

Abstract
Metastasis remains the most deadly aspect of cancer and still evades direct treatment. Clinically and experimentally, primary tumor development and metastasis are distinct processes—locally growing tumors can progress without the development of metastases. The discovery of endogenous molecules that exclusively inhibit metastasis suggests that metastasis is an amenable therapeutic target. By definition, metastasis suppressors inhibit metastasis without inhibiting tumorigenicity and are thus distinct from tumor suppressors. As the biology underlying functional mechanisms of metastasis suppressors becomes clearer, it is evident that metastasis suppressors could be harnessed as direct drug targets, prognostic markers, and to understand the fundamental biology of the metastatic process. Metastasis suppressors vary widely in their cellular localization: they are found in every cellular compartment and some are secreted. In general, metastasis suppressors appear to regulate selectively how cells respond to exogenous signals, by affecting signaling cascades which regulate downstream gene expression. This review briefly summarizes current functional and biochemical data on metastasis suppressors implicated in breast cancer. We also present a schematic integrating known mechanisms for these metastasis suppressors highlighting potential targets for therapeutic intervention.
Keywords: Breast cancer, Cancer, Metastasis, Metastasis suppressor, Signaling, Tumor, BRMS1, Cadherins, CD44, DLC1, Drg1, Gelsolin, KAI1, KISS1, MKK4, Nm23, RhoGDI2, RKIP, RRM1, SSeCKS
Cancer Metastasis
Metastasis continues to be the major cause of morbidity and mortality among cancer patients. Five-year survival rates for breast cancer drop from close to 100% when the cancer is localized to less than 25% when the cancer has colonized distant sites, underscoring the need to identify and target mediators in the metastatic process. Most therapeutic strategies include excision of the primary tumor with the anticipation that accompanying pharmaco-, radio-, and/or immuno-therapy will eradicate any remaining tumor cells that have escaped surgical removal or that may have already disseminated. Although this approach is intuitive and has been effective in many cases, re-emergence of tumors and metastases is not uncommon. In fact, recurrent metastatic growth in breast cancer has been shown to occur years after the patient has been declared cancer free [1, 2]. Approximately one-third of all breast cancer patients are positive for metastatic disease at the time of initial diagnosis [3]. This indicates that a sub-population of tumor cells had disseminated, survived, resisted therapy, and had been dormant at secondary sites only to recommence growth. Transformation from a non-malignant to a metastatic phenotype is an evolutionary process whereby cancer cells progressively acquire characteristics that result from, or are a consequence of, selection of a sub-population of cells that are eventually able to complete all steps of the metastatic cascade [4]. Heterogeneity as a consequence of genomic instability assures that different populations within the tumor will have acquired differing characteristics during tumor progression. Whether the tumor cell is inherently “hardwired” to metastasize as a result of early acquired mutations or whether the metastatic phenotype is a result of natural selection during tumor progression is still open for debate [4-7].
Primary tumor formation is a necessary requirement for metastasis and it is estimated that approximately 1×106 cells escape into circulation per gram of primary tumor per day [8]. Only a fraction of cells leaving the primary tumor survive in circulation and even fewer cells colonize secondary sites [9]. The development of a metastasis, then, involves growth of such surviving cells at a secondary site. A unified definition of a “metastasis” is however still lacking and widely differs from a single disseminated cell (so-called micrometastasis) to a macroscopic mass that grows discontinuous from the primary tumor mass. We have advocated that disseminated cells can be called metastases only if they seed and proliferate at secondary sites [6]. The arrival of a tumor cell at the secondary site implies that the cell has completed all antecedent steps in the metastatic cascade (reviewed in [10]). However, seeding at ectopic sites is by no means an indicator or guarantor of growth. Once disseminated, tumor cells are believed to be regulated by their immediate microenvironment, although there is emerging evidence that preconditioning of seeding sites may occur before tumor cells reach the secondary site. That cancer cells prepare a “fertile ground” for their arrival was elegantly demonstrated by Kaplan et al. [11] who showed that melanoma cells released soluble factors that stimulated lung fibroblasts to secrete fibronectin creating an attachment site for the arrival of α4β1/α4β7 integrins, vascular endothelial growth factor receptor (VEGFR) positive progenitor hematopoietic progenitor cells. These progenitor cells then secreted metalloproteinases which released from the matrix a gradient of factors, including stromal derived factor-1 (SDF-1) that attracted chemokine (C-X-C motif) receptor 4 (CXCR4) positive tumor cells [11].
Formation of a tumor mass at the secondary site must follow some of the same steps implicit in primary tumor growth, including proliferation, evasion of apoptosis, and angiogenesis. However, the major distinction is that these disseminated cells grow in ectopic environments. Therefore, only those cancer cells [“seeds”] that are able to adapt to the new environment [“soil”]; those that already possess the ability to respond to microenvironmental signals at the “foreign” site; or those cells that can modify the new microenvironment can be expected to thrive. This “seed and soil” hypothesis was first presented by Sir Stephen Paget when he noted that breast cancer metastases followed a pattern of growth in specific organs, including bone and liver but not others, and reasoned that such patterning may have occurred due to inherent properties of the “soil” [12]. Indeed, the proclivity of cancer cells to grow at particular sites cannot be explained by the anatomy of the vasculature and associated hemodynamics alone. Micro-vasculature does play an important role in trapping disseminated cells within organs, however, this entrapment may not always translate to growth in situ [9, 13]. Hart and Fidler confirmed Paget’s hypothesis of non-random tumor cell growth by implanting kidney, lung, and ovary tissue fragments in the thighs of mice and showed that B16 melanoma cells injected intravenously grew only in implanted lung or ovarian tissue [14]. These seminal experiments also demonstrated that disseminated cells arrested in the implants at essentially similar numbers but still showed organ selectivity for growth. Therefore, the role of vasculature in organ-selective tumor seeding and growth, although important, may be only secondary to the properties of the secondary site and the tumor cell itself.
Thus, while it can be reasoned from these data that the secondary microenvironment greatly influences eventual tumor cell growth, disseminated cancer cells must also be able to respond to the microenvironment in order to survive and grow. Hence, in theory, modulating the ability of cancer cells to respond to their microenvironment or modulating the secondary growth environment itself would be expected to alter metastasis. Collectively, the above data also reinforce the notion that metastasis is an interactive process—the tumor cell must be responsive to its particular microenvironment and the microenvironment must provide for the tumor cell to settle and eventually proliferate.
Modulation of Metastasis
The process of metastasis has theoretically been divided into a number of discrete steps including formation of the primary tumor, angiogenesis, invasion, intravasation, survival in the vasculature, extravasation, and colonization of the secondary site [10, 15]. Every step in the metastatic cascade must be completed for successful manifestation of metastasis. Therefore, the expression or loss of any gene/protein that interferes with any of the step(s) in the metastatic cascade can, in theory, modulate metastasis. Progression towards a metastatic phenotype requires a concerted effort between a variety of molecules such as MMPs, oncogenes, cell adhesion molecules, etc. that have been implicated in advancing one or more steps of the metastatic cascade. In contrast, since metastasis is a linear process, blockade of any single step in the cascade would prevent metastasis [16]. Metastasis suppressors are a class of proteins that block metastasis without inhibiting primary tumor formation. Different metastasis suppressors disrupt one or more steps in the metastatic cascade. Thus, metastasis can potentially be blocked by restoring expression of a single molecule. For the most part, metastasis suppressors have been localized to chromosomal regions frequently lost in late-stage cancers and have traditionally been identified using differential expression analysis between metastatic and non-metastatic cells for a particular cancer type. As mechanisms underlying metastasis suppressor function become clearer, metastasis suppressors can be segregated into discrete functional groups (reviewed in [17]).
Metastasis suppressors exhibit diverse cellular localization complementary to their diverse cellular functions. For example, KAI1 is localized to the cell membrane and is believed to modulate tumor cell engagement with the vasculature; MKK4, Nm23, and RKIP localize to the cytoplasm and alter transmission of cellular signals; BRMS1 localizes mainly to the nucleus and is believed to alter gene expression as part of the mammalian Sin3: histone deacetylase complexes (mSin3:HDAC) altering levels and activity of signaling molecules including phosphoinositides and nuclear factor kappa B (NF-κB). Within the cell, KISS1 co-localizes with the Golgi apparatus and is eventually secreted. Both these events are believed to be part of the metastasis suppressor action of KISS1. With restored expression of the metastasis suppressor KISS1, cells reach the secondary site without formation of overt metastases, suggesting that at least some metastasis suppressors may regulate growth at the secondary site. Thus, metastasis suppressors exhibit diversity of mechanisms of action and diversity in the steps of the metastatic cascade at which they act.
Necessary Qualifications for Being Classified as a “Metastasis Suppressor”
Metastasis suppressors suppress metastasis without inhibiting growth of the primary tumor. This criterion defines a unique class of metastasis modulators that are fundamentally different from tumor suppressors and which utilize distinct mechanisms for metastasis suppression. Thus, while tumors suppressors inhibit metastasis by virtue of inhibiting primary tumor growth, metastasis suppressors primarily affect downstream events other than primary tumor formation.
Literature exists in which molecules have been classified as metastasis suppressors based solely on in vitro data or even when primary tumor formation was suppressed [18-20]. It must be emphasized that while individual steps of the metastatic cascade may be modeled in vitro with a fair degree of physiological correlation, metastasis cannot be modeled in vitro due to the insufficient complexity of in vitro systems. Studies are also hampered by our incomplete understanding of mediators and mechanisms governing metastasis. Therefore, while demonstration of decreased invasive potential, anchorage independent growth, angiogenesis or proliferation can be used as preliminary screens for identifying candidate metastasis suppressors, these in vitro assays, either alone or in combination, are insufficient predictors of in vivo metastatic behavior.
Another question is, if the absence of a particular molecule increases metastasis, does that molecule qualify as a metastasis suppressor [21]? Although in theory, it is tempting to visualize such a molecule as a metastasis suppressor, unless experimentally proven according to the set criteria, such arbitrary classification will only add to confusion in literature and should be avoided.
Outlined below are metastasis suppressors that have been demonstrated to have an association with breast cancer metastasis. Although the precise mechanism of metastasis suppression are unknown for most, their currently known biochemical mechanisms relevant to metastasis are discussed (Tables 1, 2 and Fig. 1). Readers are encouraged to consult the primary literature for experimental details.
Table 1.
