

Abstract
The sporulation transcription factor σK of Bacillus subtilis is controlled by a signal transduction pathway that operates at the level of the proteolytic processing of the inactive precursor protein pro-σK. The conversion of pro-σK to σK requires the putative processing enzyme SpoIVFB and is governed by the regulatory proteins SpoIVFA and BofA. We engineered vegetative cells to carry out processing of pro-σK by inducing the synthesis of the proprotein, a modified form of the putative processing enzyme, and its two regulators during growth. The results showed that (i) modified SpoIVFB was the only sporulation protein necessary to achieve processing of pro-σK; (ii) SpoIVFA stimulated processing, apparently by protecting the processing enzyme from degradation; (iii) BofA inhibited processing in a manner that did not involve degradation of SpoIVFB; and (iv) the inhibition of SpoIVFB by BofA was dependent on SpoIVFA. We conclude that BofA and SpoIVFA act synergistically and are the only two sporulation proteins needed to inhibit the function of SpoIVFB. Our results are consistent with the idea that activation of pro-σK occurs by a reversal of the BofA/SpoIVFA-mediated inhibition of the processing enzyme.
Keywords: σK, proteolysis, protein localization
Proteolysis is emerging as an important theme in the regulation of transcription factors of several kinds. Examples from eukaryotic cells are the mammalian transcription factors NFκB and SREBP (sterol-regulatory element binding protein) and the Drosophila regulatory protein Ci (Cubitus interruptus). NF-κB is controlled by regulatory pathways that culminate in the proteosome-mediated destruction of an inhibitory protein IκB, which binds to NF-κB and masks its nuclear localization signal (1). The SREBP factor is derived from an integral membrane protein that is held inactive by virtue of being sequestered at the nuclear envelope and the endoplasmic reticulum. In sterol-depleted cells, the membrane-bound precursor is cleaved to generate a soluble fragment that translocates to the nucleus and activates transcription (2). Finally, full-length Ci is inactive because it is tethered in the cytoplasm. It is cleaved to generate a form that lacks the tethering domain and migrates to the nucleus through a pathway that is governed by the Hedgehog signaling protein (3, 4). The subject of the present investigation is a prokaryotic transcription factor σK that is similarly derived from an inactive precursor by regulated proteolysis.
The transcription factor σK and a closely related regulatory protein called σE play a central role in the regulation of gene expression during the process of sporulation in the bacterium Bacillus subtilis (5–7). Sporulation involves the formation of a polar septum that asymmetrically partitions the developing cell into a large mother-cell compartment and a small forespore chamber. The two cells initially lie side by side, but later in development the forespore becomes wholly engulfed by the mother cell to create a cell within a cell. The σK and σE factors are present in the mother cell but their activities are governed by signal transduction pathways that emanate from the forespore (7, 8). The σE factor is activated shortly after the formation of the polar septum by a pathway that operates at the level of the conversion of the inactive precursor protein pro-σE (9) to the mature and active form of the factor by the proteolytic removal of an N-terminal extension of 27 amino acids (10). Activation of pro-σE is governed by the integral membrane protein SpoIIGA, which is likely to be the processing enzyme (11, 12). Importantly, SpoIIGA is inactive in its default state (12, 13). To cause processing of pro-σE, SpoIIGA must be activated by a secreted signaling protein, SpoIIR, which is produced in the forespore under the control of the forespore transcription factor σF (14, 15). In keeping with this view of the σE signal transduction pathway, cells engineered to produce pro-σE and the SpoIIGA putative protease during growth are capable of generating mature σE when exposed to purified SpoIIR (16). The σE pathway is a timing device that ensures that the proprotein is not converted to σE until after the σF factor has become active in the forespore (through an unrelated pathway that does not involve regulated proteolysis) (7, 14, 17).
Herein we are concerned with the proteolytic activation of pro-σK, which occurs by the proteolytic removal of an N-terminal extension of 20 amino acids (18–20). The conversion of pro-σK to σK takes place after the engulfment stage of sporulation. Like the σE pathway, activation of pro-σK takes place in response to a signaling protein (SpoIVB) that is produced in, and presumably secreted from, the forespore under the control of the late-appearing forespore transcription factor σG (18, 21, 22). Also, like the σE pathway, the σK pathway is a timing device that delays the activation of pro-σK in the mother cell for a period of about an hour until SpoIVB is produced in the forespore (18, 21). In other respects, however, the pathway by which pro-σK is activated is strikingly different from that governing the processing of pro-σE (7).
The conversion of pro-σK to mature σK requires the integral membrane protein SpoIVFB (18, 23, 24), which is produced in the mother cell (23) and is located in the mother-cell membrane that surrounds the forespore (25). SpoIVFB is the pro-σK processing enzyme or a required component of an as yet unidentified proteolytic enzyme (18, 23, 26). Unlike the putative pro-σE processing enzyme SpoIIGA, SpoIVFB appears to be active in its default state. That is, mutants in two other genes, spoIVFA and bofA, cause SpoIVFB-mediated processing to take place without a 1-h delay and without the normal dependence on the SpoIVB signal protein (18, 23, 27). Like SpoIVFB, SpoIVFA is an integral membrane protein that is located in the mother-cell membrane that surrounds the forespore (25); BofA is also an integral membrane protein (27, 28), but its subcellular location is unknown. These results have been interpreted to indicate that the putative processing enzyme SpoIVFB is intrinsically active and is negatively regulated by SpoIVFA and BofA (18, 23, 27, 28). If so, then the SpoIVB signaling protein evidently activates the processing enzyme by overcoming the inhibition of SpoIVFB. Fig. 1 summarizes our working model for the signal transduction pathway.
