respect to the histological and molecular criteria for cancer classification, with respect to the evaluation of environmental exposures, their timeframes, quantification and covariables, with respect to study size and with respect to the rigor of multivariate analyses, are more pertinent than ever before.
References
- Benhamou S, Lee WJ, Alexandrie AK, Boffetta P, Bouchardy C, Butkiewicz D, Brockmöller J, Clapper ML, Daly A, Dolzan V, Ford J, Gaspari L, Haugen A, Hirvonen A, Husgafvel-Pursiainen K, Ingelman-Sundberg M, Kalina I, Kihara M, Kremers P, Le Marchand L, London SJ, Nazar-Stewart V, Onon-Kihara M, Rannug A, Romkes M, Ryberg D, Seidegard J, Shields P, Strange RC, Stücker I, To-Figueras J, Brennan P, Taioli E. Meta- and pooled analyses of the effects of glutathione S-transferase M1 polymorphisms and smoking on lung cancer risk. Carcmogenesis. 2002;23(8):1343–50. doi: 10.1093/carcin/23.8.1343. [DOI] [PubMed] [Google Scholar]
- Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and lung cancer: a HuGE review. Am J Epidemiol. 2005;161(1):1–14. doi: 10.1093/aje/kwi018. [DOI] [PubMed] [Google Scholar]
- Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, Chompret A, Ghiorzo P, Gruis N, Hansson J, Harland M, Hayward N, Holland EA, Mann GJ, Mantelli M, Nancarrow D, Platz A, Tucker MA. Melanoma. Genetics Consortium. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94(12):894–903. doi: 10.1093/jnci/94.12.894. [DOI] [PubMed] [Google Scholar]
- Brennan P, Lewis S, Hashibe M, Bell DA, Boffetta P, Bouchardy C, Caporaso N, Chen C, Coutelle C, Diehl SR, Hayes RB, Olshan AF, Schwartz SM, Sturgis EM, Wei Q, Zavras AI, Benhamou S. Pooled analysis of alcohol dehydrogenase genotypes and head and neck cancer: a HuGE review. Am J Epidemiol. 2004;159(1):1–16. doi: 10.1093/aje/kwh003. [DOI] [PubMed] [Google Scholar]
- Cartwright RA, Glashan RW, Rogers HJ, Ahmad RA, Barham-Hall D, Higgins E, Kahn MA. Role of N-acetyltransferase phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancer. Lancet. 1982;2(8303):842–5. doi: 10.1016/s0140-6736(82)90810-8. [DOI] [PubMed] [Google Scholar]
- Chaudru V, Laud K, Avril MF, Minière A, Chompret A, Bressac-de Paillerets B, Demenais F. Melanocortin-1 receptor (MC1R) gene variants and dysplastic nevi modify penetrance of CDKN2A mutations in French melanoma-prone pedigrees. Cancer Epidemiol Biomarkers Prev. 2005;14(10):2384–90. doi: 10.1158/1055-9965.EPI-04-0777. [DOI] [PubMed] [Google Scholar]
- Engel LS, Taioli E, Pfeiffer R, Garcia-Closas M, Marcus PM, Lan Q, Boffetta P, Vineis P, Autrup H, Bell DA, Branch RA, Brockmöller J, Daly AK, Heckbert SR, Kalina I, Kang D, Katoh T, Lafuente A, Lin HJ, Romkes M, Taylor JA, Rothman N. Pooled analysis and meta-analysis of glutathione S-transferase M1 and bladder cancer: a HuGE review. Am J Epidemiol. 2002;156(2):95–109. doi: 10.1093/aje/kwf018. [DOI] [PubMed] [Google Scholar]
- García-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardón A, Serra C, Carrato A, García-Closas R, Lloreta J, Castaño-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Torà M, Fernández F, Real FX, Rothman N. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366(9486):649–59. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, Baranova H, Bathum L, Benhamou S, Boffetta P, Bouchardy C, Breskvar K, Brockmoller J, Cascorbi I, Clapper ML, Coutelle C, Daly A, Dell’Omo M, Dolzan V, Dresler CM, Fryer A, Haugen A, Hein DW, Hildesheim A, Hirvonen A, Hsieh LL, Ingelman-Sundberg M, Kalina I, Kang D, Kihara M, Kiyohara C, Kremers P, Lazarus P, Le Marchand L, Lechner MC, van Lieshout EM, London S, Manni JJ, Maugard CM, Morita S, Nazar-Stewart V, Noda K, Oda Y, Parl FF, Pastorelli R, Persson I, Peters WH, Rannug A, Rebbeck T, Risch A, Roelandt L, Romkes M, Ryberg D, Salagovic J, Schoket B, Seidegard J, Shields PG, Sim E, Sinnet D, Strange RC, Stucker I, Sugimura H, To-Figueras J, Vineis P, Yu MC, Taioli E. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10(12):1239–48. [PubMed] [Google Scholar]