Metastasis suppressors and proposed mechanisms
| Metastasis suppressor | Chromosomal location | Proposed mechanism(s) of metastasis suppression | Selected references |
|---|---|---|---|
| BRMS1 | 11q13.1–q13.2 | Part of mSin3:HDAC complexes that regulate transcription, downregulates PtdIns(4,5)P2 and inhibits NF-κB activity and decreased osteopontin expression | [33, 37] |
| Caspase8 | 2q33–q34 | Induction of apoptosis through unligated integrins | [147] |
| E-cadherin, N-cadherin, cadherin -11 | 16q22.1, 8q11.2, 16q22.1 | Cell–cell/cell–matrix adhesion | [159, 160] |
| CD44 | 11p13 | Binds hyaluronan and osteopontin | [161] |
| DLC1 | 8p22–p21.3 | Rho-GTPase activating protein controlling cytoskeletal architecture | [162] |
| Drg1 | 8q24.3 | Unknown | [163] |
| Gelsolin | 9q33 | Actin filament dissolution; reduced motility | [164] |
| KAI1 | 11p11.2 | KAI1 on tumor cells interacts with DARC on vascular endothelium leading to tumor cell senescence | [69] |
| KISS1 | 1q32 | Maintains tumor cell dormancy at secondary site | [88] |
| MKK4 | 17p11.2 | Maintains dormancy at secondary site. May affect cell cycle by induction of p21Cip1/Waf1 and p53, activates p38 and/or JNK | [17, 165] |
| Nm23 | 17q22 | Phosphorylates KSR and prevents downstream activation of the MAPK pathway | [132] |
| RhoGDI2 | 12p12.3 | Regulates Rho; negatively modulates endothelin 1 and neuromedin U expression | [150, 151] |
| RKIP | 12q24.23 | Inhibits MAPK signaling by competitive inhibition of Raf-1:MEK interaction | [140] |
| RRM1 | 11p15.5 | Upregulates PTEN; decreases FAK phosphorylation | [166] |
| SSeCKS | 6q24–q25.2 | Scaffold protein for protein kinases A and C, inhibits osteopontin, VEGF expression; upregulates vasostatin | [154, 167] |
Chromosomal locations have been sourced from the National Center for Biotechnology Information (NCBI) Entrez Gene database updated 01/08/2007.
Table 2.
Metastasis suppressors and clinical correlations
| Metastasis suppressor | Cellular localization | Cancer type | Expression | Prognosis | Selected references | |||
|---|---|---|---|---|---|---|---|---|
| Protein | mRNA | |||||||
| Primary | Metastasis | Primary | Metastasis | |||||
| BRMS1 | Nuclear | Breast | NT | NT | NC | NC | NT | [27, 29, 30] |
| Breast | NT | NT | Increased | NT | Favorable | |||
| Breast | ↓ | NT | NT | NT | Poor | |||
| Breast | Expressed | ↓ | Expressed | ↓ | NT | |||
| Caspase-8 | Cytosol | Neuroblastoma | ↓ | NT | NT | NT | NT | [168] |
| E-cadherin, N-cadherin, cadherin-11 | Cell membrane | Breast | ↓ | NT | NT | NT | NT | [169] |
| CD44 | Cell membrane | Prostate | ↓ | NT | ↓ | NT | NT | [170] |
| DLC1 | Cytosol | NT | NT | NT | NT | NT | NT | |
| Drg1 | Cytosol | Breast | ↓ | ↓ | NT | NT | Poor | [171] |
| Prostate | ↓ | ↓ | NT | NT | Poor | |||
| Gelsolin | Cytosol | NT | NT | NT | NT | NT | NT | |
| KAI1 | Cell membrane | Breast | ↓ | NT | NT | NT | NT | [49-51, 55] |
| Colorectal | ↓ | ↓ | NT | NT | NT | |||
| Cervical | ↓ | ↓ | ↓ | ↓ | NT | |||
| Ovarian | ↓ | ↓ | NT | NT | NC | |||
| NSCLC | ↓ | ↓ | NT | NT | Poor | |||
| KISS1 | Golgi/secreted | Bladder | ↓ | ↓ | NT | NT | NC | [172-174] |
| Esophageal | NT | ↓ | NT | NT | Poor | |||
| Gastric | NT | NT | ↓ | NT | Poor | |||
| Hepatocellular | NT | NT | NC | NT | NT | |||
| Thyroid | NT | NT | Expressed | NT | NT | |||
| MKK4 | Cytosol | Breast | Expressed | ↓ | Expressed | ↓ | NT | [98, 99] |
| Pancreatic | ↓ | NT | ↑ | NT | Inconclusive | |||
| Nm23 | Cytosol/nucleus | Breast | ↓/NC | ↓/NC | ↓/NC | ↓/NC | Poor | Reviewed in [107] |
| Hepatocellular | Expressed | ↓/NC | NT | NT | Poor | |||
| Melanoma | Expressed | ↓ | NT | NT | Poor | |||
| Ovarian | Expressed | ↓ | Expressed | ↓ | Poor | |||
| RhoGDI2 | Cytosol | Bladder | ↓ | NT | NT | NT | Poor | [175] |
| RKIP | Cytosol | Breast | Expressed | ↓ | NT | NT | Poor | [139] |
| RRM1 | Cytosol/nucleus | NT | NT | NT | NT | NT | NT | |
| SSeCKS | Cytosol | Expressed | NT | NT | NT | NT | NT | [176] |
NC No change, NT Not tested
Figure 1.
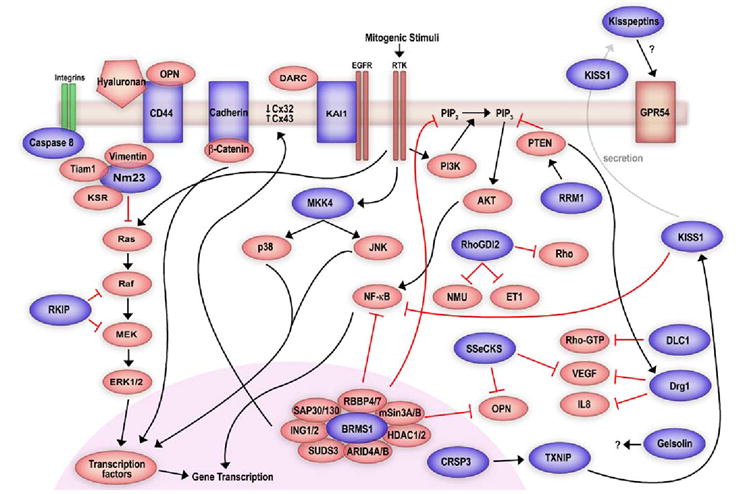
Schematic of proposed signaling by metastasis suppressors. Blue (dark) shapes indicate metastasis suppressors and red (light) shapes indicate signaling molecules. Black (dark) arrows indicate positive regulation and red (light and stunted) connections represent negative regulation. Cellular location of metastasis suppressors is according to currently known data. Only those biochemical pathways and interactions that have been experimentally confirmed are depicted, however, not all signaling pathways are shown (see text for details).
BRMS1
Metastasis suppression of MDA-MB-435 breast carcinoma cells following introduction of human chromosome 11 provided the first evidence for the presence of a metastasis suppressor on chromosome 11 [22]. BRMS1 (Breast Cancer Metastasis Suppressor 1) was subsequently identified by differential display in metastatic and metastasis suppressed MDA-MB-435 cells [23].
Experimentally, BRMS1 suppresses metastasis of breast, melanoma, bladder, and ovarian carcinoma cell lines [24-26]. However, apparently conflicting clinical data exist confounding the role of BRMS1 as a metastasis suppressor in breast cancer [27-30]. A limitation of the conflicting data appears to be the utilization of mRNA instead of protein as a measure of BRMS1 expression. The lack of antibodies against BRMS1 required the use of mRNA as a measure of BRMS1 expression in earlier studies. However, with the development of antibodies it appears mRNA and protein levels do not always correlate for BRMS1. In fact, our laboratory has observed increased BRMS1 mRNA without increased protein expression in the MCF10 breast cancer progression series (Hurst and Welch, unpublished data). Further, BRMS1 protein is stabilized by heat-shock proteins [31] and the eventual stability and localization of the BRMS1-containing complex is responsible for protein function and activity.
As with Nm23, early efforts to define BRMS1 function were to characterize its interacting proteins. Yeast two-hybrid studies revealed that BRMS1 interacted with retinoblastoma binding protein 1 (RBBP1; also known as ARID4A) and suppressor of defective silencing 3 (SUDS3; also known as mSDS3, SAP45) [31, 32]. Both interactors are components of the core mSin3:HDAC co-repressor complexes. mSin3:HDAC complexes regulate gene transcription by recruiting specific HDACs to regulatory sequences and controlling accessibility of transcription elements by chromatin modification. Traditionally, the mSin3: HDAC complex is thought to regulate transcription through HDAC1 and 2. BRMS1 was also found to interact with cytoplasmic HDACs 4, 5, and 6, suggesting that BRMS1 may recruit other HDACs to the core mSin3:HDAC complex to execute transcriptional repression. Activity of BRMS1 is dependent on its protein stability and the chaperone protein Hsp90 was found to stabilize BRMS1 protein [31].
Information obtained from interaction studies has greatly aided in the study of signaling mechanisms of BRMS1. The involvement of transcription factors as possible BRMS1 targets was deduced from its association with the mSin3: HDAC complex. The transcription factor NF-κB is known to regulate transcription of multiple pro-tumorigenic and pro-metastatic genes in human cancer. Consistent with its function as a co-repressor, BRMS1 reduces transcription by promoting the association of HDAC1 with the p65 subunit of NF-κB and decreases NF-κB transcriptional activity by deacetylation of p65 [33, 34]. Reduced NF-κB activity causes decreased expression of osteopontin (OPN), a molecule implicated in breast cancer metastasis to bone [35]. BRMS1 also selectively modulates levels of phosphatidyl inositol (4,5) bisphosphate (PtdIns(4,5)P2), the primary substrate for phosphatidyl inositoI 3-kinase (PI3K) in the PI3K-AKT pathway, which in turn, activates NF-κB downstream of AKT [36]. Downregulation of two vital components of the same signaling cascade suggests an uncanny pathway-selective regulation by BRMS1 [33, 37]. Such specific and redundant regulation by BRMS1 may be important in moderating responses from external growth factors and therefore growth at ectopic sites. Indeed, BRMS1 expression has been shown to be inversely correlated with human epidermal growth factor receptor 2 (HER2) expression in clinical samples [29, 30], strengthening the argument in favor of a growth-factor response regulatory role for BRMS1. Further, BRMS1 restores homotypic gap-junctional intercellular communication in metastatic cells and reduces colony formation in soft agar without alteration of either in vitro cell proliferation or invasion, suggesting a context-dependent regulation of tumor cell behavior [38].
As with other metastasis suppressors, the mechanism for differential regulation of tumor growth at orthotopic versus ectopic sites by BRMS1 remains unknown. Further, regulation of BRMS1 expression remains to be uncovered, and such knowledge may pave the path for the development of specific drugs that induce BRMS1 expression. That the BRMS1 gene is wild-type in all metastatic cells examined to date (Edmonds and Welch, unpublished data) and in limited clinical studies argues that treatment strategies employing restoration of endogenous expression may be a viable option.
KAI1
KAI1 (kang-ai 1; also known as CD82/C33/TIP30) was first identified following introduction of human chromosome 11 in rat prostate cancer cells by microcell-mediated chromosome transfer [39, 40]. KAI1 is part of the tetraspanin superfamily which includes 32 known members in mammals alone [41]. Tetraspanins organize and stabilize large molecular networks that support a diversity of signaling and function (reviewed in [41]). Consequently, these proteins are implicated in various cellular processes, including motility, invasion, and cell signaling, among others [42-45].