Figure 1.
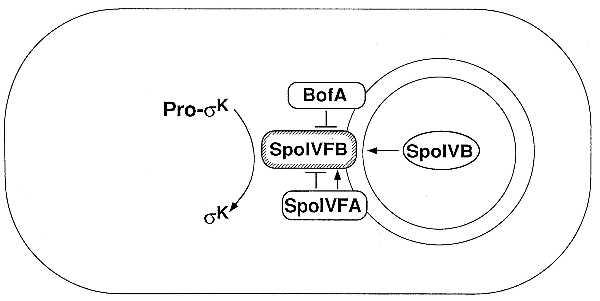
Model for the intercompartmental signal transduction pathway that activates pro-σK during sporulation (for details, see the text). The outer chamber is the mother cell, and the inner chamber is the forespore.
A spoIVFA mutant has an additional phenotype. Though not dependent on the SpoIVB signaling protein, processing in a spoIVFA mutant is temperature-sensitive (18, 23, 29). This has been interpreted to indicate that SpoIVFB is intrinsically unstable and that the putative processing enzyme is normally stabilized by direct interaction with SpoIVFA (23). Thus, SpoIVFA influences the putative processing enzyme both negatively and positively, as depicted in Fig. 1.
Because of the complexity of the σK pathway, we sought to devise a simplified system in which the contribution of BofA, SpoIVFA, and SpoIVFB to the control of pro-σK processing could be studied independently of the process of sporulation and independently of other sporulation proteins. Accordingly, we engineered the synthesis in vegetative B. subtilis cells of pro-σK and, in various combinations, BofA, SpoIVFA, and SpoIVFB. The SpoIVFB protein (or its mRNA) proved to be unstable in vegetative cells, but we succeeded in engineering the synthesis of the putative processing enzyme during growth by use of a modified form of the protein. Herein we show that this modified form of SpoIVFB is sufficient to achieve processing in vegetative cells, that SpoIVFA stimulates processing and does so by facilitating the accumulation of SpoIVFB, and that BofA and SpoIVFA act synergistically to inhibit the function of SpoIVFB. These findings are consistent with the idea that SpoIVFB is the processing enzyme or an essential component of the processing system and that the signaling protein SpoIVB triggers pro-σK activation by reversing the BofA/SpoIVFA-mediated inhibition of SpoIVFB.
MATERIALS AND METHODS
General Methods.
B. subtilis strains used were PY79 [OR9, wild-type B. subtilis] (25), OR758 [spoIVFB∷pOR260 spoIVFB-gfp] (25), RL13 [PY79 SPβ∷gerE-lacZ] (30), and RL439 [VO378, sigK trpC2] (31). Escherichia coli strains used were TG1 (RL468) and XL1-blue (Stratagene). For the routine growth of B. subtilis and E. coli at 37°C LB medium was used (32). To induce promoters under xylose control, d-xylose was added to a final concentration of 10 mM at an OD600 of 0.25–0.30. Sporulation was induced by the resuspension method at 37°C (33). Antibiotics were used at the following concentrations: chloramphenicol, 5 μg/ml; erythromycin and lincomycin together, 1 and 25 μg/ml, respectively; spectinomycin, 100 μg/ml; kanamycin, 10 μg/ml; and ampicillin, 100 μg/ml. β-Galactosidase activity was determined as described (34) by using toluene to permeabilize the cells.
Plasmid Construction.
pOR286 was created by cloning an AccI DNA segment from pSK6 (36), containing the entire sigK gene including the promoter, into the HincII site of the spc-bearing PxylA-bearing integrational plasmid pDAG8–1 (35). pOR267 (which contains a DNA segment from the SspI site upstream of the spoIVF locus and spoIVFA and spoIVFB fused to gfp) and pOR269 (which contains a DNA segment from the SacI site within spoIVFA and spoIVFB fused to gfp) were created by preparing chromosomal DNA from strain OR758. For pOR267 the chromosomal DNA was digested with SspI (for pOR269 with SacI), ligated, and used to transform E. coli. pOR277 was created by cloning an EcoRI–HindIII DNA segment from pDG1832 (a gift of P. Stragier), bearing the xylR gene and the PxylA promoter, into the EcoRI and HindIII sites of the cat-bearing amyE insertion vector pDG364 (37). pOR281 was created by cloning a HindIII–HindIII DNA segment from pOR269 into the HindIII site of pOR277. This cloning step places the spoIVFB-gfp gene fusion under the control of the xylA promoter. pOR294 was created by cloning an SspI–BamHI DNA segment from pOR267 into pOR277 digested with HindIII (rendered flush) and BamHI. This cloning step places spoIVFA and the spoIVFB-gfp gene fusion under the control of the xylA promoter. pOR305 was created by cloning a HindIII–BamHI DNA segment from pOR253 (25) into pOR294 digested with HindIII and BamHI. This results in the reconstitution of spoIVFA preceded by the xylA promoter and 46 codons of spoIVFB and insertion of the spc resistance cassette. pOR314 was created by cloning a HindIII–BamHI DNA segment (from within spoIVFA to downstream of spoIVFB) from pSC224 (23) into pOR294 digested with HindIII and BamHI. This results in the reconstitution of spoIVFA and substitution of wild-type spoIVFB for the spoIVFB-gfp gene fusion. pOR327 was created by cloning a HindIII–BamHI DNA segment from pSC224 (23) into pOR277 digested with HindIII and BamHI. This places the wild-type spoIVFB gene under the control of the xylA promoter. pOR350 was created in three steps. (i) A HindIII–BamHI DNA segment from pER44 (27) was cloned into pOR277 digested with HindIII and BamHI, resulting in pOR337. This places bofA under the control of the xylA promoter. (ii) An EcoRI–BamHI DNA segment from pOR337 (encompassing xylR, PxylA, and bofA) was cloned into pDG795 (a gift of P. Stragier; an erm-bearing thrC insertion vector) resulting in pOR343. (iii) pOR343 was digested with BamHI, and a BamHI–BamHI DNA fragment from pIC22 (38) (encompassing the phleomycin-resistance gene) was inserted downstream of the bofA gene to generate pOR350.