- Gaspari L, Marinelli D, Taioli E Collaborative Group on Genetic Susceptibility to Environmental Carcinogens. International collaborative study on genetic susceptibility to environmental carcinogens (GSEC): an update. Int J Hyg Environ Health. 2001;204(1):39–42. doi: 10.1078/1438-4639-00070. [DOI] [PubMed] [Google Scholar]
- Hashibe M, Boffetta P, Zaridze D, Shangina O, Szeszenia-Dabrowska N, Mates D, Janout V, Fabiánová E, Bencko V, Moullan N, Chabrier A, Hung R, Hall J, Canzian F, Brennan P. Evidence for an important role of alcohol- and aldehyde-metabolizing genes in cancers of the upper aerodigestive tract. Cancer Epidemiol Biomarkers Prev. 2006;15(4):696–703. doi: 10.1158/1055-9965.EPI-05-0710. [DOI] [PubMed] [Google Scholar]
- Hashibe M, Brennan P, Strange RC, Bhisey R, Cascorbi I, Lazarus P, Oude Ophuis MB, Benhamou S, Foulkes WD, Katoh T, Coutelle C, Romkes M, Gaspari L, Taioli E, Boffetta P. Meta- and pooled analyses of GSTM1, GSTT1, GSTP1, and CYP1A1 genotypes and risk of head and neck cancer. Cancer Epidemiol Biomarkers Prev. 2003;12(12):1509–17. [PubMed] [Google Scholar]
- Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22(20):3053–62. doi: 10.1038/sj.onc.1206445. [DOI] [PubMed] [Google Scholar]
- Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE review. Am J Epidemiol. 2005;162(10):925–42. doi: 10.1093/aje/kwi318. [DOI] [PubMed] [Google Scholar]
- Le Marchand L, Guo C, Benhamou S, Bouchardy C, Cascorbi I, Clapper ML, Garte S, Haugen A, Ingelman-Sundberg M, Kihara M, Rannug A, Ryberg D, Stücker I, Sugimura H, Taioli E. Pooled analysis of the CYP1A1 exon 7 polymorphism and lung cancer (United States) Cancer Causes Control. 2003;14(4):339–46. doi: 10.1023/a:1023956201228. [DOI] [PubMed] [Google Scholar]
- Manuguerra M, Saletta F, Karagas MR, Berwick M, Veglia F, Vineis P, Matullo G. XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am J Epidemiol. 2006;164(4):297–302. doi: 10.1093/aje/kwj189. [DOI] [PubMed] [Google Scholar]
- Raimondi S, Boffetta P, Anttila S, Bröckmoller J, Butkiewicz D, Cascorbi I, Clapper ML, Dragani TA, Garte S, Gsur A, Haidinger G, Hirvonen A, Ingelman-Sundberg M, Kalina I, Lan Q, Leoni VP, Le Marchand L, London SJ, Neri M, Povey AC, Rannug A, Reszka E, Ryberg D, Risch A, Romkes M, Ruano-Ravina A, Schoket B, Spinola M, Sugimura H, Wu X, Taioli E. Metabolic gene polymorphisms and lung cancer risk in non-smokers. An update of the GSEC study. Mutat Res. 2005;592(1–2):45–57. doi: 10.1016/j.mrfmmm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Vineis P, Marinelli D, Autrup H, Brockmoller J, Cascorbi I, Daly AK, Golka K, Okkels H, Risch A, Rothman N, Sim E, Taioli E. Current smoking, occupation, N-acetyltransferase-2 and bladder cancer: a pooled analysis of genotype-based studies. Cancer Epidemiol Biomarkers Prev. 2001;10(12):1249–52. [PubMed] [Google Scholar]
- Vineis P, Veglia F, Anttila S, Benhamou S, Clapper ML, Dolzan V, Ryberg D, Hirvonen A, Kremers P, Le Marchand L, Pastorelli R, Rannug A, Romkes M, Schoket B, Strange RC, Garte S, Taioli E. CYP1A1, GSTM1 and GSTT1 polymorphisms and lung cancer: a pooled analysis of gene-gene interactions. Biomarkers. 2004;9(3):298–305. doi: 10.1080/13547500400011070. [DOI] [PubMed] [Google Scholar]
- Vineis P, Veglia F, Benhamou S, Butkiewicz D, Cascorbi I, Clapper ML, Dolzan V, Haugen A, Hirvonen A, Ingelman-Sundberg M, Kihara M, Kiyohara C, Kremers P, Le Marchand L, Ohshima S, Pastorelli R, Rannug A, Romkes M, Schoket B, Shields P, Strange RC, Stucker I, Sugimura H, Garte S, Gaspari L, Taioli E. CYP1 A1 T3801 C polymorphism and lung cancer: a pooled analysis of 2451 cases and 3358 controls. Int J Cancer. 2003;104(5):650–7. doi: 10.1002/ijc.10995. [DOI] [PubMed] [Google Scholar]
- Ye Z, Song H, Higgins JP, Pharoah P, Danesh J. Five glutathione s-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: meta-analysis of 130 studies. PLoS Med. 2006;3(4):e91. doi: 10.1371/journal.pmed.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]