KAI1 expression reduces metastasis in breast, melanoma, and prostate cancer models [40, 46-48]. Studies have also shown a loss of KAI1 in poorly differentiated and late-stage cancers, and inverse correlations have been demonstrated between the loss of KAI1 and prognosis in breast [49], colorectal [50], ovarian [51], cervical [52, 53], oral [54], and non-small cell lung cancers [55]. This observed downregulation does not appear to involve KAI1 mutation or allelic loss and suggests transcriptional regulation [56].
Most studies with KAI1 have been performed with T-cells and, through these studies, a variety of mechanistic details have been uncovered. KAI1 interacts with a variety of cell surface molecules, including integrins [57-59], other tetraspans [60-62], major histocompatibility complex class 1 (MHC1) and human leucocyte antigen-DR (HLA-DR) [63, 64], and immunoglobulins. These interactions appear to modulate cellular adhesion, motility, and invasion [45, 65] of tumor as well as immune cells. KAI1 and its interactions with the cytoskeleton through integrins are well characterized [62, 66]. Palmitoylation of KAI1 appears to be essential not only for activation of p130CAS-CrkII signaling through the small GTPases (Rac, Rho, cdc42) and possibly their ability to effect cytoskeletal rearrangements, but also for the regulation of endocytosis of epidermal growth factor receptor (EGFR) [44, 67, 68]. Indeed disruption of palmitoylation disrupts motility and invasion—outcomes of downstream signaling through EGFR and the small GTPases, possibly suggesting a mechanism for KAI1-mediated metastasis suppression through the control of cellular response to external signals.
The most compelling mechanism demonstrated to date for KAI1 suggests an interaction between DARC (Duffy antigen receptor for chemokines)/gp-Fy on vascular endothelial cells and KAI1 on circulating tumor cells [69]. Although DARC is a seven-transmembrane spanning protein and binds multiple chemokines, downstream signaling does not appear to involve G-protein coupling [70]. DARC is expressed on the vascular endothelium of many organs, on erythrocytes, and epithelium [70, 71]. DARC interacts directly with KAI1 on circulating tumor cells, rendering them senescent through the induction of p21 and downregulation of the senescence opposing gene TBX2. Further, KAI1 also interacts with integrins which may be used to tether tumor cells to the vasculature, thus possibly promoting an interaction between KAI1 and DARC [69]. Together, these results suggest a multi-modal inhibition of metastasis by KAI at different steps of the metastatic cascade.
Multiple mechanisms have been proposed to explain the regulation of KAI1 expression. Marreiros and colleagues demonstrated in vitro that the synergism between p53 and junB along with activating protein 2 (AP2) function was essential for KAI1 expression [72]. Kim and colleagues propose that KAI1 expression is dependent on a balance between the expression of Tip60 and β-catenin-reptin complexes [73]. Several lines of evidence argue for and against the proposed mechanisms, suggesting a more complex or possibly cell- and cancer-type specificity in regulation of KAI1 expression [57, 74]. Phorbol myristate acetate (PMA) and etoposide have been shown to induce expression of KAI1 in vitro. Akita and colleagues showed that induction of KAI1 expression through PMA is mediated by protein kinase C [75]. On the other hand, induction of KAI1 expression through etoposide is mediated through p53 and c-Jun in concordance with previous reports suggesting a possible pathway for upregulation of KAI1 in vivo [57]. Treatment with etoposide also reduced invasion, however, etoposide is a topoisomerase II inhibitor and shown to induce cell cycle arrest and induce apoptosis. It is therefore difficult to dissect and separate mechanisms of decreased invasion and attribute the phenotype to KAI1 alone. Genes controlling metastasis suppressors have been found to be epigenetically regulated, and restoration of expression through reversal of promoter demethylation offers a possible pathway for inducing expression. Analysis of the KAI1 promoter revealed the presence of a non-hypermethylated CpG island. Predictably, treatment with 5-aza-2-deoxycytidine or trichostatin A did not upregulate KAI expression, suggesting other regulatory mechanisms [76].
KISS1
Introduction of chromosome 6 in metastatic melanoma cells suppressed metastasis to the lung and lymph nodes (>95%) without inhibiting tumor formation, suggesting the presence of a metastasis suppressor on chromosome 6 [77]. Interestingly, KISS1 was identified by subtractive hybridization and mapped to chromosome 1q32, suggesting regulation of KISS1 expression by gene(s) on chromosome 6 [78]. Further studies demonstrated that a co-factor required for Sp1 activity (CRSP3; mapped to chromosome 6) regulated thioredoxin interacting protein (TXNIP)/vitamin D upregulated protein 1 (VDUP1; mapped to chromosome 1), which, in turn regulated KISS1 expression [79]. Recent data indicate that a direct interaction between the transcription factors AP2α and Sp1 induces KISS1 expression [80, 81]. These data provide a mechanism for the regulation of KISS1 and a potential explanation for its loss in breast cancer where AP2α is frequently lost and in melanoma where CRSP3 is frequently deleted.
The metastasis suppressor function of KISS1 has been experimentally demonstrated in melanoma, ovarian, and breast cancer models [78, 82, 83] establishing KISS1 as a bona-fide metastasis suppressor in these models. Clinically, loss of KISS1 expression has been correlated with poor prognosis in thyroid, gastric, bladder, esophageal, and hepatocellular carcinomas along with melanoma and breast cancer (reviewed in [84]).
Independent laboratories have identified several cleavage sites in the amino acid sequence of KISS1 and subsequently the 54-amino acid processed form of KISS1 (KP54) was detected in human plasma samples, supporting the hypothesis that KP54 is likely to be secreted [85-87]. The fact that full length KISS1 could only be detected in cell lysates and that KP54 peptide fragments (kisspeptins) could only be detected in media from isolated trophoblasts strengthens the hypothesis that KISS1 is secreted and possibly processed at the cell surface [88]. The G-protein coupled receptor 54 (GPR54; also known as hOT7T175/AXOR12) has been identified as a cognate receptor for KP54. Activation of signaling through GPR54 abrogated SDF-1-induced AKT phosphorylation and chemotaxis, suggesting that the KISS1-GPR54 axis may be important in metastasis suppression [89].
Cells expressing KISS1 are metastasis suppressed even following direct intravascular injection, suggesting that the metastasis suppressor function of KISS1 lies at the secondary site [88, 90]. Recent evidence indicates that secretion of KISS1 is required for metastasis suppression and tumor cell dormancy in the lung. Deletion of the secretion signal of KISS1 abolished its metastasis suppressor ability in C8161 melanoma cells. Further, re-expression of KISS1 in C8161 cells causes localization to the endoplasmic reticulum-Golgi; however, deletion of the secretion signal causes redistribution to the cytoplasm [88]. Surprisingly, cell lines suppressed for metastasis by over expression of KISS1 show little or no expression of GPR54, by real-time quantitative PCR, possibly suggesting a paracrine mechanism for KISS1 action or another, as yet, unidentified receptor. The mechanistic studies above were demonstrated in melanoma cells; however, key experimental results have been replicated in pilot studies using breast carcinoma cell lines as well.
A recent study showed that kisspeptins have potent vasoconstrictor abilities; this effect requires the presence of GPR54 [91]. While little difference in microvessel density was observed in primary tumors in KISS1-expressing versus non-KISS1-expressing melanoma and breast carcinoma cells, vessel function may have been reduced via a paracrine mechanism. How selective differences would occur with respect to growth at primary versus secondary sites is a question common to all metastasis suppressors. The answer may well ultimately reside in the analysis of various tumor microenvironments.
Activation of GPR54 by kisspeptins has been shown to induce activation of mitogen activated protein kinase (MAPK) signaling [89]. KISS1 has also been shown to decrease matrix metalloproteinase activity through the negative modulation of NF-κB, suggesting activation of pathways independent of GPR54 involvement [92]. Overall, these data indicate that metastasis suppression requires secretion of KISS1 and possibly effects on vasculature at the secondary site, although the physiological relevance of myriad signaling pathways activated through the KISS1-GPR54 axis in metastasis suppression remains unclear.
As with other metastasis suppressors, the limited clinical studies to date show few mutations. That functional KISS1 was isolated from metastatic melanoma cells argues that the cellular defects of KISS1 in metastasis are largely due to defective regulation rather than mutation [93].
MKK4
MKK4 (Mitogen activated protein kinase kinase 4; also known as JNKK1/SEK1/MEK4/MAP2K4) was first identified as a metastasis suppressor in prostate cancer cells [94]. The MKK4 gene shows a low and consistent rate of mutation (5%) across tumor types, and loss of gene expression rather than mutation is believed to mediate metastasis [95]. Other than prostate cancer, MKK4 has been experimentally demonstrated to be a metastasis suppressor in ovarian carcinoma [96, 97]. An inverse correlation has been demonstrated in a subset of brain metastases from breast cancer where MKK4 mRNA as well as protein levels were reduced along with other metastasis suppressors and also in pancreatic cancer where reduced MKK4 levels were seen in primary tumors as well as in lymph node metastases [98, 99]. In contrast, Wang and colleagues showed that ectopic expression of MKK4 in a panel of breast and pancreatic cancer cell lines induced a “pro-oncogenic” phenotype in vitro opposite to that seen with clinical correlations and in prostate and ovarian carcinoma [100]. The role of MKK4 in breast and pancreatic cancer thus remains controversial and warrants further investigation.
MKK4 is a dual specificity kinase and is an intermediate of the stress-activated MAPK pathways. Three MAPK pathways are currently well-studied: (1) extracellular-signal regulated kinase (ERK), (2) p38, and (3) c-Jun NH2 terminal protein kinase (JNK; also known as SAPK). MKK4 is unique in serving as a common link between and as a dual activator of both p38 and JNK pathways in response to stress, growth factors, and cytokines. Once phosphorylated, both p38 and JNK phosphorylate downstream transcription factors, including c-jun, ATF2, and elk-1 (reviewed in [101]) resulting in an altered pattern of gene expression. These events are implicated in such cellular processes as apoptosis, differentiation, and proliferation [102]. Sustained activation of the upstream mixed lineage kinase 3 (MLK3) also activates MKK4 and downstream c-jun. Activation of c-jun acts as a negative feedback, preventing downstream activation of ERK through MEK [103]. Thus, activation of the JNK pathway downstream of MKK4 may prevent MAPK activation through various mitogenic stimuli and may contribute to cancer cell unresponsiveness towards growth factors.
The exact mechanism of MKK4 mediated metastasis suppression remains unclear. However, similar to the metastasis suppressor KISS1, cells with ectopic MKK4 expression complete all steps of the metastatic cascade and reach the secondary site but fail to form overt metastases [94]. MKK4 kinase activity is essential for metastasis suppression and is probably required to activate a particular transcription profile that mediates growth arrest or cell cycle alterations [104]. MKK6 can also activate p38, a downstream mediator of MKK4. Ectopic expression of MKK6 but not MKK7 (MKK7 activates JNK and not p38) in ovarian cancer cells also suppressed metastasis, indicating that stress-pathways selectively activating p38 may mediate metastasis suppression [97].