Strain Construction.
Strain OR837 was made by transforming pOR286 into strain RL439 at the sigK locus. To block sporulation the cells were transformed with chromosomal DNA from strain OR909 [PY79 spoIIEΔ∷kn] to generate strain OR910 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn]. Strain OR910 was used as the basis for further strain constructions described below. To put various alleles or gene fusions of the spoIVF locus at the amyE locus under the control of the xylose promoter into strain OR910 the following strains were first made. Competent cells of OR9 were transformed with the following linearized plasmids: pOR305 to generate strain OR903; pOR281 to generate strain OR839; pOR294 to generate strain OR885; pOR314 to generate strain OR913; pOR327 to generate strain OR914. Chromosomal DNAs from strains OR903, OR839, OR885, OR913, and OR914 were individually transformed into competent cells of strain OR910 selecting for chloramphenicol resistance to generate the following strains: OR920 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn, amyE∷Pxyl-spoIVFA (cm, spc)]; OR916 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn, amyE∷Pxyl-spoIVFB-gfp (cm)]; OR918 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn, amyE∷Pxyl-spoIVFA, spoIVFB-gfp (cm)]; OR922 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn, amyE∷Pxyl-spoIVFA, spoIVFB (cm)]; OR932 [trpC2, sigK∷pOR286 Pxyl-sigK (spc), spoIIEΔ∷kn, amyE∷Pxyl-spoIVFB (cm)]. To be able to assay for σK-directed gene expression in strains OR910 (936), 920 (930), 916 (926), 918 (928), 922 (924), and 932 (934), specialized transductions were performed on each strain by using a gerE-lacZ gene fusion-bearing derivative of phage SPβ from strain RL13 as described (39) and selecting for erythromycin and lincomycin resistance. (The numbers in parentheses indicate the numbers of the strains containing the gerE-lacZ gene fusion.) To put bofA at the thrC locus under the control of the xylose promoter into strains OR910, 916, and 918, strain OR9 was transformed with linearized pOR350 to generate strain OR948. Chromosomal DNA from OR948 was transformed into strains OR910, 916, and 918 selecting for erythromycin and lincomycin resistance to generate strains OR952, OR954, and OR956.
Western Blot Analysis.
Samples (1 ml) were collected at the indicated intervals during growth or sporulation. Whole cells extracts were prepared as described (25), except that each resuspended cell pellet was first put at 37°C for 15 min, followed by an incubation on ice for 15 min. Whole cell extracts were separated by SDS/PAGE on 12.5% polyacrylamide gels and electroblotted to an Immobilon-P membrane (Millipore). For immunodetection the blots were blocked in TBS (20 mM Tris⋅HCl, pH 7.5/0.5M NaCl) with 2% Carnation dry milk (for 1–4 hr) at room temperature with shaking. The blots were incubated overnight in TBS/2% milk and the appropriate antibody. Two methods were used as indicated to detect the binding of the primary antibody: a chemiluminescent detection system (Amersham, ECL) or a colorimetric detection system (Promega, ProtoBlot Western blot AP system). The same filter was probed with multiple antibodies as described (25). The antibodies used were rabbit polyclonal antiserum against SpoIVFB [affinity-purified as described (25)], σK [polyclonal antiserum (25)], or green fluorescent protein (GFP; CLONTECH, product 8361–2). The antibodies were used at 1:250, 1:10,000 and 1:2,000 dilutions, respectively.
RESULTS
Use of a Modified Form of the Putative Processing Enzyme SpoIVFB.
Our strategy for studying the regulation of the pro-σK signal transduction pathway was to construct a system in which the function of the putative processing enzyme SpoIVFB and its regulators, SpoIVFA and BofA, could be investigated independently of the process of sporulation and other sporulation proteins. Accordingly, we constructed strains in which expression of the genes for pro-σK, the putative processing enzyme, and its regulators could be induced during growth.
An obstacle in these experiments was that we could not achieve accumulation of the putative processing enzyme in cells engineered to express the spoIVF operon during growth. As presented in Fig. 2, Western blot analysis using antibodies to SpoIVFB showed that the putative processing enzyme could be readily detected in an extract of sporulating cells in which the operon was transcribed from its normal sporulation promoter (Fig. 2 Upper, lanes q–t) but no SpoIVFB could be detected in an extract of growing cells engineered to transcribe the operon from an inducible promoter (Pxyl) (Fig. 2 Upper, lanes m–p). We solved this problem through the use of a modified form of spoIVFB that had been fused at its 3′ terminus to the coding sequence for the GFP of Aquorea victoria (25). Serendipitously, the modified form of SpoIVFB (SpoIVFB-GFP) was able to accumulate in cells engineered to express the gene fusion during growth whereas the wild-type protein could not.
Figure 2.
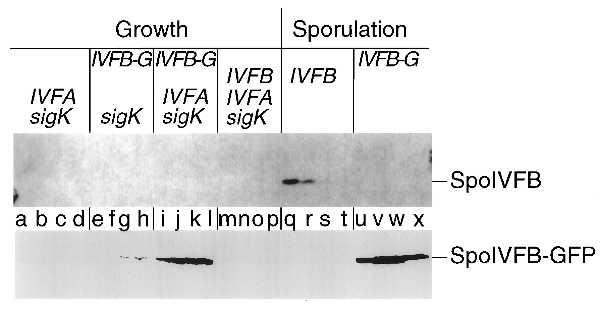
Accumulation during growth of a modified form of the putative processing enzyme SpoIVFB. A Western blot of cell extracts from B. subtilis cells is shown in which the genes or gene fusion that are listed above the blot were artificially induced during growth (lanes a–p) or were turned on in response to sporulation (lanes q–x) and analyzed with an affinity-purified antibody that recognizes the wild-type SpoIVFB protein (Upper) or a polyclonal antibody that recognizes GFP (Lower), to visualize the SpoIVFB-GFP protein fusion. The binding of the primary antibody was detected by using a chemiluminescent detection system (Upper) and a colorimetric detection system (Lower). sigK refers to the gene encoding pro-σK, IVFA refers to the gene encoding SpoIVFA, IVFB-G refers to the gene fusion encoding the SpoIVFB-GFP protein fusion, and IVFB refers to the gene encoding the wild-type SpoIVFB protein. Lowercase type (shown between the two panels) refers to the individual lanes. The lanes are divided into sets of four, with the first letter corresponding to 1 hr after induction, and each subsequent letter in the set indicating an additional hour of induction, up to 4 hr of induction (lanes a–p). Details of the induction protocol during growth and the protocol used to initiate sporulation are included in Materials and Methods. For lanes q–x, the lanes are divided into sets of four, with the first letter corresponding to the second hour of sporulation and each subsequent letter in the set indicating an additional hour of sporulation, up to the fifth hour of sporulation. Cell extracts were prepared and an amount corresponding to 0.1 ml of cells was separated by SDS/PAGE. Lanes correspond to the following strains: a–d, OR920; e–h, OR916; i–l, OR918; m–p, OR922; q–t, OR9; u–x, OR758. Note that the cell extracts used in lanes a–p are the same extracts used in Fig. 3, lanes e–p and r–u.
Western blot analysis showed that SpoIVFB-GFP could be readily detected in extracts of sporulating cells expressing the spoIVF operon with the gene fusion substituted for the wild-type spoIVFB gene from its normal sporulation promoter (Fig. 2 Lower, lanes u–x). Importantly, SpoIVFB-GFP could also be detected in extracts of vegetative cells artificially expressing the spoIVF operon with spoIVFB-gfp substituted for the wild-type spoIVFB gene from the Pxyl (Fig. 2 Lower, lanes i–l). A complication in this analysis was that the fusion protein could not be detected with antibodies to SpoIVFB itself. Evidently, the joining of SpoIVFB to GFP destroyed or obscured the principal epitope(s) to which the anti-SpoIVFB antibodies reacted. Instead, we used antibodies against GFP to detect the fusion protein. Nevertheless, relative to the level of accumulation observed in sporulating cells, the fusion protein accumulated more readily in growing cells than did the unmodified wild-type protein. The failure of unmodified SpoIVFB to accumulate detectably in vegetative cells could explain why only a very low level processing was observed previously in cells engineered to express spoIVF during growth (26).
We presume that wild-type SpoIVFB (or its mRNA) is unstable in vegetative cells and is rapidly degraded during growth and that the SpoIVFB-GFP fusion protein (or the fusion mRNA) somehow resists degradation (or inactivation). Aside from its apparently enhanced stability in growing cells, however, the fusion protein behaved indistinguishably from wild-type SpoIVFB in sporulating cells with regard to the regulation of pro-σK processing as judged by three criteria. (i) spoIVFB-gfp was fully functional in substituting for wild-type spoIVFB with respect to spore formation (25). (ii) The timing of pro-σK processing in sporulating cells harboring spoIVFB-gfp was similar to that observed in sporulating cells harboring the unmodified spoIVFB gene (data not shown). (iii) The activation of pro-σK in cells harboring the gene fusion was dependent upon the SpoIVB signaling protein (data not shown).