Nm23
The first metastasis suppressor discovered and the most widely studied, Nm23 (Non-metastatic clone # 23) was originally identified in murine melanoma cell lines of varying metastatic potential by differential colony hybridization [105]. The human nm23 family now comprises eight members; however, to date only Nm23-H1 and Nm23-H2 have been experimentally demonstrated to suppress metastasis [106].
Nm23 expression has been studied in a variety of tumor types to determine clinical utility as a prognostic marker. Decreased expression of Nm23 was correlated with poor patient survival and tumor grade in subsets of breast, ovarian, hepatocellular carcinomas and melanoma, among other cancer types (reviewed in [107]). In contrast, increased Nm23 expression in neuroblastoma was correlated with poor patient prognosis and survival and was attributed to gene amplification. In addition to gene amplification, mutations in nm23 have been noted in neuroblastoma; however, the incidence is rare and has been observed only in late stage tumors [108]. Such disparity underscores the fundamental differences in biology of histologically distinct cancer types and argues caution when extrapolating findings from one type to another.
Establishment of metastasis involves cross-talk between the tumor cell and its microenvironment, and disruption of such communication would be expected to reduce metastatic potential. Consistent with this idea, transfection of Nm23 reduces in vitro motility and/or colony formation in response to a variety of growth factors, including transforming growth factor-β (TGF-β) [109, 110], IGF-1, and PDGF [111]. Several studies in clinical specimens and in experimental systems have demonstrated an inverse correlation between Nm23 and cell surface receptors, such as EGFR [112] and HER2 [113-116] in multiple tumor types [110]. Although these studies point towards a potential mechanism of altered responsiveness towards exogenous growth signals in Nm23 mediated metastasis suppression, these data are only correlative and over interpretation should be avoided.
Another approach utilized to characterize the mechanism of metastasis suppression involved identification of Nm23-interacting proteins. Using co-immunoprecipitation and yeast-two hybrid analyses, Nm23 was associated with a large cohort of proteins with distinct functions including Tiam1 (nucleotide exchange factor for Rac1) [117], Rad (GTPase) [118, 119], glyceraldehyde 3-phosphate dehydrogenase [120], vimentin [121], G-proteins [122], and casein kinase 2 (CK2) [123] among others (reviewed in [124]). Interaction with other proteins was presumed to either alter Nm23 function (e.g. CK2, GAPDH) or vice versa (e.g. Tiam1, Rad). However, the physiological relevance of many interactors remains speculative in most cases.
Nm23 has three reported activities: NDP kinase [125], histidine kinase [126] and exonuclease [127]. Mutants that disrupt all of the activities are unable to suppress metastasis. CK2 was reported to phosphorylate Nm23 and reduce its NDPK activity; however, metastasis suppression by Nm23 has been shown to be independent of its NDPK activity [128, 129]. Further, Kaetzel and colleagues have also shown that point mutants that disrupt either the exonuclease activity alone or the NDPK activity alone fail to suppress metastasis in 1205Lu melanoma cells (Kaetzel, personal communication). Thus, the definitive mechanism of action for Nm23 remains uncertain.
Mechanistically, the most convincing lead appears to be the interaction of Nm23-H1 with the kinase suppressor of Ras (KSR). KSR is a scaffold protein that binds members of the MAPK pathway (Raf-1, MEK1/2, and ERK1/2) [130]. Under resting conditions, KSR is retained in the cytoplasm by binding to 14-3-3 proteins; this binding is mediated by Ser392 phosphorylation of KSR. Mitogenic stimuli induce dephosphorylation of KSR, resulting in translocation to the plasma membrane and facilitation of signaling between Raf, MEK and ERK [131]. Nm23-H1 phosphorylates KSR at Ser392, possibly sequestering KSR and preventing further downstream activation of the MAPK pathway. This hypothesis is strengthened by the observation that Nm23-H1 transfectants show reduced basal and stimulated MAPK phosphorylation [132].
The eventual aim is to utilize Nm23 and other metastasis suppressors in the clinical setting either as prognostic markers or as targets for drug development. To this end, devising strategies to restore expression and determining whether restored expression is sufficient to suppress metastasis in vivo remains a continual challenge. Some headway has been made with Nm23 with the identification of transcription factor binding sites (MAF/Ets, a CTF/NF1 half site and an ACAAAG enhancer) in the Nm23 promoter contributing to differential expression in breast carcinoma cells [133-136]. These promoter elements were determined to be identical to those in the MMTV-LTR and implicated in the regulation of MMTV through glucocorticoids. Both dexamethasone and medroxyprogesterone acetate (MPA) induced Nm23 expression in vitro, and MPA treated mice showed a significant reduction in frequency and incidence of metastases, indicating possible manipulation of metastasis suppressor expression through external drug administration [136, 137].
RKIP
Raf kinase inhibitor protein (RKIP) was first identified as a metastasis suppressor in prostate cancer [138]. Only correlative evidence of RKIP as a metastasis suppressor in breast cancer exists. RKIP was shown to be expressed in human breast tumors but largely absent in matched lymph node metastases [139].
Biochemically, RKIP functions as a negative modulator of MAPK signaling by preventing activation of MEK by Raf-1. Although Raf-1, MEK, and ERK associate with RKIP, neither are substrates of RKIP. RKIP primarily competitively inhibits Raf-1 interaction with MEK, indicating that its major function may be to sequester MEK and control downstream signal transmission by serving as a checkpoint [140]. Both Raf-1 and B-Raf are required for maximal activation of the MAPK pathway; however, apparently conflicting data exists regarding the ability of RKIP to inhibit B-Raf [141, 142]. Such apparent contradiction may be a result of different cell line models used in these studies. Activation of the MAPK pathway prevents induction of apoptosis. In line with this paradigm, reduction of RKIP expression increases basal MAPK activation and confers resistance to apoptosis. Conversely, enforced RKIP expression sensitizes breast cancer cells to apoptosis [143].
A recent study indicates that RKIP mRNA binds and potently activates 5′-phosphorylated, 2, 5′-linked oligoadenylates [144]. RNAse L binds to these activated oligoadenylates and induces apoptosis through caspases [145]. These results provide a novel function for RKIP and possibly may be important in its metastasis suppressor function.
Tumor or Metastasis Suppressor, Depending upon Cellular Context
A number of candidates molecules suppress metastasis in vivo, however, they do not adhere to the strict definition of a metastasis suppressor, i.e., they suppress tumor formation in some models. Molecules that display such dual functionality (DLC1, DRG1) are outlined in Tables 1, 2 and in Fig. 1, but are not discussed further here because of space limitations.
There are a growing number of metastasis suppressors identified [146]. Since most have only recently been discovered, they have not been tested yet in breast cancer. Multiple cadherins are implicated in cell–cell adhesion and are thought to prevent the initial detachment of tumor cells from the primary site. Recently, Stupack and colleagues demonstrated that cellular adhesion to non-preferred substrates could induce caspase-8-dependent apoptosis [147]. In their neuroblastoma model, caspase-8 selectively inhibited metastasis without blocking tumor formation. CD44, the receptor for hyaluronic acid and osteopontin, has complex roles in cellular adhesion. Depending upon which of the many splice variants is studied, expression can modulate adhesion, cellular homing, metastasis and cellular growth. Gelsolin, which belongs to a superfamily of calcium-dependent actin-interacting proteins regulates cellular motility, but also contributes to regulation of cell division, invasion and adhesion.
Rho-guanine nucleotide dissociation inhibitor (RhoGDI2, also known as RhoGDIβ, LyGDI, GDID4) was first identified as a metastasis suppressor in bladder cancer [148, 149]. RhoGDIs sequester Rho proteins in an inactive state and transport them to effector sites. Two targets of RhoGDI2 are currently known, endothelin1 (ET1) and neuromedin U (NMU). Both ET1 and NMU promote metastasis; so, RhoGDI2 is suspected to regulate metastasis promoters [150, 151]. Interestingly, ET1 has been shown by Guise and colleagues to regulate metastasis in bone [152, 153], the most frequent site of breast cancer metastasis.
Ribonucleotide reductase M1 (RRM1) converts ribonucleotides to deoxyribonucleotides required for DNA synthesis, but its mechanism of action in controlling metastasis is still unknown. Src-suppressed C Kinase substrate (SSeCKS, pronounced essex) is the rodent ortholog of human A-kinase anchoring protein 12 (AKAP12)/Gravin. SSeCKS is a major substrate for protein kinase C and also functions as a scaffolding protein for protein kinases A and C. Recent evidence indicates that re-expression of SSeCKS inhibits expression of VEGF, suggesting a key role in regulating angiogenesis [154].
Perspective
Currently, the greatest challenges facing clinical cancer medicine are (1) to be able to detect cancer at the earliest possible stage and, (2) managing cancer once it has spread beyond the primary tumor. Early detection of tumors is extremely desirable since the tumor burden is relatively small, which not only decreases the statistical likelihood of metastatic cells developing within the primary tumor mass, but also means that fewer cells are likely to have escaped from the primary tumor. Both parameters correspond to a decreased likelihood of metastasis, in addition to a decreased likelihood of metastatic variants within a heterogeneous tumor. Early detection also decreases the chances of outgrowth of drug-resistant populations. The diagnostic challenge is largely a matter of limit of detection. Currently, even with the most advanced imaging techniques, small tumors escape identification in random screens. Although still in the developmental stage and as a possible future complement to imaging, it is now possible to detect minimal disease early through analysis of bone marrow aspirates and serum samples for genetic markers of tumor progression [155, 156]. Early detection is desirable for all patients, but particularly those considered to be at “increased risk.” Also, early detection of disseminated cells before they escape dormancy should reduce the unnecessary adjuvant therapy for patients with exclusively localized tumors. For patients with micro-metastases, the considerations are more complex. The most convincing evidence that maintenance of dormancy may be clinically achievable was demonstrated in mice treated with medroxyprogesterone; these mice showed elevated Nm23 expression in metastases and a significant decrease in metastatic burden compared to controls [137]. Although these results are too preliminary to warrant clinical implementation, they nonetheless highlight potential clinical utility.
Adjuvant and neo-adjuvant chemotherapy are mainly aimed at metastases, if any. Cells that escape the initial adjuvant treatment can recur as metastases. Too often, such cells have further acquired a resistant phenotype or characteristics of cancer stem cells are manifest. Importantly, a recent study demonstrated that administration of the plant-derived sesquiterpene lactone—parthenolide to mice (using a murine osteosarcoma cell line LM8) led to lung metastasis suppression. However, in this model, subcutaneous injection of LM8 cells followed by treatment with parthenolide did not suppress tumorigenicity [157]. Conceptually, the drug mimics the activities of metastasis suppressors. While gene replacement approaches may ultimately be possible, the plausibility of using targeted molecules to achieve the same end point is more immediately feasible. In addition, granulocyte macrophage-colony stimulating factor (GM-CSF) is important for osteolytic breast cancer metastasis. Inhibition of GM-CSF suppresses bone metastasis without affecting primary tumor growth [158]. Although results with parthenolide and GM-CSF are preliminary, could these results point to potentially identifying “metastasis suppressor drugs?” Mechanistic studies with another metastasis suppressor RhoGDI2 uncovered NMU as a target and a novel metastasis promoter [151]. Studies of mechanisms underlying metastasis suppression therefore not only show the potential to identify novel “drugable targets” but also novel mediators in the metastatic process.