SpoIVFB-GFP Is Sufficient to Cause a Low Level of Pro-σK Processing During Growth.
Next, we asked whether SpoIVFB-GFP would cause activation of pro-σK in cells engineered to produce the modified processing enzyme during growth. As shown in Fig. 3, Western blot analysis using antibodies against σK revealed the presence of a low but significant level of an immunoreactive polypeptide that had an electrophoretic mobility indistinguishable from that of mature σK in extracts of cells producing proprotein and SpoIVFB-GFP (Fig. 3, lanes j–l; note protein at the position of bone fide σK, as shown in lane q). As a control, no σK could be detected in cells producing pro-σK but not SpoIVFB-GFP (Fig. 3, lanes a–d).
Figure 3.
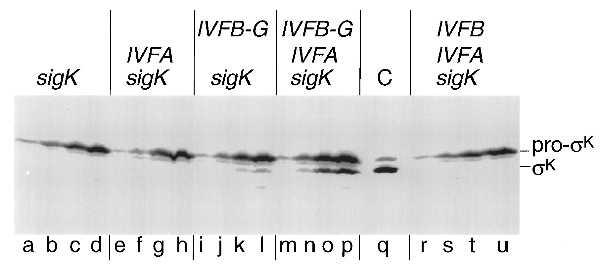
Pro-σK processing in cells engineered to produce a modified form of SpoIVFB during growth. A Western blot of cell extracts from B. subtilis cells is shown in which the genes or gene fusion that are listed above the blot were artificially induced during growth (except for lane C) and analyzed with a polyclonal antibody that recognizes both pro-σK and σK. The binding of the primary antibody was detected by using a colorimetric detection system. sigK refers to the gene encoding pro-σK, IVFA refers to the gene encoding SpoIVFA, IVFB refers to the gene encoding SpoIVFB, and IVFB-G refers to the gene fusion encoding the SpoIVFB-GFP protein fusion. Lowercase type (shown below the blot) refers to the individual lanes. Except for lane q, the lanes are divided into sets of four, with the first letter corresponding to 1 hr after induction, and each subsequent letter in the set indicating an additional hour of induction, up to 4 hr of induction. Details of the induction protocol are included in Materials and Methods. Lane q is a control lane of a cell extract from sporulating cells (at 6 hr after the initiation of sporulation) to indicate the electrophoretic mobility of both pro-σK and σK in this gel system (their relative positions are shown on the right). Cell extracts were prepared and an amount corresponding to 0.1 ml of cells was separated by SDS/PAGE. Lanes correspond to the following strains: a–d, OR910; e–h, OR920; i–l, OR916; m–p, OR918; r–u, OR922; q, OR9.
As a demonstration that the SpoIVFB-GFP-dependent product of pro-σK processing was functional, the experiment of Fig. 4 shows that cells producing pro-σK and the putative processing enzyme supported a low level of expression of lacZ fused to a sporulation gene (gerE) under the control of σK, and this was correlated with the appearance of mature σK (compare with Fig. 3, lanes j–l). Once again, as a control, gerE-lacZ was not significantly expressed in cells producing pro-σK but not SpoIVFB-GFP (Fig. 4).
Figure 4.
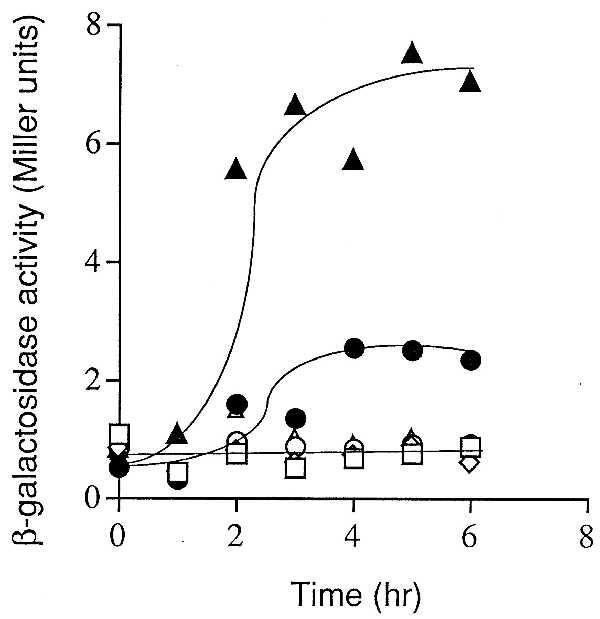
σK-directed β-galactosidase synthesis during growth. Cells were grown in LB medium and induced. Samples were collected at the indicated times after induction and assayed for β-galactosidase activity. Symbols correspond to the following strains (each strain contains gerE-LacZ and produces pro-σk as well as the proteins indicated in parentheses): ▴, OR928 (SpoIVFA, SpoIVFB-GFP); •, OR926 (SpoIVFB-GFP); □, OR936 (σK); ⋄, OR930 (SpoIVFA); ○, OR934 (SpoIVFB); ▵, OR924 (SpoIVFA, SpoIVFB).
SpoIVFA Stimulates Pro-σK Processing.
To investigate the role of SpoIVFA, we monitored processing in cells engineered to produce SpoIVFA, SpoIVFB-GFP, and pro-σK during growth or only the processing enzyme and its proprotein substrate. As judged both by Western blot analysis with anti-σK antibodies and expression of gerE-lacZ, processing (Fig. 3, compare lanes j–l to lanes n–p) and activation of pro-σK (Fig. 4) was severalfold greater in the presence of SpoIVFA than in its absence. As controls, cells engineered to produce SpoIVFA and pro-σK or SpoIVFA, wild-type (unmodified) SpoIVFB, and pro-σK during growth did not convert pro-σK to σK (Fig. 3, lanes e–h and lanes r–u, respectively) and did not activate gerE-lacZ (Fig. 4). It should be noted that even when stimulated by SpoIVFA, the maximal level of σK-directed expression of gerE-lacZ (7 Miller units) achieved during growth was much lower than that observed in wild-type sporulating cells (∼80 Miller units, data not shown). Conceivably, in vegetative cells σK is only partially effective in competing with the primary σ factor σA, whereas late in sporulation σK is the principal σ factor present in the mother-cell compartment.
An explanation for the stimulatory effect of SpoIVFA is provided by Fig. 2 Lower, which shows that the level of SpoIVFB-GFP was severalfold higher in the presence of SpoIVFA (Fig. 2 Lower, lanes i–l) than in its absence (Fig. 2 Lower, lanes e–h). Indeed, the level of processing (Fig. 3) and the level of expression of gerE-lacZ (Fig. 4) were approximately correlated with the relative amount of the fusion protein. These observations reinforce the view that SpoIVFB is the processing enzyme or its regulator and suggest that SpoIVFA exerts its effect, at least in part, by helping to protect the processing enzyme from degradation during growth. [SpoIVFA may play a similar role in sporulating cells because previous experiments had shown that the level of wild-type SpoIVFB during sporulation is lower in the absence than in the presence of SpoIVFA (25).] Thus, in our experiments the presence of SpoIVFA and the use of a fusion with GFP contributed to maximizing the cellular concentration of SpoIVFB and hence to enhancing the level of pro-σK processing.
BofA Inhibits Pro-σK Processing in a Manner That Depends on SpoIVFA.
To investigate the role of BofA in the control of pro-σK processing, cells engineered to produce pro-σK, SpoIVFA, and SpoIVFB-GFP were additionally engineered to express the gene for BofA during growth. Western blot analysis with anti-σK antibodies showed that the presence of BofA strongly inhibited processing (Fig. 5 Upper, compare lanes q–t representing the absence of BofA with lanes u–x representing the presence of BofA). We conclude that BofA blocks the conversion of pro-σK to mature σK. Revealingly, BofA did not cause destruction of the processing enzyme; the level of SpoIVFB-GFP was similar in the presence and absence of BofA (Fig. 5 Lower, compare lanes q–t representing the absence of BofA to lanes u–x representing the presence of BofA). Rather, BofA evidently acts to inhibit the activity of SpoIVFB.
Figure 5.
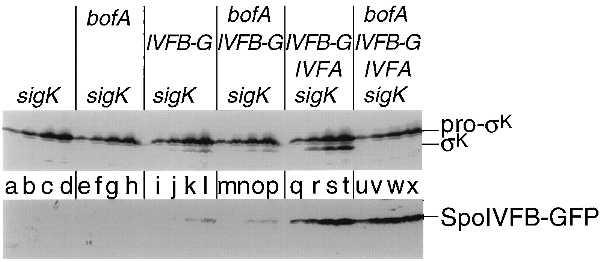
Processing of pro-σK is inhibited in cells engineered to produce BofA during growth. A Western blot of cell extracts from B. subtilis cells is shown in which the genes or gene fusion that are listed above the blot were artificially induced during growth and analyzed with a polyclonal antibody that recognizes both pro-σK and σK (Upper) or GFP (Lower), to visualize the SpoIVFB-GFP protein fusion. The binding of the primary antibody was detected using a colorimetric detection system (Upper) and a chemiluminescent detection system (Lower). sigK refers to the gene encoding pro-σK, IVFA refers to the gene encoding SpoIVFA, IVFB-G refers to the gene fusion encoding the SpoIVFB-GFP protein fusion, and bofA refers to the gene encoding BofA. Lowercase type (shown between the two panels) refers to the individual lanes. The lanes are divided into sets of four, with the first letter corresponding to 1 hr after induction and each subsequent letter in the set indicating an additional hour of induction, up to 4 hr of induction. Details of the induction protocol are included in Materials and Methods. Cell extracts were prepared and an amount corresponding to 0.1 ml of cells was separated by SDS/PAGE. Lanes correspond to the following strains: a–d, OR910; e–h, OR952; i–l, OR916; m–p, OR954; q–t, OR918; u–x, OR956.
Finally, we investigated the effect of BofA on processing during growth in the presence and absence of SpoIVFA. As noted above, the level of processing (and of SpoIVFB-GFP) is severalfold lower in the absence of SpoIVFA than in its presence. Nevertheless, the low level of SpoIVFA-independent processing was substantially immune to the inhibitory effect of BofA (Fig. 5 Upper, compare lanes i–l representing the absence of BofA with lanes m–p representing the presence of BofA). Note also that in the absence of SpoIVFA (as in its presence; see above), BofA had little effect on the level of SpoIVFB-GFP (Fig. 5 Lower, compare lanes i–l representing the absence of BofA with lanes m–p representing the presence of BofA). We conclude that SpoIVFA and BofA act synergistically to inhibit pro-σK processing by SpoIVFB-GFP during growth and that this inhibition is exerted at the level of the activity of the processing enzyme rather than its stability.