Acknowledgments
KSV is supported by a post-doctoral fellowship (PDF1122006) from the Susan G. Komen For The Cure. Work from the Welch laboratory has been generously supported by US Public Health Service grants CA62168, CA87728, CA89019, U.S. Army Medical Research and Materiel Command grants DAMD-17-01-0358, DAMD-17-02-1-0541, and DAMD17-03-01-0584 and the National Foundation for Cancer Research—Center for Metastasis Research. We wish to thank members of the Welch lab for their critical review of this manuscript and excellent suggestions.
Abbreviations
- AKT
v-akt murine thymoma viral oncogene homolog 1
- AP2
Activating protein 2
- ARID4A
AT rich interactive domain 4A
- CK2
Casein kinase 2
- CRSP3
Co-factor required for Sp1 activity
- CXCR4
Chemokine (C-X-C motif) receptor 4
- EGFR
Epidermal growth factor receptor
- ET1
Endothelin 1
- GM-CSF
Granulocyte macrophage-colony stimulating factor
- GPR54
G-protein coupled receptor 54
- GSK1
Glycogen synthase kinase 1
- GTPase
Guanine tri-phosphatase
- HER2
Human epidermal growth factor receptor 2
- HLA-DR
Human leucocyte antigen-DR
- IL8
Interleukin 8
- JNK
c-Jun NH2 terminal protein kinase
- KSR
Kinase suppressor of Ras
- MAPK
Mitogen activated protein kinase
- MHC1
Major histocompatibility complex class 1
- MLK3
Mixed lineage kinase 3
- MPA
Medroxyprogesterone acetate
- HDAC
Histone deacetylase
- NF-κB
Nuclear factor-kappa B
- NMU
Neuromedin U
- PI3K
Phosphatidyl inositol 3-kinase
- PtdIns(4,5)P2
Phosphatidyl inositol (4,5) bisphosphate
- PMA
Phorbol myristate acetate
- PTEN
Phosphatase and tensin homolog deleted on chromosome 10
- RBBP1
Retinoblastoma binding protein 1
- RhoGAP
Rho GTPase-activating protein
- SDF-1
Stromal derived factor-1
- SUDS3
Suppressor of defective silencing 3
- TGF-β
Transforming growth factor-β
- TXNIP
Thioredoxin interacting protein
- VDUP1
Vitamin D upregulated protein 1
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor 1
Contributor Information
Kedar. S. Vaidya, Department of Pathology, University of Alabama at Birmingham, Birmingham, AL 35294, USA
Danny. R. Welch, Departments of Pathology, Cell Biology, Pharmacology and Toxicology, Comprehensive Cancer Center, National Foundation for Cancer Research—Center for Metastasis Research, University of Alabama at Birmingham, Birmingham, AL 35294, USA
References
- 1.Page DL, Kidd TE, Jr, Dupont WD, Simpson JF, Rogers LW. Lobular neoplasia of the breast: higher risk for subsequent invasive cancer predicted by more extensive disease. Human Pathol. 1991;22(12):1232–9. doi: 10.1016/0046-8177(91)90105-x. [DOI] [PubMed] [Google Scholar]
- 2.Rosen PP, Kosloff C, Lieberman PH, Adair F, Braun DW., Jr Lobular carcinoma in situ of the breast. Detailed analysis of 99 patients with average follow-up of 24 years. Am J Surg Pathol. 1978;2(3):225–51. doi: 10.1097/00000478-197809000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 4.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–35. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Welch DR. Do we need to redefine a cancer metastasis and staging definitions? Breast Dis. 2007;26(1):3–12. doi: 10.3233/bd-2007-26102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hynes RO. Metastatic potential: generic predisposition of the primary tumor or rare, metastatic variants—or both? Cell. 2003;113(7):821–3. doi: 10.1016/s0092-8674(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 8.Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35(3):512–6. [PubMed] [Google Scholar]
- 9.Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst. 1970;45(4):773–82. [PubMed] [Google Scholar]
- 10.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–3. [PubMed] [Google Scholar]
- 13.Fidler IJ, Gersten DM, Riggs CW. Relationship of host immune status to tumor cell arrest, distribution and survival in experimental metastasis. Cancer. 1977;40(1):46–55. doi: 10.1002/1097-0142(197707)40:1<46::aid-cncr2820400110>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 14.Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980;40(7):2281–7. [PubMed] [Google Scholar]
- 15.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 16.Fidler IJ, Radinsky R. Search for genes that suppress cancer metastasis. J Natl Cancer Inst. 1996;88(23):1700–3. doi: 10.1093/jnci/88.23.1700. [DOI] [PubMed] [Google Scholar]
- 17.Rinker-Schaeffer CW, O’Keefe JP, Welch DR, Theodorescu D. Metastasis suppressor proteins: discovery, molecular mechanisms and clinical application. Clin Cancer Res. 2006;12(13):3382–9. doi: 10.1158/1078-0432.CCR-06-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanai J, Mammoto T, Seth P, Mori K, Karumanchi SA, Barasch J, et al. Lipocalin 2 diminishes invasiveness and metastasis of Ras-transformed cells. J Biol Chem. 2005;280(14):13641–7. doi: 10.1074/jbc.M413047200. [DOI] [PubMed] [Google Scholar]
- 19.Venkatesha S, Hanai J, Seth P, Karumanchi SA, Sukhatme VP. Lipocalin 2 antagonizes the proangiogenic action of ras in transformed cells. Mol Cancer Res. 2006;4(11):821–9. doi: 10.1158/1541-7786.MCR-06-0110. [DOI] [PubMed] [Google Scholar]
- 20.Urtreger AJ, Werbajh SE, Verrecchia F, Mauviel A, Puricelli LI, Kornblihtt AR, et al. Fibronectin is distinctly downregulated in murine mammary adenocarcinoma cells with high metastatic potential. Oncol Rep. 2006;16(6):1403–10. [PubMed] [Google Scholar]
- 21.Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, Shaw LM. Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol Cell Biol. 2006;26(24):9338–51. doi: 10.1128/MCB.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips KK, Welch DR, Miele ME, Lee J-H, Wei LL, Weissman BE. Suppression of MDA-MB-435 breast carcinoma cell metastasis following the introduction of human chromosome 11. Cancer Res. 1996;56(10):1222–6. [PubMed] [Google Scholar]
- 23.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60(11):2764–9. [PubMed] [Google Scholar]
- 24.Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin Exp Metastasis. 2001;18(6):519–25. doi: 10.1023/a:1011819621859. [DOI] [PubMed] [Google Scholar]
- 25.Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, et al. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273(2):229–39. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Lin QD, DI W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006;16(2):522–31. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 27.Kelly LM, Buggy Y, Hill A, O’Donovan N, Duggan C, McDermott EW, et al. Expression of the breast cancer metastasis suppressor gene, BRMS1, in human breast carcinoma: lack of correlation with metastasis to axillary lymph nodes. Tumor Biol. 2005;26(4):213–6. doi: 10.1159/000086955. [DOI] [PubMed] [Google Scholar]
- 28.Lombardi G, Di CC, Capodanno A, Iorio MC, Aretini P, Isola P, et al. High level of messenger RNA for BRMS1 in primary breast carcinomas is associated with poor prognosis. Int J Cancer. 2006;120(6):1169–78. doi: 10.1002/ijc.22379. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Yamashita H, Toyama T, Yamamoto Y, Kawasoe T, Iwase H. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin Cancer Res. 2006;12(21):6410–4. doi: 10.1158/1078-0432.CCR-06-1347. [DOI] [PubMed] [Google Scholar]
- 30.Hicks DG, Yoder BJ, Short S, Tarr S, Prescott N, Crowe JP, et al. Loss of BRMS1 protein expression predicts reduced disease-free survival in hormone receptor negative and HER2 positive subsets of breast cancer. Clin Cancer Res. 2006;12(22):6702–8. doi: 10.1158/1078-0432.CCR-06-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hurst DR, Mehta A, Moore BP, Phadke PA, Meehan WJ, Accavitti MA, et al. Breast cancer metastasis suppressor 1 (BRMS1) is stabilized by the Hsp90 chaperone. Biochem Biophys Res Commun. 2006;348(4):1429–35. doi: 10.1016/j.bbrc.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279(2):1562–9. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 33.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 2005;65(9):3586–95. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26(23):8683–96. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samant RS, Clark DW, Fillmore RA, Cicek M, Metge BJ, Chandramouli KH, et al. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-Kappa B activation. Mol Cancer. 2007;6(1):6–14. doi: 10.1186/1476-4598-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature. 1999;401(6748):82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 37.DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, et al. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65(3):713–7. [PubMed] [Google Scholar]
- 38.Samant RS, Seraj MJ, Saunders MM, Sakamaki T, Shevde LA, Harms JF, et al. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metastasis. 2001;18(8):683–93. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 39.Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268(5212):884–6. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- 40.Ichikawa T, Ichikawa Y, Dong J, Hawkins AL, Griffin CA, Isaacs WB, et al. Localization of metastasis suppressor gene(s) for prostatic cancer to the short arm of human chromosome 11. Cancer Res. 1992;52(12):3486–90. [PubMed] [Google Scholar]
- 41.Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol. 2005;6(10):801–11. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 42.Sugiura T, Berditchevski F. Function of alpha3beta1-tetraspanin protein complexes in tumor cell invasion. Evidence for the role of the complexes in production of matrix metalloproteinase 2 (MMP-2) J Cell Biol. 1999;146(6):1375–89. doi: 10.1083/jcb.146.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longo N, Yanez-Mo M, Mittelbrunn M, de la RG, Munoz ML, Sanchez-Madrid F, et al. Regulatory role of tetraspanin CD9 in tumor-endothelial cell interaction during transendothelial invasion of melanoma cells. Blood. 2001;98(13):3717–26. doi: 10.1182/blood.v98.13.3717. [DOI] [PubMed] [Google Scholar]
- 44.Zhou B, Liu L, Reddivari M, Zhang XA. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004;64(20):7455–63. doi: 10.1158/0008-5472.CAN-04-1574. [DOI] [PubMed] [Google Scholar]
- 45.Sridhar SC, Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25(16):2367–78. doi: 10.1038/sj.onc.1209269. [DOI] [PubMed] [Google Scholar]