DISCUSSION
We have devised a system for studying the contribution of the putative processing enzyme SpoIVFB and its two regulators, SpoIVFA and BofA, to pro-σK processing independently of the process of sporulation and independently of other sporulation proteins. Our system is based on the artificial synthesis of pro-σK, SpoIVFB, SpoIVFA, and BofA in vegetatively growing cells. An obstacle in these experiments was the fact that the putative processing enzyme SpoIVFB failed to accumulate in cells engineered to express spoIVFB during growth, evidently because SpoIVFB (or its mRNA) is unstable and subject to rapid degradation (or inactivation) in vegetative cells. Instead, a modified form of SpoIVFB, SpoIVFB-GFP, was used that was sufficiently resistant to degradation (or whose mRNA was sufficiently resistant to inactivation) to allow the accumulation of the fusion protein in growing cells.
With this system, we systematically engineered the synthesis of the four components of the processing system in various combinations and made the following observations. First, the presence of SpoIVFB-GFP was sufficient to cause the conversion of pro-σK to mature σK, and hence SpoIVFB is the only sporulation protein necessary for processing. From this we conclude that SpoIVFB is the processing enzyme or it is an essential component of a proteolytic system that is present both during growth and sporulation. Second, SpoIVFA stimulated processing and did so, at least in part, by facilitating the accumulation of the putative protease. The simplest interpretation of this is that the SpoIVFB-GFP fusion protein is only partially protected from degradation and that SpoIVFA directly interacts with the fusion protein, thereby further protecting it from proteolysis. A direct interaction between SpoIVFA and SpoIVFB could also explain the temperature sensitivity of a spoIVFA mutant during sporulation if we assume that in sporulating cells SpoIVFB is thermolabile in the absence of its SpoIVFA partner. [An alternative interpretation involving the effect of spoIVFA translation on the efficiency of translation of spoIVFB in the two-cistron spoIVF operon is not excluded, although earlier work has shown that null mutations in spoIVFA do not exert a polar effect on spoIVFB (23).]
The third and most important observation was that BofA inhibited processing and did so in a manner that depended on SpoIVFA. From this we infer that BofA and SpoIVFA act synergistically to inhibit SpoIVFB-mediated processing and further that BofA and SpoIVFA are the only sporulation proteins necessary to achieve inhibition of SpoIVFB. Importantly, inhibition was not due to the destruction of the processing enzyme (at least not overtly). Rather, BofA and SpoIVFA evidently somehow block the function of SpoIVFB. Because BofA, SpoIVFA, and SpoIVFB are integral membrane proteins (23, 25, 27, 28), it would not be surprising if the three sporulation proteins form a heteromeric complex in the membrane that holds the processing enzyme in an inactive state (18, 23).
The demonstration that BofA and SpoIVFA are necessary and sufficient to block the function of SpoIVFB is the most significant contribution of the present investigation because it underscores the view that processing is negatively regulated and that pro-σK is activated by a reversal of the inhibition of the processing enzyme. In sporulating cells this reversal is caused by the signaling protein SpoIVB, which is the only protein produced under the control of the forespore transcription factor σG that is required to activate pro-σK (22). A principal challenge for future studies should be to use the vegetative processing system to understand how the SpoIVB signaling protein triggers SpoIVFB-mediated processing. In light of present and past observations, we speculate that during sporulation the SpoIVB signaling protein is secreted into the space between the forespore and mother-cell membranes, where it interacts with the proposed heteromeric processing complex to reverse the inhibitory effect of BofA/SpoIVFA on the SpoIVFB processing enzyme.
Acknowledgments
We thank K. Pogliano for DNA from strain MO1566, D. Garsin for pDAG8–1, P. Stragier for pDG795 and pDG1832, R. Pruitt for advice on image processing, and Lee Kroos for critical reading of the manuscript. O.R. thanks B. Weisberg and National Institute of Child Health and Human Development for their hospitality during the writing of this manuscript. This work was supported by National Institutes of Health Grant GM18568 to R.L.