- 46.Takaoka A, Hinoda Y, Sato S, Itoh F, Adachi M, Hareyama M, et al. Reduced invasive and metastatic potentials of KAI1-transfected melanoma cells. Jpn J Cancer Res. 1998;89(4):397–404. doi: 10.1111/j.1349-7006.1998.tb00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang XH, Welch DR, Phillips KK, Weissman BE, Wei LL. KAI1, a putative marker for metastatic potential in human breast cancer. Cancer Lett. 1997;119(2):149–55. doi: 10.1016/s0304-3835(97)00273-5. [DOI] [PubMed] [Google Scholar]
- 48.Phillips KK, White AE, Hicks DJ, Welch DR, Barrett JC, Wei LL, et al. Correlation between reduction of metastasis in the MDA-MB-435 model system and increased expression of the Kai-1 protein. Mol Carcinog. 1998;21(3):111–20. [PubMed] [Google Scholar]
- 49.Yang X, Wei L, Tang C, Slack R, Montgomery E, Lippman M. KAI1 protein is down-regulated during the progression of human breast cancer. Clin Cancer Res. 2000;6(9):3424–9. [PubMed] [Google Scholar]
- 50.Lombardi DP, Geradts J, Foley JF, Chiao C, Lamb PW, Barrett JC. Loss of KAI1 expression in the progression of colorectal cancer. Cancer Res. 1999;59(22):5724–31. [PubMed] [Google Scholar]
- 51.Liu FS, Dong JT, Chen JT, Hsieh YT, Ho ES, Hung MJ. Frequent down-regulation and lack of mutation of the KAI1 metastasis suppressor gene in epithelial ovarian carcinoma. Gynecol Oncol. 2000;78(1):10–5. doi: 10.1006/gyno.2000.5801. [DOI] [PubMed] [Google Scholar]
- 52.Liu FS, Chen JT, Dong JT, Hsieh YT, Lin AJ, Ho ESC, et al. KAI1 metastasis suppressor gene is frequently down-regulated in cervical carcinoma. Am J Pathol. 2001;159(5):1629–34. doi: 10.1016/s0002-9440(10)63009-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu FS, Dong JT, Chen JT, Hsieh YT, Ho ES, Hung MJ, et al. KAI1 metastasis suppressor protein is down-regulated during the progression of human endometrial cancer. Clin Cancer Res. 2003;9(4):1393–8. [PubMed] [Google Scholar]
- 54.Farhadieh RD, Smee R, Ow K, Yang JL, Russell PJ, Crouch R, et al. Down-regulation of KAI1/CD82 protein expression in oral cancer correlates with reduced disease free survival and overall patient survival. Cancer Lett. 2004;213(1):91–8. doi: 10.1016/j.canlet.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Goncharuk VN, del-Rosario A, Kren L, Anwar S, Sheehan CE, Carlson JA, et al. Co-downregulation of PTEN, KAI-1, and nm23-H1 tumor/metastasis suppressor proteins in non-small cell lung cancer. Ann Diagn Pathol. 2004;8(1):6–16. doi: 10.1016/j.anndiagpath.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Dong JT, Suzuki H, Pin SS, Bova GS, Schalken JA, Isaacs WB, et al. Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Res. 1996;56(19):4387–90. [PubMed] [Google Scholar]
- 57.Mashimo T, Bandyopadhyay S, Goodarzi G, Watabe M, Pai SK, Gross SC, et al. Activation of the tumor metastasis suppressor gene, KAI1, by etoposide is mediated by p53 and c-Jun genes. Biochem Biophys Res Commun. 2000;274(2):370–6. doi: 10.1006/bbrc.2000.3139. [DOI] [PubMed] [Google Scholar]
- 58.Shibagaki N, Hanada K, Yamashita H, Shimada S, Hamada H. Overexpression of CD82 on human T cells enhances LFA-1/ICAM-1-mediated cell–cell adhesion: functional association between CD82 and LFA-1 in T cell activation. Eur J Immunol. 1999;29(12):4081–91. doi: 10.1002/(SICI)1521-4141(199912)29:12<4081::AID-IMMU4081>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 59.Mannion BA, Berditchevski F, Kraeft SK, Chen LB, Hemler ME. Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63, and CD53 specifically associated with integrin alpha 4 beta 1 (CD49d/CD29) J Immunol. 1996;157(5):2039–2047. [PubMed] [Google Scholar]
- 60.Horvath G, Serru V, Clay D, Billard M, Boucheix C, Rubinstein E. CD19 is linked to the integrin-associated tetraspans CD9, CD81, and CD82. J Biol Chem. 1998;273(46):30537–43. doi: 10.1074/jbc.273.46.30537. [DOI] [PubMed] [Google Scholar]
- 61.Lee JH, Park SR, Chay KO, Seo YW, Kook H, Ahn KY, et al. KAI1 COOH-terminal interacting tetraspanin (KITENIN), a member of the tetraspanin family, interacts with KAI1, a tumor metastasis suppressor, and enhances metastasis of cancer. Cancer Res. 2004;64(12):4235–43. doi: 10.1158/0008-5472.CAN-04-0275. [DOI] [PubMed] [Google Scholar]
- 62.Delaguillaumie A, Harriague J, Kohanna S, Bismuth G, Rubinstein E, Seigneuret M, et al. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J Cell Sci. 2004;117(Pt 22):5269–82. doi: 10.1242/jcs.01380. [DOI] [PubMed] [Google Scholar]
- 63.Angelisova P, Hilgert I, Horejsi V. Association of four antigens of the tetraspans family (CD37, CD53, TAPA-1, and R2/C33) with MHC class II glycoproteins. Immunogenetics. 1994;39(4):249–56. doi: 10.1007/BF00188787. [DOI] [PubMed] [Google Scholar]
- 64.Szollosi J, Horejsi V, Bene L, Angelisova P, Damjanovich S. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J Immunol. 1996;157(7):2939–46. [PubMed] [Google Scholar]
- 65.Zhang XA, Lane WS, Charrin S, Rubinstein E, Liu L. EWI2/PGRL associates with the metastasis suppressor KAI1/CD82 and inhibits the migration of prostate cancer cells. Cancer Res. 2003;63(10):2665–74. [PubMed] [Google Scholar]
- 66.Delaguillaumie A, Lagaudriere-Gesbert C, Popoff MR, Conjeaud H. Rho GTPases link cytoskeletal rearrangements and activation processes induced via the tetraspanin CD82 in T lymphocytes. J Cell Sci. 2002;115(Pt 2):433–43. doi: 10.1242/jcs.115.2.433. [DOI] [PubMed] [Google Scholar]
- 67.Odintsova E, Sugiura T, Berditchevski F. Attenuation of EGF receptor signaling by a metastasis suppressor, the tetraspanin CD82/KAI-1. Curr Biol. 2000;10(16):1009–12. doi: 10.1016/s0960-9822(00)00652-7. [DOI] [PubMed] [Google Scholar]
- 68.Odintsova E, Voortman J, Gilbert E, Berditchevski F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. J Cell Sci. 2003;116(Pt 22):4557–66. doi: 10.1242/jcs.00793. [DOI] [PubMed] [Google Scholar]
- 69.Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med. 2006;12(8):933–8. doi: 10.1038/nm1444. [DOI] [PubMed] [Google Scholar]
- 70.Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261(5125):1182–4. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- 71.Chaudhuri A, Nielsen S, Elkjaer ML, Zbrzezna V, Fang F, Pogo AO. Detection of Duffy antigen in the plasma membranes and caveolae of vascular endothelial and epithelial cells of nonerythroid organs. Blood. 1997;89(2):701–12. [PubMed] [Google Scholar]
- 72.Marreiros A, Dudgeon K, Dao V, Grimm MO, Czolij R, Crossley M, et al. KAI1 promoter activity is dependent on p53, junB and AP2: evidence for a possible mechanism underlying loss of KAI1 expression in cancer cells. Oncogene. 2005;24(4):637–49. doi: 10.1038/sj.onc.1208216. [DOI] [PubMed] [Google Scholar]
- 73.Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C. Transcriptional regulation of a metastasis suppressor gene by Tip60 and β-catenin complexes. Nature. 2005;434(7035):921–6. doi: 10.1038/nature03452. [DOI] [PubMed] [Google Scholar]
- 74.Jackson P, Ow K, Yardley G, Delprado W, Quinn DI, Yang JL, et al. Downregulation of KAI1 mRNA in localised prostate cancer and its bony metastases does not correlate with p53 overexpression. Prostate Cancer Prostatic Dis. 2003;6(2):174–81. doi: 10.1038/sj.pcan.4500634. [DOI] [PubMed] [Google Scholar]
- 75.Akita H, Iizuka A, Hashimoto Y, Kohri K, Ikeda K, Nakanishi M. Induction of KAI-1 expression in metastatic cancer cells by phorbol esters. Cancer Lett. 2000;153(1–2):79–83. doi: 10.1016/s0304-3835(00)00352-9. [DOI] [PubMed] [Google Scholar]
- 76.Sekita N, Suzuki H, Ichikawa T, Kito H, Akakura K, Igarashi T, et al. Epigenetic regulation of the KAI1 metastasis suppressor gene in human prostate cancer cell lines. Jpn J Cancer Res. 2001;92(9):947–51. doi: 10.1111/j.1349-7006.2001.tb01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Welch DR, Chen P, Miele ME, McGary CT, Bower JM, Weissman BE, et al. Microcell-mediated transfer of chromosome 6 into metastatic human C8161 melanoma cells suppresses metastasis but does not inhibit tumorigenicity. Oncogene. 1994;9(1):255–62. [PubMed] [Google Scholar]
- 78.Lee J-H, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996;88(23):1731–7. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 79.Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub CA, Freedman LP, et al. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003;63(2):432–40. [PubMed] [Google Scholar]
- 80.Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M. Transcriptional regulation of KiSS-1 gene regulation in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene. 2007;26(12):1739–47. doi: 10.1038/sj.onc.1209963. [DOI] [PubMed] [Google Scholar]
- 81.Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M. Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene. 2006 doi: 10.1038/sj.onc.1209963. [DOI] [PubMed] [Google Scholar]
- 82.Lee J-H, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57(12):2384–7. [PubMed] [Google Scholar]
- 83.Jiang Y, Berk M, Singh LS, Tan HY, Yin LH, Powell CT, et al. KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin Exp Metastasis. 2005;22(5):369–76. doi: 10.1007/s10585-005-8186-4. [DOI] [PubMed] [Google Scholar]
- 84.Nash KT, Welch DR. The KISS1 metastasis suppressor: mechanistic insights and clinical utility. Front Biosci. 2006;11(1):647–59. doi: 10.2741/1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Horikoshi Y, Matsumoto H, Takatsu Y, Ohtaki T, Kitada C, Usuki S, et al. Dramatic elevation of plasma metastin concentrations in human pregnancy: metastin as a novel placentaderived hormone in humans. J Clin Endocrinol Metab. 2003;88(2):914–9. doi: 10.1210/jc.2002-021235. [DOI] [PubMed] [Google Scholar]
- 86.Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276(37):34631–6. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- 87.Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, et al. Metastasis suppressor gene KiSS1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411(6837):613–7. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 88.Nash KT, Phadke PA, Navenot J-M, Hurst DR, Accavitti-Loper MA, Sztul E, et al. KISS1 metastasis suppressor secretion, multiple organ metastasis suppression, and maintenance of tumor dormancy. J Natl Cancer Inst. 2007;99(4):309–21. doi: 10.1093/jnci/djk053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Navenot JM, Wang Z, Chopin M, Fujii N, Peiper SC. Kisspeptin-10-induced signaling of GPR54 negatively regulates chemotactic responses mediated by CXCR4: a potential mechanism for the metastasis suppressor activity of kisspeptins. Cancer Res. 2005;65(22):10450–6. doi: 10.1158/0008-5472.CAN-05-1757. [DOI] [PubMed] [Google Scholar]
- 90.Goldberg SF, Harms JF, Quon K, Welch DR. Metastasis-suppressed C8161 melanoma cells arrest in lung but fail to proliferate. Clin Exp Metastasis. 1999;17(7):601–7. doi: 10.1023/a:1006718800891. [DOI] [PubMed] [Google Scholar]
- 91.Mead EJ, Maguire JJ, Kuc RE, Davenport AP. Kisspeptins are novel potent vasoconstrictors in humans, with a discrete localization of their receptor, G protein-coupled receptor 54, to atherosclerosis-prone vessels. Endocrinology. 2007;148(1):140–7. doi: 10.1210/en.2006-0818. [DOI] [PubMed] [Google Scholar]
- 92.Yan CH, Wang H, Boyd DD. KiSS-1 represses 92-kDa type IV collagenase expression by down-regulating NFκB binding to the promoter as a consequence of IκBα-induced block of p65/p50 nuclear translocation. J Biol Chem. 2001;276(2):1164–72. doi: 10.1074/jbc.M008681200. [DOI] [PubMed] [Google Scholar]
- 93.Goldberg SF, Lee JH, Welch DR. KiSS-1: a suppressor of metastasis. Jikken Igaku (Experimental Medicine—Japanese) 1998;16(16):37–46. [Google Scholar]
- 94.Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Res. 1999;59(21):5483–7. [PubMed] [Google Scholar]
- 95.Su GH, Song JJ, Repasky EA, Schutte M, Kern SE. Mutation rate of MAP2K4/MKK4 in breast carcinoma. Human Mutat. 2002;19(1):81–5. doi: 10.1002/humu.9002. [DOI] [PubMed] [Google Scholar]
- 96.Yamada SD, Hickson JA, Hrobowski Y, Vander Griend DJ, Benson D, Montag A, et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62(22):6717–23. [PubMed] [Google Scholar]
- 97.Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 Kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66(4):2264–70. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 98.Stark AM, Tongers K, Maass N, Mehdorn HM, Held-Feindt J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J Cancer Res Clin Oncol. 2004;131(3):191–8. doi: 10.1007/s00432-004-0629-9. [DOI] [PubMed] [Google Scholar]
- 99.Couvelard A, Hu J, Steers G, O’Toole D, Sauvanet A, Belghiti J, et al. Identification of potential therapeutic targets by gene-expression profiling in pancreatic endocrine tumors. Gastroenterology. 2006;131(5):1597–610. doi: 10.1053/j.gastro.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 100.Wang L, Pan Y, LeDai J. Evidence of MKK4 pro-oncogenic activity in breast and pancreatic tumors. Oncogene. 2004;23(35):5978–85. doi: 10.1038/sj.onc.1207802. [DOI] [PubMed] [Google Scholar]
- 101.Robinson VL, Hickson JA, Vander Griend DJ, Dubauskas Z, Rinker-Schaeffer CW. MKK4 and metastasis suppression: a marriage of signal transduction and metastasis research. Clin Exp Metastasis. 2003;20(1):25–30. doi: 10.1023/a:1022586318678. [DOI] [PubMed] [Google Scholar]
- 102.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)-from inflammation to development. Curr Opin Cell Biol. 1998;10(2):205–19. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 103.Shen YH, Godlewski J, Zhu J, Sathyanarayana P, Leaner V, Birrer MJ, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem. 2003;278(29):26715–21. doi: 10.1074/jbc.M303264200. [DOI] [PubMed] [Google Scholar]
- 104.Vander Griend DJ, Kocherginsky M, Hickson JA, Stadler WM, Lin AN, Rinker-Schaeffer CW. Suppression of metastatic colonization by the context-dependent activation of the c-jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65(23):10984–91. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- 105.Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, et al. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988;80(3):200–4. doi: 10.1093/jnci/80.3.200. [DOI] [PubMed] [Google Scholar]
- 106.Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO. The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembranes. 2000;32(3):247–58. doi: 10.1023/a:1005584929050. [DOI] [PubMed] [Google Scholar]
- 107.Hartsough MT, Steeg PS. Nm23/nucleoside diphosphate kinase in human cancers. J Bioenerg Biomembranes. 2000;32(3):301–8. doi: 10.1023/a:1005597231776. [DOI] [PubMed] [Google Scholar]
- 108.Leone A, Seeger RC, Hong CM, Hu YY, Arboleda J, Brodeur GM, et al. Evidence for nm23 RNA overexpression, DNA amplification and mutation in aggressive childhood neuroblastomas. Oncogene. 1993;8(4):855–65. [PubMed] [Google Scholar]
- 109.Leone A, Flatow U, King CR, Sandeen MA, Margulies IMK, Liotta LA, et al. Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell. 1991;65(1):25–35. doi: 10.1016/0092-8674(91)90404-m. [DOI] [PubMed] [Google Scholar]
- 110.Suzuki E, Ota T, Tsukuda K, Okita A, Matsuoka K, Murakami M, et al. nm23-H1 reduces in vitro cell migration and the liver metastatic potential of colon cancer cells by regulating myosin light chain phosphorylation. Int J Cancer. 2004;108(2):207–11. doi: 10.1002/ijc.11546. [DOI] [PubMed] [Google Scholar]
- 111.Kantor JD, McCormick B, Steeg PS, Zetter BR. Inhibition of cell motility after nm23 transfection of human and murine tumor cells. Cancer Res. 1993;53(9):1971–3. [PubMed] [Google Scholar]
- 112.Scambia G, Ferrandina G, Marone M, Benedetti PP, Giannitelli C, Piantelli M, et al. nm23 in ovarian cancer: correlation with clinical outcome and other clinicopathologic and biochemical prognostic parameters. J Clin Oncol. 1996;14(2):334–42. doi: 10.1200/JCO.1996.14.2.334. [DOI] [PubMed] [Google Scholar]
- 113.Yamashita H, Kobayashi S, Iwase H, Itoh Y, Kuzishima T, Iwata H, et al. Analysis of oncogenes and tumor suppressor genes in human breast cancer. Jpn J Cancer Res. 1993;84(8):871–8. doi: 10.1111/j.1349-7006.1993.tb02060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tommasi S, Fedele V, Crapolicchio A, Bellizzi A, Paradiso A, Reshkin SJ. ErbB2 and the antimetastatic nm23/NDP kinase in regulating serum induced breast cancer invasion. Int J Mol Med. 2003;12(1):131–4. [PubMed] [Google Scholar]
- 115.Nakopoulou LL, Tsitsimelis D, Lazaris AC, Tzonou A, Gakiopoulou H, Dicoglou CC, et al. Nm-23, c-erbB-2, and progesterone receptor expression in invasive breast cancer: correlation with clinicopathologic parameters. Cancer Detect Prev. 1999;23(4):297–308. doi: 10.1046/j.1525-1500.1999.99035.x. [DOI] [PubMed] [Google Scholar]
- 116.Ohba K, Miyata Y, Koga S, Kanda S, Kanetake H. Expression of nm23-H1 gene product in sarcomatous cancer cells of renal cell carcinoma: correlation with tumor stage and expression of matrix metalloproteinase-2, matrix metalloproteinase-9, sialyl Lewis X, and c-erbB-2. Urology. 2005;65(5):1029–34. doi: 10.1016/j.urology.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 117.Kuppers DA, Lan K, Knight JS, Robertson ES. Regulation of matrix metalloproteinase 9 expression by Epstein-Barr virus nuclear antigen 3C and the suppressor of metastasis Nm23-H1. J Virol. 2005;79(15):9714–24. doi: 10.1128/JVI.79.15.9714-9724.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu JH, Tseng YH, Kantor JD, Rhodes CJ, Zetter BR, Moyers JS, et al. Interaction of the Ras-related protein associated with diabetes Rad and the putative tumor metastasis suppressor NM23 provides a novel mechanism of GTPase regulation. Proc Natl Acad Sci. 1999;96(26):14911–8. doi: 10.1073/pnas.96.26.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tseng YH, Vicent D, Zhu JH, Niu YL, Adeyinka A, Moyers JS, et al. Regulation of growth and tumorigenicity of breast cancer cells by the low molecular weight GTPase Rad and Nm23. Cancer Res. 2001;61(5):2071–9. [PubMed] [Google Scholar]
- 120.Engel M, Seifert M, Theisinger B, Seyfert U, Welter C. Glyceraldehyde-3-phosphate dehydrogenase and Nm23-H1/Nucleoside diphosphate kinase A—Two old enzymes combine for the novel Nm23 protein phosphotransferase function. J Biol Chem. 1998;273(32):20058–65. doi: 10.1074/jbc.273.32.20058. [DOI] [PubMed] [Google Scholar]
- 121.Pinon VP, Millot G, Munier A, Vassy J, Linares-Cruz G, Capeau J, et al. Cytoskeletal association of the A and B nucleoside diphosphate kinases of interphasic but not mitotic human carcinoma cell lines: specific nuclear localization of the B subunit. Exp Cell Res. 1999;246(2):355–67. doi: 10.1006/excr.1998.4318. [DOI] [PubMed] [Google Scholar]
- 122.Kimura N, Shimada N. Evidence for complex formation between GTP binding protein(Gs) and membrane-associated nucleoside diphosphate kinase. Biochem Biophys Res Commun. 1990;168(1):99–106. doi: 10.1016/0006-291x(90)91680-q. [DOI] [PubMed] [Google Scholar]
- 123.Biondi RM, Engel M, Sauane M, Welter C, Issinger OG, Jimenez de AL, et al. Inhibition of nucleoside diphosphate kinase activity by in vitro phosphorylation by protein kinase CK2. Differential phosphorylation of NDP kinases in HeLa cells in culture. FEBS Lett. 1996;399(1–2):183–7. doi: 10.1016/s0014-5793(96)01299-9. [DOI] [PubMed] [Google Scholar]
- 124.Salerno M, Ouatas T, Palmieri D, Steeg PS. Inhibition of signal transduction by the nm23 metastasis suppressor: possible mechanisms. Clin Exp Metastasis. 2003;20(1):3–10. doi: 10.1023/a:1022578000022. [DOI] [PubMed] [Google Scholar]
- 125.Biggs J, Hersperger E, Steeg PS, Liotta LA, Shearn A. A Drosophila gene that is homologous to a mammalian gene associated with tumor metastasis codes for a nucleoside diphosphate kinase. Cell. 1990;63(5):933–40. doi: 10.1016/0092-8674(90)90496-2. [DOI] [PubMed] [Google Scholar]
- 126.Freije JMP, Blay P, MacDonald NJ, Manrow RE, Steeg PS. Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J Biol Chem. 1997;272(9):5525–32. doi: 10.1074/jbc.272.9.5525. [DOI] [PubMed] [Google Scholar]
- 127.Ma DQ, McCorkle JR, Kaetzel DM. The metastasis suppressor NM23-H1 possesses 3′–5′ exonuclease activity. J Biol Chem. 2004;279(17):18073–84. doi: 10.1074/jbc.M400185200. [DOI] [PubMed] [Google Scholar]
- 128.MacDonald NJ, de la Rosa A, Benedict MA, Freije JM, Krutsch H, Steeg PS. A serine phosphorylation of Nm23, and not its nucleoside diphosphate kinase activity, correlates with suppression of tumor metastatic potential. J Biol Chem. 1993;268(34):25780–9. [PubMed] [Google Scholar]
- 129.Steeg PS, de la Rosa A, Flatow U, MacDonald NJ, Benedict M, Leone A. Nm23 and breast cancer metastasis. Breast Cancer Res Treat. 1993;25(2):175–87. doi: 10.1007/BF00662142. [DOI] [PubMed] [Google Scholar]