ABBREVIATION
- GFP
green fluorescent protein
References
- 1.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 2.Brown M S, Goldstein J L. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 3.Aza-Blanc P, Ramirez-Weber F A, Laget M P, Schwartz C, Kornberg T B. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- 4.Hammerschmidt M, Brook A, McMahon A P. Trends Genet. 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- 5.Losick R, Youngman P, Piggot P J. Annu Rev Genet. 1986;20:625–669. doi: 10.1146/annurev.ge.20.120186.003205. [DOI] [PubMed] [Google Scholar]
- 6.Piggot P J, Coote J G. Bacteriol Rev. 1976;40:908–962. doi: 10.1128/br.40.4.908-962.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stragier P, Losick R. Annu Rev Genet. 1996;30:297–241. doi: 10.1146/annurev.genet.30.1.297. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser D, Losick R. Cell. 1993;73:873–885. doi: 10.1016/0092-8674(93)90268-u. [DOI] [PubMed] [Google Scholar]
- 9.LaBell T L, Trempy J E, Haldenwang W G. Proc Natl Acad Sci USA. 1987;84:1784–1788. doi: 10.1073/pnas.84.7.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyao A, Theeragool G, Takeuchi M, Kobayashi Y. J Bacteriol. 1993;175:4081–4086. doi: 10.1128/jb.175.13.4081-4086.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonas R M, Weaver E A, Kenney T J, Moran C P., Jr J Bacteriol. 1988;170:507–511. doi: 10.1128/jb.170.2.507-511.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stragier P, Bonamy C, Karmazyn-Campelli C. Cell. 1988;52:697–704. doi: 10.1016/0092-8674(88)90407-2. [DOI] [PubMed] [Google Scholar]
- 13.Shazand K, Frandsen N, Stragier P. EMBO J. 1995;14:1439–1445. doi: 10.1002/j.1460-2075.1995.tb07130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karow M L, Glaser P, Piggot P J. Proc Natl Acad Sci USA. 1995;92:2010–2016. doi: 10.1073/pnas.92.6.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Londono-Vallejo J-A, Stragier P. Genes Dev. 1995;9:503–508. doi: 10.1101/gad.9.4.503. [DOI] [PubMed] [Google Scholar]
- 16.Hofmeister A E M, Londono-Vallejo A, Harry E, Stragier P, Losick R. Cell. 1995;83:219–226. doi: 10.1016/0092-8674(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 17.Pogliano K, Hofmeister A E, Losick R. J Bacteriol. 1997;179:3331–3341. doi: 10.1128/jb.179.10.3331-3341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cutting S, Oke V, Driks A, Losick R, Lu S, Kroos L. Cell. 1990;62:239–250. doi: 10.1016/0092-8674(90)90362-i. [DOI] [PubMed] [Google Scholar]
- 19.Kroos L, Kunkel B, Losick R. Science. 1989;243:526–529. doi: 10.1126/science.2492118. [DOI] [PubMed] [Google Scholar]
- 20.Stragier P, Kunkel B, Kroos L, Losick R. Science. 1989;243:507–512. doi: 10.1126/science.2536191. [DOI] [PubMed] [Google Scholar]
- 21.Cutting S, Driks A, Schmidt R, Kunkel B, Losick R. Genes Dev. 1991;5:456–466. doi: 10.1101/gad.5.3.456. [DOI] [PubMed] [Google Scholar]
- 22.Gomez M, Cutting S, Stragier P. J Bacteriol. 1995;177:4825–4827. doi: 10.1128/jb.177.16.4825-4827.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cutting S, Roels S, Losick R. J Mol Biol. 1991;221:1237–1256. doi: 10.1016/0022-2836(91)90931-u. [DOI] [PubMed] [Google Scholar]
- 24.Lu S, Halberg R, Kroos L. Proc Natl Acad Sci USA. 1990;87:9722–9726. doi: 10.1073/pnas.87.24.9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Resnekov O, Alper S, Losick R. Genes Cells. 1996;1:529–542. doi: 10.1046/j.1365-2443.1996.d01-262.x. [DOI] [PubMed] [Google Scholar]
- 26.Lu S, Cutting S, Kroos L. J Bacteriol. 1995;177:1082–1085. doi: 10.1128/jb.177.4.1082-1085.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricca E, Cutting S, Losick R. J Bacteriol. 1992;174:3177–3184. doi: 10.1128/jb.174.10.3177-3184.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varcamonti M, Marasco R, De Felice M, Sacco M. Microbiology. 1997;143:1053–1058. doi: 10.1099/00221287-143-4-1053. [DOI] [PubMed] [Google Scholar]
- 29.Lamont I L, Mandelstam J. J Gen Micro. 1984;130:1263–1269. doi: 10.1099/00221287-130-5-1263. [DOI] [PubMed] [Google Scholar]
- 30.Cutting S, Panzer S, Losick R. J Mol Biol. 1989;207:393–404. doi: 10.1016/0022-2836(89)90262-3. [DOI] [PubMed] [Google Scholar]
- 31.Oke V, Losick R. J Bacteriol. 1993;175:7341–7347. doi: 10.1128/jb.175.22.7341-7347.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 33.Sterlini J M, Mandelstam J. Biochem J. 1969;113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 35.Webb C, Teleman A, Gordon S, Straight A, Belmont A, Lin D C, Grossman A D, Wright A, Losick R. Cell. 1997;88:667–674. doi: 10.1016/s0092-8674(00)81909-1. [DOI] [PubMed] [Google Scholar]
- 36.Kunkel B, Losick R, Stragier P. Genes Dev. 1990;4:525–535. doi: 10.1101/gad.4.4.525. [DOI] [PubMed] [Google Scholar]
- 37.Harwood C R, Cutting S M. Molecular Biological Methods for Bacillus. New York: Wiley; 1990. [Google Scholar]
- 38.Steinmetz M, Richter R. Gene. 1994;142:79–83. doi: 10.1016/0378-1119(94)90358-1. [DOI] [PubMed] [Google Scholar]
- 39.Sandman K, Kroos L, Cutting S, Youngman P, Losick R. J Mol Biol. 1988;200:461–473. doi: 10.1016/0022-2836(88)90536-0. [DOI] [PubMed] [Google Scholar]