- 130.Sacks DB. The role of scaffold proteins in MEK/ERK signalling. Biochem Soc Trans. 2006;34(Pt 5):833–6. doi: 10.1042/BST0340833. [DOI] [PubMed] [Google Scholar]
- 131.Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8(5):983–93. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 132.Hartsough MT, Morrison DK, Salerno M, Palmieri D, Ouatas T, Mair M, et al. Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of ras via a histidine protein kinase pathway. J Biol Chem. 2002;277(35):32389–99. doi: 10.1074/jbc.M203115200. [DOI] [PubMed] [Google Scholar]
- 133.Mink S, Hartig E, Jennewein P, Doppler W, Cato AC. A mammary cell-specific enhancer in mouse mammary tumor virus DNA is composed of multiple regulatory elements including binding sites for CTF/NFI and a novel transcription factor, mammary cell-activating factor. Mol Cell Biol. 1992;12(11):4906–18. doi: 10.1128/mcb.12.11.4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Welte T, Garimorth K, Philipp S, Jennewein P, Huck C, Cato AC, et al. Involvement of Ets-related proteins in hormone-independent mammary cell-specific gene expression. Eur J Biochem. 1994;223(3):997–1006. doi: 10.1111/j.1432-1033.1994.tb19078.x. [DOI] [PubMed] [Google Scholar]
- 135.Malewski T, Zwierzchowski L. Changes of tissue-specific transcription factors in the rabbit mammary gland during pregnancy and lactation. Tsitol Genet. 1997;31(4):58–69. [PubMed] [Google Scholar]
- 136.Ouatas T, Clare SE, Hartsough MT, De L, Steeg PS. MMTV-associated transcription factor binding sites increase nm23-H1 metastasis suppressor gene expression in human breast carcinoma cell lines. Clin Exp Metastasis. 2002;19(1):35–42. doi: 10.1023/a:1013897022827. [DOI] [PubMed] [Google Scholar]
- 137.Palmieri D, Halverson DO, Ouatas T, Horak CE, Salerno M, Johnson J, et al. Medroxyprogesterone acetate elevation of Nm23-H1 metastasis suppressor expression in hormone receptor-negative breast cancer. J Natl Cancer Inst. 2005;97(9):632–42. doi: 10.1093/jnci/dji111. [DOI] [PubMed] [Google Scholar]
- 138.Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, et al. Effects of Raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95(12):878–89. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 139.Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, et al. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11(20):7392–7. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 140.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401(6749):173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 141.Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005;24(21):3535–40. doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- 142.Trakul N, Menard RE, Schade GR, Qian ZH, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280(26):24931–40. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- 143.Chatterjee D, Bai Y, Wang Z, Beach S, Mott S, Roy R, et al. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004;279(17):17515–23. doi: 10.1074/jbc.M313816200. [DOI] [PubMed] [Google Scholar]
- 144.Molinaro RJ, Jha BK, Malathi K, Varambally S, Chinnaiyan AM, Silverman RH. Selection and cloning of poly(rC)-binding protein 2 and Raf kinase inhibitor protein RNA activators of 2′,5′-oligoadenylate synthetase from prostate cancer cells. Nucleic Acids Res. 2006;34(22):6684–95. doi: 10.1093/nar/gkl968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rusch L, Zhou A, Silverman RH. Caspase-dependent apoptosis by 2′,5′-oligoadenylate activation of RNase L is enhanced by IFN-beta. J Interferon Cytokine Res. 2000;20(12):1091–100. doi: 10.1089/107999000750053762. [DOI] [PubMed] [Google Scholar]
- 146.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369(9574):1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439(7072):95–9. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 148.Adra CN, Kobayashi H, Rowley JD, Lim B. Assignment of the human GDID4 gene, a GDP/GTP-exchange regulator, to chromosome 12p12.3. Genomics. 1994;24(1):188–90. doi: 10.1006/geno.1994.1601. [DOI] [PubMed] [Google Scholar]
- 149.Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, et al. RhoGD12 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002;62(22):6418–23. [PubMed] [Google Scholar]
- 150.Titus B, Frierson HF, Conaway M, Ching K, Guise T, Chirgwin J, et al. Endothelin axis is a target of the lung metastasis suppressor gene RhoGD12. Cancer Res. 2005;65(16):7320–7. doi: 10.1158/0008-5472.CAN-05-1403. [DOI] [PubMed] [Google Scholar]
- 151.Wu Y, McRoberts K, Berr SS, Frierson HF, Jr, Conaway M, Theodorescu D. Neuromedin U is regulated by the metastasis suppressor RhoGDI2 and is a novel promoter of tumor formation, lung metastasis and cancer cachexia. Oncogene. 2006;26(5):765–73. doi: 10.1038/sj.onc.1209835. [DOI] [PubMed] [Google Scholar]
- 152.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97(3):779–84. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 153.Mohammad KS, Guise TA. Mechanisms of osteoblastic metastases: role of endothelin-1. Clin Orthop Relat Res. 2003;415:S67–S74. doi: 10.1097/01.blo.0000093047.96273.4e. [DOI] [PubMed] [Google Scholar]
- 154.Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis-suppressing activity in MatLyLu prostate cancer cells correlates with vascular endothelial growth factor inhibition. Cancer Res. 2006;66(11):5599–607. doi: 10.1158/0008-5472.CAN-05-4123. [DOI] [PubMed] [Google Scholar]
- 155.Taback B, Giuliano AE, Lai R, Hansen N, Singer FR, Pantel K, et al. Epigenetic analysis of body fluids and tumor tissues: application of a comprehensive molecular assessment for early-stage breast cancer patients. Ann N Y Acad Sci. 2006:1075211–21. doi: 10.1196/annals.1368.029. [DOI] [PubMed] [Google Scholar]
- 156.Schwarzenbach H, Chun FK, Lange I, Carpenter S, Gottberg M, Erbersdobler A, et al. Detection of tumor-specific DNA in blood and bone marrow plasma from patients with prostate cancer. Int J Cancer. 2007;120(7):1457–63. doi: 10.1002/ijc.22470. [DOI] [PubMed] [Google Scholar]
- 157.Kishida Y, Yoshikawa H, Myoui A. Parthenolide, a natural inhibitor of nuclear factor-kappab, inhibits lung colonization of murine osteosarcoma cells. Clin Cancer Res. 2007;13(1):59–67. doi: 10.1158/1078-0432.CCR-06-1559. [DOI] [PubMed] [Google Scholar]
- 158.Park BK, Zhang HL, Zeng QH, Dai JL, Keller ET, Giordano T, et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13(1):62–9. doi: 10.1038/nm1519. [DOI] [PubMed] [Google Scholar]
- 159.Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148(4):779–90. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61(9):3819–25. [PubMed] [Google Scholar]
- 161.Gao AC, Lou W, Dong JT, Isaacs JT. CD44 is a metastasis suppressor gene for prostatic cancer located on human chromosome 11p13. Cancer Res. 1997;57(5):846–9. [PubMed] [Google Scholar]
- 162.Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;58(10):2196–9. [PubMed] [Google Scholar]
- 163.Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, et al. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003;63(8):1731–6. [PubMed] [Google Scholar]
- 164.Fujita H, Okada F, Hamada J, Hosokawa M, Moriuchi T, Koya RC, et al. Gelsolin functions as a metastasis suppressor in B16-BL6 mouse melanoma cells and requirement of the carboxyl-terminus for its effect. Int J Cancer. 2001;93(6):773–80. doi: 10.1002/ijc.1413. [DOI] [PubMed] [Google Scholar]
- 165.Nuntharatanapong N, Chen K, Sinhaseni P, Keaney JF., Jr EGF receptor-dependent JNK activation is involved in arsenite-induced p21Cip1/Waf1 upregulation and endothelial apoptosis. Am J Physiol Heart Circ Physiol. 2005;289(1):H99–H107. doi: 10.1152/ajpheart.00901.2004. [DOI] [PubMed] [Google Scholar]
- 166.Gautam A, Bepler G. Suppression of lung tumor formation by the regulatory subunit of ribonucleotide reductase. Cancer Res. 2006;66(13):6497–502. doi: 10.1158/0008-5472.CAN-05-4462. [DOI] [PubMed] [Google Scholar]
- 167.Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, et al. SSeCKS regulates angiogenesis and tight junction formation in blood–brain barrier. Nat Med. 2003;9(7):900–6. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 168.Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, et al. CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm014. [DOI] [PubMed] [Google Scholar]
- 169.Jeschke U, Mylonas I, Shabani N, Kunert-Keil C, Schindlbeck C, Gerber B, et al. Expression of sialyl lewis X, sialyl Lewis A, Ecadherin and cathepsin-D in human breast cancer: immunohistochemical analysis in mammary carcinoma in situ, invasive carcinomas and their lymph node metastasis. Anticancer Res. 2005;25(3A):1615–22. [PubMed] [Google Scholar]
- 170.Kallakury BV, Yang F, Figge J, Smith KE, Kausik SJ, Tacy NJ, et al. Decreased levels of CD44 protein and mRNA in prostate carcinoma. Correlation with tumor grade and ploidy. Cancer. 1996;78(7):1461–9. doi: 10.1002/(sici)1097-0142(19961001)78:7<1461::aid-cncr13>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 171.Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004;64(21):7655–60. doi: 10.1158/0008-5472.CAN-04-1623. [DOI] [PubMed] [Google Scholar]
- 172.Sanchez-Carbayo M, Capodieci P, Cordon-Cardo C. Tumor suppressor role of KiSS-1 in bladder cancer—Loss of KiSS-1 expression is associated with bladder cancer progression and clinical outcome. Am J Pathol. 2003;162(2):609–17. doi: 10.1016/S0002-9440(10)63854-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ikeguchi M, Yamaguchi K, Kaibara N. Clinical significance of the loss of KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10(4):1379–83. doi: 10.1158/1078-0432.ccr-1519-02. [DOI] [PubMed] [Google Scholar]
- 174.Ringel MD, Hardy E, Bernet VJ, Burch HB, Schuppert F, Burman KD, et al. Metastin receptor is overexpressed in papillary thyroid cancer and activates MAP Kinase in thyroid cancer cells. J Clin Endocrinol Metab. 2002;87(5):2399. doi: 10.1210/jcem.87.5.8626. [DOI] [PubMed] [Google Scholar]
- 175.Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF., Jr Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res. 2004;10(11):3800–6. doi: 10.1158/1078-0432.CCR-03-0653. [DOI] [PubMed] [Google Scholar]
- 176.Xia W, Unger P, Miller L, Nelson J, Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 2001;61(14):5644–51. [PubMed] [Google Scholar]